

Abstract
Scope
Green tea extract (GTE) reduces liver steatosis and inflammation during nonalcoholic steatohepatitis (NASH). We hypothesized GTE would mitigate NASH in a nuclear factor erythroid-2-related-factor-2 (Nrf2)-dependent manner in a high fat (HF)-induced model.
Methods and results
Nrf2-null and wild-type (WT) mice were fed a HF diet containing 0 or 2% GTE for 8 wk prior to assessing parameters of NASH. Compared to WT mice, Nrf2-null mice had increased serum alanine aminotransferase, hepatic triglyceride, expression of free fatty acid uptake and lipogenic genes, malondialdehyde, and NFκB phosphorylation and expression of pro-inflammatory genes. In WT mice, GTE increased Nrf2 and NADPH:quinone oxidoreductase-1 mRNA, and lowered hepatic steatosis, lipid uptake and lipogenic gene expression, malondialdehyde, and NFκB-dependent inflammation. In Nrf2-null mice, GTE lowered NFκB phosphorylation and TNFα and MCP1 mRNA to levels observed in WT mice fed GTE whereas hepatic triglyceride and lipogenic genes were lowered only to those of WT mice fed no GTE. Malondialdehyde was lowered in Nrf2-null mice fed GTE, but not to levels of WT mice, and without improving the hepatic antioxidants α-tocopherol, ascorbic acid, and uric acid.
Conclusion
Nrf2 deficiency exacerbates NASH whereas anti-inflammatory and hypolipidemic activities of GTE likely occur largely independent of Nrf2 signaling.
Keywords: green tea, NASH, Nrf2, oxidative stress, inflammation
Graphical abstract
Green tea extract (GTE) protects against nonalcoholic steatohepatitis (NASH) through hypolipidemic and antiinflammatory activities. In wild-type and Nrf2-null mice that were fed a high-fat diet with or without GTE for 8 wk, Nrf2 deficient mice had an exacerbated pathology of NASH including greater lipid accumulation and NFκB-dependent inflammatory responses. GTE mitigated liver steatosis and inflammation in wild-type mice while also increasing Nrf2 and Nqo1 expression. However, GTE also significantly lowered NFκB phosphorylation and liver steatosis in Nrf2-null mice. Thus, GTE provided extensive Nrf2-independent hepatoprotection to limit liver injury and inflammation during NASH.
1 Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disorder in the United States [1]. It is a progressive disorder that advances in severity from liver steatosis to nonalcoholic steatohepatitis (NASH), fibrosis, and cirrhosis to increase hepatocellular carcinoma risk [2]. An estimated 70 million Americans have NAFLD [3], and its prevalence is expected to increase due to its close association with obesity and diabetes [4] and a lack of validated therapies that mitigate its development and progression.
The pathogenesis from steatosis to NASH is often described by the “two-hit” mechanism [5]. The “first-hit”, mediated by obesity, insulin resistance, and dysregulated lipid metabolism, results in excess hepatic lipid accumulation [5]. Consequently, steatotic livers are vulnerable to “second-hits”, mediated by oxidative stress and inflammation, that exacerbate liver injury [5]. The involvement of reactive oxygen species (ROS) during NASH [5] supports a role of nuclear factor erythroid-2-related factor-2 (Nrf2) in limiting oxidative stress that otherwise induces liver injury [6]. Indeed, transcriptional activities of Nrf2 regulate xenobiotic metabolism and antioxidant defenses [6]. They also reduce inflammation and fatty acid synthesis [7-10], suggesting that its cytoprotective activities could attenuate NASH through multiple mechanisms.
Nrf2-dependent hepatoprotection has been demonstrated in several models of NASH. Nrf2-null mice fed a methionine- and choline-deficient (MCD) diet had exacerbated pathology of NASH, including greater expression of pro-inflammatory and fatty acid metabolism genes as well as histological and biochemical markers of liver injury and lipid peroxidation [11]. Conversely, Kelch-like ECH-associated protein 1 (Keap1) knockout mice fed a MCD diet are protected from NASH due to their constitutive activation of Nrf2 [12]. Nrf2-null mice fed a high-fat (HF) diet also show that Nrf2 reduces steatosis and oxidative stress by suppressing lipogenic and cholesterogenic pathways [13]. Nrf2 may also mitigate oxidative stress during NASH by improving redox status, consistent with its role in regulating glutathione biosynthesis and expression of antioxidant defenses (e.g. NADPH:quinone oxidoreductase 1 (Nqo1)) [6].
NASH patients and rodent models of NASH have greater nuclear factor κB (NFκB) activation and expression of pro-inflammatory genes including tumor necrosis factor-α (TNFα) and monocyte chemoattractant protein-1 (MCP-1) [14, 15]. NFκB activation occurs through intracellular and extracellular pathways that converge at the phosphorylation of IκB kinase (IKK), which promotes IκB phosphorylation and nuclear translocation of NFκB [16]. Nrf2 deficiency increases NFκB activation during NASH [17], consistent with evidence from a pro-inflammatory experimental model that Nrf2 activation decreases IKK activity and phosphorylation and degradation of IκBα [18]. Thus, therapies that induce Nrf2 would be expected to attenuate NFκB-dependent pro-inflammatory responses otherwise promoting liver injury during NASH.
High dietary intakes of green tea are associated with a lower risk of liver injury, hyperlipidemia, and inflammation [19]. Although no clinical studies have examined green tea during NASH, green tea extract (GTE) protects against NASH, likely through its major catechin epigallocatechin gallate (EGCG), in genetic and diet-induced models through hypolipidemic, antioxidant, and anti-inflammatory mechanisms [20]. For example, GTE attenuated NASH in genetically obese mice by lowering lipogenic gene expression and reducing lipid peroxidation in association with an upregulation in enzymatic activities of several Nrf2-dependent enzymatic antioxidant defenses that detoxify ROS [21]. In a dietary HF-induced model of NASH, GTE lowered NFκB binding activity by reducing IκBα phosphorylation while also decreasing protein and mRNA levels of TNFα and MCP-1 [22]. Consistent with these findings and evidence that GTE improves endogenous antioxidant defenses under the regulation of Nrf2 [21], we hypothesized in the present study that hepatoprotective activities of GTE during NASH are mediated in a Nrf2-dependent manner. We therefore fed Nrf2-null mice and their wild-type (WT) controls a HF diet containing GTE at 0 or 2%, and then assessed markers of liver injury and oxidative stress, expression of genes involved in fatty acid uptake and lipogenesis, and NFκB-dependent inflammatory responses to define Nrf2-dependent hypolipidemic and anti-inflammatory activities of GTE during NASH.
2 Materials and Methods
2.1 Materials
All solvents were HPLC-grade or higher and were purchased from Thermo Scientific (Waltham, MA) along with the following chemicals: citric acid, EDTA, and PBS. BSA, EGCG, thiobarbituric acid (TBA), Tris-buffered saline with Tween-20 (TBST), and 1,1,3,3-tetramethoxypropane (TPP) were from Sigma (St. Louis, MO).
2.2 In Vitro Study
HC-04 human hepatocytes, a cell line well-suited for biomechanistic studies of xenobiotics [23], were used to examine dose- and time-dependent effects of EGCG, the predominant polyphenolic catechin in green tea [20], on Nrf2 activation and expression. HC-04 cells were obtained from BEI Resources (American Type Culture Collection; Manassas, VA). In all experiments, cells were incubated (5% CO2; 37°C) in low glucose Dulbecco's Modified Eagle Medium (DMEM; Sigma) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate, and 0.25 μg/mL amphotericin B). EGCG at physiologically relevant concentrations (0-5 μM) [20] was dissolved in 0.05% citric acid and all experiments were performed in serum-free DMEM containing 1% antibiotic-antimycotic. Cells were seeded at a density of 4×105 for mRNA expression studies or 8×105 for all other studies and were harvested at specific time points up to 2 h following incubation with EGCG. Nuclear accumulation of Nrf2 protein in response to EGCG (0-5 μM) or the pro-oxidant positive control t-butylhydroperoxide (100 μM) was assessed by Western Blot, as described below, after isolating the nuclear fraction using a kit (Active Motif, Carlsbad, CA). Nrf2 mRNA expression was assessed at 0-5 μM EGCG at 0-2 h post-treatment using RT-PCR, as described below, after total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA). Cytotoxicity was assessed using the lactate dehydrogenase (LDH) assay (Sigma) and the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; Promega, Madison, WI) assay at 2 h post-treatment according to the manufacturers' instructions.
