

Abstract
Anticonvulsant drugs, when given during vulnerable periods of brain development, can have long-lasting consequences on nervous system function. In rats, the second postnatal week approximately corresponds to the late third trimester of gestation/early infancy in humans. Exposure to phenobarbital during this period has been associated with deficits in learning and memory, anxiety-like behavior, and social behavior, among other domains. Phenobarbital is the most common anticonvulsant drug used in neonatology. Several other drugs, such as lamotrigine, phenytoin, and clonazepam have also been reported to trigger behavioral changes. A new generation anticonvulsant drug, retigabine, has not previously been evaluated for long-term effects on behavior. Retigabine acts as an activator of KCNQ channels, a mechanism that is unique among anticonvulsants. Here, we examined the effects retigabine exposure from postnatal day (