2.3 Mouse Study
The protocol for this study was approved by the Institutional Animal Care and Use Committee (2012A00000156) at The Ohio State University. Homozygous Nrf2-null mice on a C57BL/6 background provided by Dr. Angela Slitt (University of Rhode Island) were bred at University of Connecticut, and then male Nrf2-null mice (n = 23) were supplied to The Ohio State University. Male C57BL6 WT mice (n = 20) were purchased from Charles River Laboratories (Wilmington, MA). All mice were acclimated to the temperature-, light-, and humidity-controlled facility for at least 2 wk until WT and Nrf2-null mice were 11-12 wk old. Nrf2-null and WT mice were housed individually while receiving a HF diet containing powdered GTE at 0 or 2% (w/w) for 8 wk. The HF diet containing 60% of energy from fat (primarily as lard), as detailed previously [22, 24], was formulated to contain 0 or 2% GTE, which was kindly provided by Unilever BestFoods.
Powdered GTE contained 30% total catechins (w/w), which consisted of 48% EGCG, 31% epigallocatechin, 13% epicatechin gallate, and 8% epicatechin as verified by HPLC-UV [25]. GTE at 2% was chosen based on our studies showing that it reduces liver steatosis and NFκB-dependent inflammation in genetic and HF-induced models of NASH [21, 22, 25]. GTE at 2% is also closely equivalent to 10 servings/d (120 mL/serving) in humans [25], a dietary level associated with a lower risk of liver injury in Japanese adults [19]. Mice had free access to food and water throughout the study. Body mass was measured weekly and food intake daily. After the feeding period, mice were sacrificed in the fasted state (10-12 h) under isoflurane. Blood was collected from the retroorbital sinus and centrifuged to obtain serum. Tissues were excised, rinsed in PBS, blotted, and frozen in liquid nitrogen before storing at -80°C. A portion of the central hepatic lobe was collected into formalin for histological assessment or into RNAlater (Sigma) for gene expression studies.
2.4 Liver Histology
Paraffin-embedded liver sections (4-5 μm) were stained with hematoxylin and eosin. Images were captured using an Olympus IX50 microscope from 10 randomly selected fields (200× magnification) to assess NASH using established histologic criteria that score steatosis, hepatocellular ballooning, and lobular inflammation [26]. In brief, steatosis was scored as: grade 0 for no fatty hepatocytes, grade 1 for fatty hepatocytes occupying <33% of the hepatic parenchyma, grade 2 for fatty hepatocytes occupying 33-66% of the hepatic parenchyma, or grade 3 for fatty hepatocytes occupying >66% of the hepatic parenchyma. Hepatocellular ballooning was scored as grade 0 for none, grade 1 for few ballooned cells, and grade 2 for predominant ballooning. Hepatic inflammatory infiltrates were scored as grade 0 for none, grade 1 for <2 foci/field, grade 2 for 2-4 foci/field, or grade 3 for >4 foci/field.
2.5 Lipids and Clinical Assays
Hepatic total lipid was determined gravimetrically [25], and solubilized to determine triglyceride, cholesterol (Pointe Scientific, Canton, MI) and nonesterified fatty acid (NEFA; Wako Diagnostics, Mountain View, CA) using spectrophotometric kits. Serum lipids were determined directly with these kits. Serum glucose was measured by clinical assay (Pointe Scientific) and insulin by ELISA (Crystal Chem, Downers Grove, IL). Serum alanine aminotransferase (ALT) activity was measured by clinical assay (Pointe Scientific).
2.6 RT-PCR
Gene expression studies were performed as described [21], with minor modifications. In brief, total RNA was extracted using TRIzol (Invitrogen). RT-PCR was performed using a SYBR Green PCR Kit, a Bio-Rad CFX384 instrument (Hercules, CA), and primer sequences purchased from Sigma (Table 1). mRNA expression of human Nrf2 in HC-04 cells were quantified relative to β-actin using the 2−ΔΔCT method [27]. For studies in mice, hepatic expression of genes involved in antioxidant defense [Nrf2; Nqo1], lipid metabolism [cluster of differentiation-36 (CD36); sterol regulatory element binding protein-1c (SREBP-1c); fatty acid synthase (FAS); stearoyl-CoA desaturase-1 (SCD1); diglyceride acyltransferase (DGAT1/2)] and inflammation [MCP-1, TNFα, inducible nitric oxide synthase (iNOS); toll-like receptor-4 (TLR4); tumor necrosis factor receptor-1 (TNFR1)] were quantified relative to hypoxanthine-guanine phosphoribosyl transferase (HPRT).
Table 1. Primers used for RT-PCR gene expression studiesa,b.
| Gene | Accession Number | Forward Primer | Reverse Primer |
|---|---|---|---|
| β-actin* | NM_001101 | CCATCGAGCACGGCATC | ATTGTAGAAGGTGTGGTGCAGA |
| CD36 | NM_007643 | CCGAGGACCACACTGTGTC | AACCCCACAAGAGTTCTTTCAAA |
| DGAT1 | NM_010046 | TCCGCCTCTGGGCATTC | GAATCGGCCCACAATCCA |
| DGAT2 | NM_026384 | AGTGGCAATGCTATCATCATCGT | AAGGAATAAGTGGGAACCAGATCA |
| FAS | NM_007988 | GGAGGTGGTGATAGCCGGTAT | TGGGTAATCCATAGAGCCCAG |
| HPRT | NM_013556 | CACAGGACTAGAACACCTGC | GCTGGTGAAAAGGACCTCT |
| iNOS | NM_010927 | TTCTGTGCTGTCCCAGTGAG | TGAAGAAAACCCCTTGTGCT |
| MCP-1 | NM_011333 | TGATCCCAATGAGTAGGCTGGAG | ATGTCTGGACCCATTCCTTCTTG |
| Nrf2* | NM_006164 | GCTCATACTCTTTCCGTCGC | ATCATGATGGACTTGGAGCTG |
| Nrf2 | NM_010902 | CGAGATATACGCAGGAGAGGTAAGA | GCTCGACAATGTTCTCCAGCTT |
| Nqo1 | NM_008706 | TTCTGTGGCTTCCAGGTCTT | AGGCTGCTTGGAGCAAAATA |
| SCD1 | NM_009127 | TTCTTGCGATACACTCTGGTGC | CGGGATTGAATGTTCTTGTCGT |
| SREBP-1c | NM_011480 | GCAGCCACCATCTAGCCTG | CAGCAGTGAGTCTGCCTTGAT |
| TLR4 | NM_021297 | CCTCTGCCTTCACTACAGAGACTTT | TGTGGAAGCCTTCCTGGATG |
| TNFα | NM_013693 | CTCCAGGCGGTGCCTATG | GGGCCATAGAACTGATGAGAGG |
| TNFR1 | NM_011609 | GGGCACCTTTACGGCTTCC | GGTTCTCCTTACAGCCACACA |
All primer sequences are for mice except for human sequences that are annotated by an asterisk.
Abbreviations: CD36, cluster of differentiation-36; DGAT, diglyceride acyltransferase; FAS, fatty acid synthase; HPRT, hypoxanthine-guanine phosphoribosyl transferase; iNOS, inducible nitric oxide synthase; MCP-1, monocyte chemoattractant protein-1; Nrf2, nuclear factor erythroid-2-related factor-2; Nqo1, NAD(P)H dehydrogenase:quinone 1; SCD1, stearoyl-CoA desaturase-1; SREBP-1c, sterol regulatory element binding protein-1c; TLR4, toll-like receptor-4; TNFα, tumor necrosis factor-α; TNFR1, tumor necrosis factor receptor-1
2.7 Western Blotting
Total cellular proteins were extracted from liver using a Pierce T-PER kit containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Proteins were denatured under reducing conditions, separated on a 10% SDS-PAGE, and transferred to a nitrocellulose membrane. For studies in HC-04 cells, membranes were probed overnight with primary antibodies against Nrf2 (#12721; Cell Signaling Technology; Danvers, MA) or TFIIB (#SC-225; Santa Cruz Biotechnology; Dallas, Texas) that were diluted 1:2000 in 2% non-fat dried milk in TBST. For studies in mice, membranes were probed for 1 h at room temperature with primary antibody against β-actin (#4967; Cell Signaling Technology) that was diluted 1:1000 in TBST, or they were probed overnight at 4°C with antibodies against phosphorylated-p65 (phospho-p65; 1:500; #3033) and p65 (1:1000; #8242) prepared in TBST containing 5% (w/v) BSA. After washing in TBST, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (1:1000; #SC-2056; Santa Cruz Biotechnology). Detection was accomplished by enhanced chemiluminescence and densitometry analysis on a Kodak 2000RT imaging station using 1D software.
2.8 Hepatic Antioxidants and Lipid Peroxidation
Hepatic ascorbic acid and uric acid were measured by HPLC as described [25], with minor modification to use an UltiMate 3000 HPLC system (Thermo Scientific) equipped with two 6011RS coulometric cells programmed to 25, 150, 250, and 350 mV. Hepatic α-tocopherol was measured by LC-MS as we described [28] following saponification and hexane extraction. Hepatic malondialdehyde (MDA) was extracted and measured by HPLC-FL as described [21], with minor modifications. In brief, liver was homogenized in lysis buffer (Gold Biotechnology; St. Louis, MO) containing 5 mM DL-dithiothreitol (Sigma), 5 mM EDTA disodium salt, and protease and phosphatase inhibitor cocktail (Thermo Scientific). Following centrifugation, the supernatant was mixed with TBA, incubated, and the butanol extract was injected on a HPLC-FL system set to 532/553 nm (excitation/emission). Separation was performed at 1 mL/min on an Alltima C18 column (250 × 4.6 mm, 5 μm; Columbia, MD) using 50:50 methanol and phosphate buffer (pH 6.5). MDA was quantified from standards prepared in parallel from TPP, and normalized to hepatic protein.
2.9 Statistical Analysis
Data (means ± SEM) were analyzed using GraphPad Prism, version 6 (GraphPad Software; La Jolla, CA). 1-way or 2-way ANOVA was used as appropriate for studies performed in HC-04 cells. For mouse studies, 2-way ANOVA was used to evaluate main effects due to genotype and GTE, and their interaction. Variables with unequal variance were log-transformed to achieve a normal distribution. Newman-Keuls post-hoc test was used to evaluate group mean differences following statistically significant main or interactive effects as appropriate. Groups without a common superscript were statistically significant different at P≤0.05.
3 Results
3.1 EGCG increases nuclear Nrf2 accumulation in vitro and dietary GTE increases hepatic Nrf2 expression
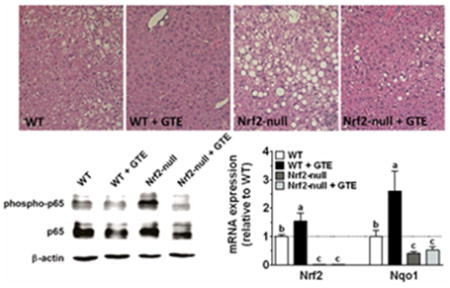
Studies in HC-04 cells were conducted to define the effect of physiologic concentrations of EGCG on nuclear accumulation of Nrf2. Western blotting studies indicated that 2 h treatment with EGCG (0.1-5 μM) increased nuclear accumulation of Nrf2 at all concentrations tested (Figure 1A). This occurred without any time (0-2 h)- or concentration (0-5 μM)-dependent changes in Nrf2 mRNA expression (Figure 1B). Increases in nuclear Nrf2 accumulation by EGCG also occurred without affecting cytotoxicity (Figure 1C-D). Further corroborating, livers of mice chronically fed a HF diet containing GTE showed increased (P<0.05) mRNA expression of Nrf2 and its transcriptional target Nqo1 compared to mice fed the HF diet containing no GTE (Figure 2). Expression of these genes in Nrf2-null mice were lower than WT mice regardless of GTE. Collectively, data support that hepatoprotective activities of green tea observed in rodent models of NASH [20] may be mediated through Nrf2.
Figure 1. Time- and dose-response effects of EGCG on cell cytotoxicity and nuclear accumulation and mRNA expression of Nrf2 in HC-04 human hepatocytes.

HC-04 were treated with 0-5 μM EGCG for up to 2 h as described under Methods. A) Representative Western blot of Nrf2 from isolated nuclear fractions of HC-04 cells following 2 h incubation with EGCG or 100 μM t-butylhydroperoxide (positive control). B) HC-04 cells were treated for up to 2 h with 0.1-5 μM EGCG before isolating total RNA using Trizol and reverse transcribing for RT-PCR analysis of Nrf2 mRNA expression. C) LDH leakage and D) cell viability were assessed using spectrophotometric kits as described under Methods following treatment of HC-04 for 2 h with 0-5 μM EGCG. Shown are mean responses of experiments performed in triplicate. Data were analyzed by 1- or 2-way ANOVA as appropriate. There were no statistically significant time- or concentration-dependent effects of EGCG. Abbreviations: EGCG, epigallocatechin gallate; LDH, lactate dehydrogenase; Nrf2, nuclear factor erythroid-2-related factor-2.
Figure 2. Hepatic mRNA expression of Nrf2 (A) and Nqo1 (B) in Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0 or 2% for 8 wk.

RNA was isolated using Trizol and reverse transcribed for RT-PCR analysis using the primers described in Table 1. Data (means ± SEM, n = 10-12 mice per group) were analyzed by 2-way ANOVA with Newman–Keuls post-test to evaluate main and interactive effects. Groups without a common letter are significantly different (P<0.05). Abbreviations: GTE, green tea extract; Nrf2, nuclear factor erythroid-2-related factor-2; Nqo1, NADPH:quinone oxidoreductase 1; WT, wild-type.
3.2. GTE reduces body mass independent of food intake and Nrf2 status, but reductions in adiposity by GTE are partly Nrf2-dependent
At study onset, body mass was not different between Nrf2-null and WT mice (Table 2). Following the 8 wk feeding period, body mass of Nrf2-null mice was lower than those of WT mice despite no significant difference in food intake (P>0.05). Body masses of WT and Nrf2-null mice fed GTE were lower than those of their respective controls, but without any genotype differences. Liver and adipose mass did not differ between Nrf2-null and WT mice. However, GTE lowered liver mass to a similar extent regardless of genotype, whereas adiposity was lowered by GTE to a greater extent in WT mice compared to Nrf2-null mice (Table 2).
Table 2. Body composition, food intake, and serum and liver metabolic parameters in Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0% or 2% for 8 wka,b,c.
| WT | WT + GTE | Nrf2-null | Nrf2-null + GTE | |
|---|---|---|---|---|
| Initial Body Weight (g) | 26.9 ± 0.4 | 27.0 ± 0.5 | 28.4 ± 0.8 | 28.9 ± 0.6 |
| Final Body Weight (g) | 43.1 ± 0.9a | 29.5 ± 1.1c | 38.8 ± 1.1b | 32.0 ± 1.3c |
| Food Intake (g/d) | 2.9 ± 0.1 | 2.5 ± 0.3 | 2.3 ± 0.1 | 2.4 ± 0.4 |
| Liver Mass (g) | 1.40 ± 0.1a | 1.05 ± 0.0bc | 1.16 ± 0.1ab | 0.87 ± 0.1c |
| Total Adipose Tissue (g) | 3.93 ± 0.2a | 1.03 ± 0.2c | 3.34 ± 0.1a | 2.12 ± 0.3b |
| Hepatic Total Lipid (mg/g liver) | 231.7 ± 16.1b | 156.9 ± 13.4c | 312.5 ± 19.0a | 219.2 ± 17.9b |
| Hepatic Triglyceride (μmol/g liver) | 72.9 ± 10.5b | 34.4 ± 4.0c | 122.7 ± 13.4a | 71.1 ± 10.5b |
| Hepatic Cholesterol (μmol/g liver) | 8.4 ± 0.3b | 8.5 ± 0.4b | 12.3 ± 0.8a | 10.9 ± 1.4a |
| Hepatic NEFA (μmol/g liver) | 34.5 ± 28.3 | 35.4 ± 5.4 | 31.5 ± 3.9 | 34.4 ± 2.2 |
| Serum ALT (U/L) | 28.5 ± 1.6b | 11.0 ± 0.6c | 85.8 ± 13.9a | 23.5 ± 6.9bc |
| Serum Glucose (mmol/L) | 17.6 ± 1.2a | 13.0 ± 0.9b | 15.7 ± 0.6a | 11.4 ± 1.2b |
| Serum Insulin (pmol/L) | 90.2 ± 8.1b | 52.9 ± 7.4c | 123.7 ± 9.2a | 63.8 ± 11.4c |
| Serum NEFA (mmol/L) | 0.94 ± 0.0a | 0.68 ± 0.0b | 0.93 ± 0.1a | 0.93 ± 0.0a |
Total adipose mass represents the sum of epididymal and retroperitoneal fat pads.
Data are means ± SEM, n = 10-12 in each group. Data were analyzed by 2-way ANOVA with Newman-Keuls post-test to assess group mean differences. Means in a row not sharing a common letter are significantly different, P≤0.05.
Abbreviations: ALT, alanine aminotransferase; GTE, green tea extract; HF, NEFA, non-esterified fatty acids; Nrf2, nuclear factor erythroid-2-related factor-2; WT, wild-type.
3.3 GTE reduces hepatic steatosis and injury regardless of Nrf2 deficiency
Histologic evaluation provided clear visual evidence that Nrf2-null mice had exacerbated liver steatosis and hepatocellular ballooning compared to WT mice (Figure 3). Although GTE reduced these parameters in both genotypes, their reduction was more pronounced in WT mice fed GTE in which there was little or no histologic evidence of steatosis or ballooning. In contrast, inflammatory infiltration was minimal regardless of genotype or GTE.
Figure 3. Histologic evaluation of livers from Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0 or 2% for 8 wk.

A) Representative hematoxylin and eosin-stained liver sections (original magnification 200x) in WT and Nrf2-null mice fed a high fat with or without GTE. Nrf2-null mice had exacerbated macrovesicular steatosis and hepatocyte ballooning relative to WT mice. GTE reduced these parameters regardless of genotype, but the effects were more pronounced in WT mice. Histologic scoring of liver steatosis (B), hepatocyte ballooning (C), and inflammatory infiltrates. Data were analyzed by 2-way ANOVA with Newman–Keuls post-test to evaluate main and interactive effects. Groups without a common letter are significantly different (P<0.05). Data are means ± SEM, n = 10-12 mice per group). Abbreviations: GTE, green tea extract; Nrf2, nuclear factor erythroid-2-related factor-2; WT, wild-type.
Hepatic lipid was quantified to corroborate histologic evidence of steatosis (Table 2). A genotype × GTE interaction (P<0.05) indicated that hepatic total lipid was increased in Nrf2-null mice compared with WT mice, but that GTE reduced it to lower levels in WT mice compared to Nrf2-null mice. Higher hepatic total lipid in Nrf2-null mice was attributed to greater hepatic triglyceride and cholesterol (Table 2). However, hepatic cholesterol within each genotype was unaffected by GTE, whereas hepatic triglyceride was lowered by GTE regardless of genotype, and more greatly lowered in WT mice fed GTE. Hepatic NEFA was unaffected by Nrf2 or GTE.
Serum ALT activity, a biomarker of liver injury, was greater in Nrf2-null mice compared to WT mice (Table 2). Serum ALT was lower in WT and Nrf2-null mice fed GTE compared to their respective controls with no difference in ALT between WT and Nrf2-null mice fed GTE. Serum glucose was unaffected by genotype, but was reduced to a similar extent by GTE in both WT and Nrf2-null mice (Table 2). Serum insulin was higher in Nrf2-null mice compared to WT mice. GTE lowered insulin levels in both genotypes relative to respective controls with no differences between WT and Nrf2-null mice fed GTE. Lastly, serum NEFA was unaffected in Nrf2-null mice, but was lowered by GTE in WT mice only (Table 2). Together, a HF diet exacerbates liver steatosis and injury, and insulinemia in Nrf2-null mice, but GTE-mediated improvements in these parameters occurred largely independent of Nrf2 status.
3.4 GTE lowers mRNA expression of hepatic lipid uptake and lipogenic genes regardless of Nrf2 status
To assess hepatic lipid-lowering activities of GTE, expression of genes involved in free fatty acid uptake and lipogenesis were examined (Figure 4). Nrf2-null mice compared with WT mice had increased mRNA of the fatty acid uptake transporter CD36 as well as the lipogenic transcription factor SREBP-1c and its downstream targets FAS and SCD1. Nrf2-null mice also had greater DGAT-1 and -2 mRNA compared with WT mice. Although GTE reduced expression of each gene, GTE lowered their expression to a greater extent in WT mice fed GTE compared to Nrf2-null mice fed GTE with the exception of DGAT-1 and -2, which were reduced to a similar extent by GTE in both genotypes. Thus, liver steatosis in Nrf2-null mice is likely exacerbated by greater hepatic uptake of free fatty acids and lipid synthesis, but GTE likely only provides limited Nrf2-dependent hepatoprotection on these parameters.
Figure 4. Hepatic mRNA expression of genes involved in free fatty acid uptake and lipid synthesis in Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0 or 2% for 8 wk.

RNA was isolated using Trizol and reverse transcribed for RT-PCR analysis using the primers described in Table 1. Data (means ± SEM, n = 10-12 mice per group) were analyzed by 2-way ANOVA with Newman–Keuls post-test to evaluate main and interactive effects. Groups without a common letter are significantly different (P<0.05). Abbreviations: CD36, cluster of differentiation-36; DGAT, diglyceride acyltransferase; FAS, fatty acid synthase; GTE, green tea extract; Nrf2, nuclear factor erythroid-2-related factor-2; SCD1, stearoyl-CoA desaturase-1; SREBP-1c, sterol regulatory element binding protein-1c; WT, wild-type.
3.5 GTE lowers lipid peroxidation otherwise increased by Nrf2 deficiency without improving antioxidant status
Consistent with greater liver steatosis (Figure 2), Nrf2-null mice had greater hepatic MDA compared to WT controls (Figure 5A). Although MDA was significantly lower in both WT and Nrf2-null mice fed GTE compared to their respective controls, MDA in Nrf2-null mice fed GTE remained significantly higher than those of WT mice fed no GTE (Figure 5A). Endogenous and dietary antioxidants known to regulate oxidative stress were then assessed to better define the interaction of GTE and Nrf2 status on hepatic lipid peroxidation. Ascorbic acid, which is devoid from the purified diet because rodents synthesize it, was lower in Nrf2-null mice and unaffected by GTE regardless of genotype (Figure 5B). Uric acid, a byproduct of purine catabolism that has antioxidant activity at physiologic levels [29], was lower in Nrf2-null mice and unaffected by GTE in both WT and Nrf2-null mice (Figure 5C). To the contrary, Nrf2-null mice had greater accumulation of hepatic α-tocopherol compared to WT mice (Figure 5D). Nrf2-null mice fed GTE had α-tocopherol levels lowered to the extent of WT controls fed no GTE, whereas α-tocopherol of WT mice fed GTE was lower than that of WT mice. However, normalization of α-tocopherol to total hepatic lipid resulted in Nrf2-null mice having greater α-tocopherol, but without any effect of GTE in either genotype (data not shown). Collectively, Nrf2 deficiency exacerbates hepatic MDA, but GTE-mediated improvements in lipid peroxidation occur without improving hepatic antioxidants.
Figure 5. Hepatic lipid peroxidation and antioxidant markers in Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0 or 2% for 8 wk.

A) Hepatic MDA was measured by HPLC-FL following incubation of liver homogenates with thiobarbituric acid and extraction with butanol. B-C) Hepatic ascorbic acid and uric acid were measured by HPLC-ECD following deproteinization of liver homogenates. D) Hepatic α-tocopherol was measured by HPLC-ECD following alcoholic saponification and extraction with hexane. Data (means ± SEM, n = 10-12 mice per group) were analyzed by 2-way ANOVA with Newman-Keuls post-test to evaluate main and interactive effects. Groups without a common letter are significantly different (P<0.05). Abbreviations: GTE, green tea extract; MDA, malondialdehyde; Nrf2, nuclear factor erythroid-2-related factor-2; WT, wild-type.
3.6 GTE reduces inflammatory responses otherwise increased by Nrf2 deficiency
NFκB activation increases with oxidative stress and contributes to hepatic injury during NASH [22]. Consistent with GTE reducing NFκB-dependent inflammatory responses during NASH in association with lowered oxidative stress [21, 22], we hypothesized that anti-inflammatory activities of GTE would occur in a Nrf2-dependent manner. Consistent with our hypothesis, Nrf2-null mice had greater protein levels of phospho-p65, a transcriptionally active form of NFκB [30], compared with WT mice (Figure 6). Phospho-p65 levels were lowered in WT mice fed GTE. However, and contrary to our hypothesis, GTE also lowered phospho-p65 levels in Nrf2-null mice to the extent observed in WT mice fed GTE. These decreases in phospho-p65 occurred without any Nrf2- or GTE-mediated alterations in total p65 protein (Figure 6).
Figure 6. Hepatic protein of the phosphorylated p65 subunit of NFκB or total p65 in Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0 or 2% for 8 wk.

A) Representative Western blot of phospho-p65 and p65 and the loading control β-actin. B) Quantitative densitometry analysis of target protein accumulation in total cellular extracts. Data (means ± SEM, n = 10-12 mice per group) were analyzed by 2-way ANOVA with Newman-Keuls post-test to evaluate main and interactive effects. Groups without a common letter are significantly different (P<0.05). Abbreviations: GTE, green tea extract; Nrf2, nuclear factor erythroid-2-related factor-2; phospho-p65; phosphorylated p65 subunit of NFκB; WT, wild-type.
To corroborate the opposing activities of Nrf2 status and GTE on NFκB activation, expression of NFκB-dependent target genes implicated in NASH were assessed. Consistent with Nrf2-null mice having increased phospho-p65 protein, they also had greater TNFα, MCP-1, and iNOS mRNA compared with WT mice (Figure 7). GTE lowered mRNA levels of each of these pro-inflammatory targets regardless of genotype. However, iNOS mRNA in Nrf2-null mice fed GTE was only lowered to the extent observed in WT mice, whereas TNFα and MCP-1 were lowered to levels not different from WT mice fed GTE. Thus, while Nrf2 deficiency increases NFκB-dependent inflammation, anti-inflammatory activities of GTE during NASH appear to be mediated largely in a Nrf2-independent manner. Consistent with this notion, we considered an alternative hypothesis that anti-inflammatory activities of GTE could be mediated by cellular receptors that trigger NFκB activation during NASH, specifically TNFR1 and TLR4 [31, 32]. Indeed, hepatic mRNA expression of both TNFR1 and TLR4 was higher in Nrf2-null mice compared with WT mice (Figure 8). GTE reduced TNFR1 and TLR4 mRNA expression regardless of genotype with no difference in mRNA levels between genotypes.
Figure 7. Hepatic mRNA expression of pro-inflammatory genes in Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0 or 2% for 8 wk.

RNA was isolated using Trizol and reverse transcribed for RT-PCR analysis for the NFκB-dependent targets TNFα, MCP-1, and iNOS using the primers described in Table 1. Data (means ± SEM, n = 10-12 mice per group) were analyzed by 2-way ANOVA with Newman-Keuls post-test to evaluate main and interactive effects. Groups without a common letter are significantly different (P<0.05). Abbreviations: GTE, green tea extract; iNOS, inducible nitric oxide synthase; MCP-1, monocyte chemoattractant protein-1; Nrf2, nuclear factor erythroid-2-related factor-2; TNFα, tumor necrosis factor-α: phospho-p65; WT, wild-type.
Figure 8. Hepatic mRNA expression of TNFR1 and TLR4 in Nrf2-null and wild-type mice fed a high-fat diet containing GTE at 0 or 2% for 8 wk.

RNA was isolated using Trizol and reverse transcribed for RT-PCR analysis for TNFR1 and TLR4 using the primers described in Table 1. Data (means ± SEM, n = 10-12 mice per group) were analyzed by 2-way ANOVA with Newman-Keuls post-test to evaluate main and interactive effects. Groups without a common letter are significantly different (P<0.05). Abbreviations: GTE, green tea extract; Nrf2, nuclear factor erythroid-2-related factor-2; TLR4, toll-like receptor 4; TNFR1, tumor necrosis factor receptor-1; WT, wild-type.
4 Discussion
Consistent with our hypothesis, and the work of others [6], Nrf2 deficient mice fed a HF diet had exacerbated NASH pathology as evidenced by greater NFκB-dependent pro-inflammatory responses, along with increased hepatic lipid peroxidation, liver steatosis, and expression of hepatic free fatty acid uptake and lipogenic genes. Contrary to our hypothesis, and despite evidence suggesting Nrf2-mediated hepatoprotective activities of GTE during NASH [21, 22] that are likely mediated through EGCG [20]. Our data suggest that GTE provides limited Nrf2-dependent protection against liver steatosis and injury, lipogenic gene expression, and activation of NFκB and expression of its pro-inflammatory mediators. In addition, although GTE lowers hepatic lipid peroxidation regardless of Nrf2 status, this occurred without any GTE-mediated improvements in hepatic antioxidants in Nrf2-null mice. Collectively, anti-inflammatory and hypolipidemic activities of GTE during NASH likely occur largely through a mechanism independent of Nrf2 activation.
High dietary green tea intakes (≥10 servings/d) are associated with lower biomarker levels of liver injury, inflammation, and hyperlipidemia [19]. In agreement, evidence in genetic and diet-induced rodent models of NASH support that dietary GTE or its principal catechin EGCG reduces liver steatosis and injury, insulin resistance, oxidative stress, and inflammation [20]. Although the mechanism underlying these health benefits remains unclear, hepatoprotective mechanisms of GTE likely occur through both Nrf2-independent and -dependent mechanisms. For example, EGCG reduces intestinal lipid absorption by inhibiting pancreatic phospholipase A2 activity [33] and may mitigate insulin resistance by reducing postprandial glucose resulting from starch ingestion by inhibiting α-amylase activity [34]. In contrast, studies in ob/ob mice and rats fed a HF diet support a Nrf2-dependent protective mechanism consistent with GTE upregulating enzymatic activities of antioxidant defenses under the transcriptional control of Nrf2 (e.g. glutathione S-transferase, glutathione peroxidase, catalase) [21]. GTE also reduced NFκB activation and expression of TNFα and MCP-1 that were otherwise increased by a HF diet, and their lowering occurred in association with improved glutathione status [22], consistent with studies in ob/ob mice showing that GTE increases the expression of Nrf2-dependent genes regulating glutathione biosynthesis [21].
Supporting our hypothesis that GTE would protect against NASH in a Nrf2-dependent manner, we show that physiologically relevant concentrations (0.1-5 μM) of EGCG [20], a prominent constituent of GTE, increase nuclear protein accumulation of Nrf2 in HC-04 cells in agreement with studies in vitro and in rodent models indicating EGCG-mediated activation of Nrf2-dependent targets that detoxify ROS [35]. This cytoprotective response is thought to be mediated through the electrophilic potential of EGCG [35], which is readily auto-oxidized at near-neutral pH similar to that occurring during digestion, and absorbed by Caco-2 cells [36]. The EGCG electrophile is then expected to promote oxidative modification of cysteine residues on Keap1. This induces conformation changes of the Keap1-Nrf2 complex to suppress Nrf2 degradation by limiting ubiquitination, and facilitates nuclear translocation of newly synthesized Nrf2 where it binds to antioxidant response elements (ARE) to regulate the expression of antioxidant and xenobiotic defenses [37, 38]. Consistent with these findings, we show that GTE in WT mice fed a HF diet, but not in Nrf2-null mice, increases mRNA expression of Nrf2 and its downstream target gene Nqo1, which assists in lowering oxidative stress by maintaining cellular antioxidants in their reduced form, scavenging superoxide, and catalyzing reductive metabolism of xenobiotics [6]. Although numerous antioxidant defenses largely regulated by Nrf2 are also regulated by other transcription factors [39], the present study aimed to define the extent to which antioxidant activities of GTE protect against NASH in a Nrf2-dependent manner by lowering NFκB-dependent inflammation and hepatic lipid accumulation, which have been shown to be attenuated by dietary GTE or EGCG in obese models of NASH [21, 22, 24, 25, 40, 41].
NFκB can be activated in a redox-dependent manner by oxidative stress. In agreement, Nrf2-null mice fed a MCD diet to induce NASH have greater lipid peroxidation along with nuclear accumulation of the p65 subunit of NFκB and mRNA expression of its pro-inflammatory mediators [17]. In our study, livers of Nrf2-null mice fed a HF diet have increased phospho-p65 protein and TNFα, MCP-1, and iNOS mRNA expression along with higher MDA compared to WT controls. GTE lowered hepatic NFκB binding activity in rats fed a HF diet [22]. This is corroborated in our study by GTE-mediated decreases in phospho-p65 protein accumulation and TNFα and MCP-1 mRNA expression in WT mice, consistent with EGCG reducing circulating MCP-1 in mice fed a HF diet [24]. However, these inflammatory responses in Nrf2-null mice fed GTE were also lowered, and to levels no different from WT mice fed GTE, suggesting that anti-inflammatory activities of GTE occur in a Nrf2-independent manner. Furthermore, the hepatic antioxidants ascorbic acid and uric acid, a byproduct of purine metabolism known to be regulated by Nrf2 [42], were also lowered by Nrf2 deficiency, and otherwise unaffected by GTE regardless of Nrf2 status.
Insulin resistance is an early event leading to NASH [5], although direct evidence also shows that TNFα-mediated inflammation accelerates the induction of liver steatosis in mice [43]. In agreement with others [44], and the present evidence that Nrf2 deficiency increases NFκB-mediated inflammation, we also show that Nrf2-null mice have exacerbated histologic and biochemical markers of liver steatosis compared to WT mice. Greater liver steatosis in Nrf2-null mice is consistent with a mechanism of enhanced fatty acid uptake by CD36 and greater lipid synthesis mediated by SREBP1c and its downstream lipogenic genes FAS and SCD1 that are upregulated in association with hyperinsulinemia. However, these Nrf2-dependent activities that likely promote hepatic steatosis occurred without affecting hepatic NEFA, which may reflect their rapid esterification to form triglyceride that is mediated by the observed increase in DGAT expression and needed to protect against free fatty acid-induced hepatic lipotoxicity [45].
Consistent with our studies showing GTE-mediated improvements in liver steatosis in association with lowered serum insulin and expression of lipogenic genes [21, 40], the present study shows that GTE reduced liver steatosis regardless of Nrf2 deficiency. Although biochemical measures of hepatic total lipid and triglyceride in Nrf2-null mice fed GTE were lowered compared to Nrf2-null controls, they were not lowered to the extent observed in WT mice fed GTE. This suggests that a modest proportion of the hypolipidemic activities of GTE occur in a Nrf2-dependent manner. The observed lipid-lowering activity is corroborated by our findings that GTE in Nrf2-null mice lowered the expression of CD36 and lipogenic genes (i.e. SREBP1c, FAS, SCD-1) only to the extent observed in WT controls, but not WT mice fed GTE. Although this study was not designed to define events downstream of Nrf2 activation responsible for these hypolipidemic activities, nor could it isolate the anti-inflammatory activities of GTE from its hepatic lipid-lowering activities, Nrf2 maintains energy metabolism by regulating genes for lipid biosynthesis and mitochondrial biogenesis [46] and warrants further investigation. Likewise, consideration for examining Nrf1-dependent hepatoprotection is warranted consistent with Nrf1 transcriptional activities similarly mediated through its binding to ARE to regulate lipid metabolism [47]. However, Nrf1 limits steady-state cellular stress whereas Nrf2 provides secondary defense against unscheduled stress [47], and may have a role in mitigating NASH. Indeed, studies in liver-specific Nrf1 knockout mice show that Nrf1 regulates hepatic lipid metabolism [48] consistent with Nrf1 regulating the expression of PPARα and PPARγ coactivators [49]. Thus, GTE may attenuate NASH through Nrf1 consistent with EGCG reducing liver steatosis in HF-fed mice while upregulating skeletal muscle mRNA expression of Nrf1 and fatty acid oxidation genes [50].
Inflammation contributes to NASH consistent with NASH patients and rodents models of NASH having greater NFκB activation [14, 22, 51]. Although our findings implicate Nrf2 deficiency in increasing NFκB-dependent inflammation, they also suggest that anti-inflammatory activities of GTE on NFκB activation occur independent of Nrf2 responses that suppress ROS. Indeed, HF feeding is known to induce Nrf2 cytoprotective responses that limit ROS accumulation [52]. Thus, GTE may be limited in further upregulating Nrf2-dependent defenses, and supports examining Nrf2-dependent protection by GTE in other NASH models, consistent with the flavone baicalein, a Nrf2 activator, suppressing liver steatosis and inflammation in mice fed a MCD diet [53].
Mechanisms other than Nrf2-mediated alterations in intracellular redox status, such as receptor-mediated pathways, are also known to regulate NFκB activation during NASH [54]. Greater inflammation in NASH patients is associated with small intestinal bacteria overgrowth and greater TLR4 expression on CD14 positive cells that is correlated to plasma IL-8 [31]. Alternatively, studies in TNFR1 knockout mice [32], TNFR1 knockin mice that are unable to down-regulate TNFα-dependent inflammation [55], or treatment with anti-TNFα antibodies [56] support that inhibiting TNFR signaling attenuates NASH. Consistent with these lines of study, we show that hepatic TNFR1 and TLR4 expression that are otherwise increased by Nrf2 deficiency are normalized by GTE in Nrf2-null mice to the extent observed in WT mice fed GTE. Thus, these receptor-mediated pathways warrant consideration to better define anti-inflammatory activities of GTE associated with lowered hepatic TNFα by GTE in obese models of NASH [21, 22] as well as studies showing that EGCG suppresses TLR4 signaling in LPS-stimulated adipocytes [57] and reduces hepatic TLR4 protein in a mouse model of viral hepatitis [58]. Evidence also supports that polyphenols favorably improve the intestinal microbiota composition to mitigate inflammatory-related disorders [59].
In conclusion, GTE protects against HF-induced liver injury and NASH by lowering hepatic lipid accumulation and NFκB-dependent inflammation. Suppression of NFκB-dependent inflammation by GTE is likely independent of Nrf2 signaling and these anti-inflammatory activities may contribute, at least in part, to its hepatic lipid-lowering activities. Identifying the mechanism by which GTE mitigates NFκB-dependent inflammation, such as that mediated through TNFR1 and/or TLR4, warrants further investigation. Furthermore, it is likely that pleotropic effects of GTE may protect against NASH through multiple mechanisms, consistent with EGCG regulating lipid metabolism by activating PPARα and inhibiting PPARγ [60]. Regardless of the mechanism, green tea is among the most commonly used dietary supplements, it is considered safe when ingested as recommended [61], and it or EGCG may provide therapeutic potential in protecting obese individuals from NASH.
Acknowledgments
J Li, TN Sapper, JE Manautou and RS Bruno designed the study; J Li, TN Sapper, E Mah, KE Schill, C Chitchumroonchokchai, MV Moller, JD McDonald, PR Rohrer, S Rudraiah, and RS Bruno conducted experiments and analyzed data; J Li and RS Bruno wrote the manuscript. All authors read, provided substantial intellectual input, and approved the final manuscript.
This work was supported by grants to RSB from USDA-NIFA (2014-67017-21761), The Ohio State University (OSU) Food Innovation Center, the Molecular Carcinogenesis and Chemoprevention Program of the OSU Comprehensive Cancer (National Institutes of Health, National Cancer Institute P30CA16058) and the OSU Center for Advanced Functional Foods Research and Entrepreneurship as well as state and federal funds appropriated to the OSU Ohio Agricultural Research and Development Center.
Abbreviations
- ANOVA
analysis of variance
- CD-36
cluster of differentiation-36
- DGAT
diglyceride acyltransferase
- DMEM
Dulbecco's Modified Eagle Medium
- EGCG
epigallocatechin gallate
- FAS
fatty acid synthase
- GTE
green tea extract
- HPRT
hypoxanthine-guanine phosphoribosyl transferase
- IKK
IκB kinase
- iNOS
inducible nitric oxide synthase
- Keap1
Kelch-like ECH-associated protein 1
- LDH
lactate dehydrogenase
- MCD
methionine choline deficient
- MCP-1
monocyte chemoattractant protein-1
- MDA
malondialdehyde
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NEFA
nonesterified fatty acid
- NFκB
nuclear factor κB
- Nqo1, NADPH
quinone oxidoreductase 1
- Nrf2
nuclear factor erythroid-2-related factor-2
- phospho-p65
phosphorylated-p65
- ROS
reactive oxygen species
- SCD1
stearoyl-CoA desaturase-1
- SREBP-1c
sterol regulatory element binding protein-1c
- TBA
thiobarbituric acid
- TBST
Tris-buffered saline with Tween-20
- TLR4
toll-like receptor-4
- TPP
1,1,3,3-tetramethoxypropane
- TNFα
tumor necrosis factor-α
- TNFR1
tumor necrosis factor receptor-1
- WT
wild-type
Footnotes
The authors have declared no conflict of interest.
References
- 1.Sass DA, Chang P, Chopra KB. Nonalcoholic fatty liver disease: a clinical review. Dig Dis Sci. 2005;50:171–180. doi: 10.1007/s10620-005-1267-z. [DOI] [PubMed] [Google Scholar]
- 2.Wong VW, Wong GL, Choi PC, Chan AW, et al. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59:969–974. doi: 10.1136/gut.2009.205088. [DOI] [PubMed] [Google Scholar]
- 3.Angulo P. Obesity and nonalcoholic fatty liver disease. Nutr Rev. 2007;65:S57–63. doi: 10.1111/j.1753-4887.2007.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 4.Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology. 1990;12:1106–1110. doi: 10.1002/hep.1840120505. [DOI] [PubMed] [Google Scholar]
- 5.Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:663–678. doi: 10.1053/bega.2002.0333. [DOI] [PubMed] [Google Scholar]
- 6.Bataille AM, Manautou JE. Nrf2: a potential target for new therapeutics in liver disease. Clin Pharmacol Ther. 2012;92:340–348. doi: 10.1038/clpt.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy NM, Potteti HR, Mariani TJ, Biswal S, Reddy SP. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am J Respir Cell Mol Biol. 2011;45:1161–1168. doi: 10.1165/rcmb.2011-0144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yates MS, Tran QT, Dolan PM, Osburn WO, et al. Genetic versus chemoprotective activation of Nrf2 signaling: overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis. 2009;30:1024–1031. doi: 10.1093/carcin/bgp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu KC, Cui JY, Klaassen CD. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci. 2011;123:590–600. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitteringham NR, Abdullah A, Walsh J, Randle L, et al. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J Proteomics. 2010;73:1612–1631. doi: 10.1016/j.jprot.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugimoto H, Okada K, Shoda J, Warabi E, et al. Deletion of nuclear factor-E2-related factor-2 leads to rapid onset and progression of nutritional steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2010;298:G283–294. doi: 10.1152/ajpgi.00296.2009. [DOI] [PubMed] [Google Scholar]
- 12.Zhang YK, Yeager RL, Tanaka Y, Klaassen CD. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol Appl Pharmacol. 2010;245:326–334. doi: 10.1016/j.taap.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, et al. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J Pharmacol Exp Ther. 2008;325:655–664. doi: 10.1124/jpet.107.135822. [DOI] [PubMed] [Google Scholar]
- 14.Videla LA, Tapia G, Rodrigo R, Pettinelli P, et al. Liver NF-kappaB and AP-1 DNA binding in obese patients. Obesity (Silver Spring) 2009;17:973–979. doi: 10.1038/oby.2008.601. [DOI] [PubMed] [Google Scholar]
- 15.Dela Pena A, Leclercq I, Field J, George J, et al. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663–1674. doi: 10.1053/j.gastro.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdhry S, Nazmy MH, Meakin PJ, Dinkova-Kostova AT, et al. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic Biol Med. 2010;48:357–371. doi: 10.1016/j.freeradbiomed.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai K, Nakachi K. Cross sectional study of effects of drinking green tea on cardiovascular and liver diseases. BMJ. 1995;310:693–696. doi: 10.1136/bmj.310.6981.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masterjohn C, Bruno RS. Therapeutic potential of green tea in nonalcoholic fatty liver disease. Nutr Rev. 2012;70:41–56. doi: 10.1111/j.1753-4887.2011.00440.x. [DOI] [PubMed] [Google Scholar]
- 21.Park HJ, DiNatale DA, Chung MY, Park YK, et al. Green tea extract attenuates hepatic steatosis by decreasing adipose lipogenesis and enhancing hepatic antioxidant defenses in ob/ob mice. J Nutr Biochem. 2011;22:393–400. doi: 10.1016/j.jnutbio.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Park HJ, Lee JY, Chung MY, Park YK, et al. Green tea extract suppresses NFkappaB activation and inflammatory responses in diet-induced obese rats with nonalcoholic steatohepatitis. J Nutr. 2012;142:57–63. doi: 10.3945/jn.111.148544. [DOI] [PubMed] [Google Scholar]
- 23.Lim PL, Tan W, Latchoumycandane C, Mok WC, et al. Molecular and functional characterization of drug-metabolizing enzymes and transporter expression in the novel spontaneously immortalized human hepatocyte line HC-04. Toxicol In Vitro. 2007;21:1390–1401. doi: 10.1016/j.tiv.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Bose M, Lambert JD, Ju J, Reuhl KR, et al. The major green tea polyphenol, (-)-epigallocatechin-3-gallate, inhibits obesity, metabolic syndrome, and fatty liver disease in high-fat-fed mice. J Nutr. 2008;138:1677–1683. doi: 10.1093/jn/138.9.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruno RS, Dugan CE, Smyth JA, DiNatale DA, Koo SI. Green tea extract protects leptin-deficient, spontaneously obese mice from hepatic steatosis and injury. J Nutr. 2008;138:323–331. doi: 10.1093/jn/138.2.323. [DOI] [PubMed] [Google Scholar]
- 26.Brunt EM, Kleiner DE, Wilson LA, Belt P, et al. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53:810–820. doi: 10.1002/hep.24127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Mah E, Pei R, Guo Y, Ballard KD, et al. gamma-Tocopherol-rich supplementation additively improves vascular endothelial function during smoking cessation. Free Radic Biol Med. 2013;65:1291–1299. doi: 10.1016/j.freeradbiomed.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 29.Bruno RS, Ramakrishnan R, Montine TJ, Bray TM, Traber MG. {alpha}-Tocopherol disappearance is faster in cigarette smokers and is inversely related to their ascorbic acid status. Am J Clin Nutr. 2005;81:95–103. doi: 10.1093/ajcn/81.1.95. [DOI] [PubMed] [Google Scholar]
- 30.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Shanab AA, Scully P, Crosbie O, Buckley M, et al. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci. 2011;56:1524–1534. doi: 10.1007/s10620-010-1447-3. [DOI] [PubMed] [Google Scholar]
- 32.Kanuri G, Spruss A, Wagnerberger S, Bischoff SC, Bergheim I. Role of tumor necrosis factor alpha (TNFalpha) in the onset of fructose-induced nonalcoholic fatty liver disease in mice. J Nutr Biochem. 2011;22:527–534. doi: 10.1016/j.jnutbio.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Koo SI, Noh SK. Green tea as inhibitor of the intestinal absorption of lipids: potential mechanism for its lipid-lowering effect. J Nutr Biochem. 2007;18:179–183. doi: 10.1016/j.jnutbio.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forester SC, Gu Y, Lambert JD. Inhibition of starch digestion by the green tea polyphenol, (-)-epigallocatechin-3-gallate. Mol Nutr Food Res. 2012;56:1647–1654. doi: 10.1002/mnfr.201200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Na HK, Surh YJ. Modulation of Nrf2-mediated antioxidant and detoxifying enzyme induction by the green tea polyphenol EGCG. Food Chem Toxicol. 2008;46:1271–1278. doi: 10.1016/j.fct.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Neilson AP, Song BJ, Sapper TN, Bomser JA, Ferruzzi MG. Tea catechin auto-oxidation dimers are accumulated and retained by Caco-2 human intestinal cells. Nutr Res. 2010;30:327–340. doi: 10.1016/j.nutres.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 38.Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des. 2003;9:2499–2511. doi: 10.2174/1381612033453730. [DOI] [PubMed] [Google Scholar]
- 40.Chung MY, Mah E, Masterjohn C, Noh SK, et al. Green Tea Lowers Hepatic COX-2 and Prostaglandin E2 in Rats with Dietary Fat-Induced Nonalcoholic Steatohepatitis. J Med Food. 2014 doi: 10.1089/jmf.2014.0048. [DOI] [PubMed] [Google Scholar]
- 41.Chen YK, Cheung C, Reuhl KR, Liu AB, et al. Effects of green tea polyphenol (-)-epigallocatechin-3-gallate on newly developed high-fat/Western-style diet-induced obesity and metabolic syndrome in mice. J Agric Food Chem. 2011;59:11862–11871. doi: 10.1021/jf2029016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayes JD, Ashford ML. Nrf2 orchestrates fuel partitioning for cell proliferation. Cell Metab. 2012;16:139–141. doi: 10.1016/j.cmet.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 43.Endo M, Masaki T, Seike M, Yoshimatsu H. TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c) Exp Biol Med (Maywood) 2007;232:614–621. [PubMed] [Google Scholar]
- 44.Chambel SS, Santos-Goncalves A, Duarte TL. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. Biomed Res Int. 2015;2015:597134. doi: 10.1155/2015/597134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamaguchi K, Yang L, McCall S, Huang J, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 46.Vomhof-Dekrey EE, Picklo MJ., Sr The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J Nutr Biochem. 2012;23:1201–1206. doi: 10.1016/j.jnutbio.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, et al. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283:33554–33562. doi: 10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsujita T, Peirce V, Baird L, Matsuyama Y, et al. Transcription factor Nrf1 negatively regulates the cystine/glutamate transporter and lipid-metabolizing enzymes. Mol Cell Biol. 2014;34:3800–3816. doi: 10.1128/MCB.00110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirotsu Y, Hataya N, Katsuoka F, Yamamoto M. NF-E2-related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the Lipin1 and PGC-1beta genes. Mol Cell Biol. 2012;32:2760–2770. doi: 10.1128/MCB.06706-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sae-Tan S, Grove KA, Kennett MJ, Lambert JD. (-)-Epigallocatechin-3-gallate increases the expression of genes related to fat oxidation in the skeletal muscle of high fat-fed mice. Food Funct. 2011;2:111–116. doi: 10.1039/C0FO00155D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Velayudham A, Dolganiuc A, Ellis M, Petrasek J, et al. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology. 2009;49:989–997. doi: 10.1002/hep.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, et al. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol. 2014;34:3305–3320. doi: 10.1128/MCB.00677-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xin HG, Zhang BB, Wu ZQ, Hang XF, et al. Treatment with baicalein attenuates methionine-choline deficient diet-induced non-alcoholic steatohepatitis in rats. Eur J Pharmacol. 2014;738:310–318. doi: 10.1016/j.ejphar.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 54.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aparicio-Vergara M, Hommelberg PP, Schreurs M, Gruben N, et al. Tumor necrosis factor receptor 1 gain-of-function mutation aggravates nonalcoholic fatty liver disease but does not cause insulin resistance in a murine model. Hepatology. 2013;57:566–576. doi: 10.1002/hep.26046. [DOI] [PubMed] [Google Scholar]
- 56.Li Z, Yang S, Lin H, Huang J, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology. 2003;37:343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 57.Bao S, Cao Y, Zhou H, Sun X, et al. Epigallocatechin gallate (EGCG) suppresses lipopolysaccharide-induced Toll-like receptor 4 (TLR4) activity via 67 kDa laminin receptor (67LR) in 3T3-L1 adipocytes. J Agric Food Chem. 2015;63:2811–2819. doi: 10.1021/jf505531w. [DOI] [PubMed] [Google Scholar]
- 58.Liu D, Zhang X, Jiang L, Guo Y, Zheng C. Epigallocatechin-3-gallate (EGCG) attenuates concanavalin A-induced hepatic injury in mice. Acta Histochem. 2014;116:654–662. doi: 10.1016/j.acthis.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Marchesi JR, Adams DH, Fava F, Hermes GD, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2015 doi: 10.1136/gutjnl-2015-309990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee SJ, Jia Y. The effect of bioactive compounds in tea on lipid metabolism and obesity through regulation of peroxisome proliferator-activated receptors. Curr Opin Lipidol. 2015;26:3–9. doi: 10.1097/MOL.0000000000000145. [DOI] [PubMed] [Google Scholar]
- 61.Sarma DN, Barrett ML, Chavez ML, Gardiner P, et al. Safety of green tea extracts : a systematic review by the US Pharmacopeia. Drug Saf. 2008;31:469–484. doi: 10.2165/00002018-200831060-00003. [DOI] [PubMed] [Google Scholar]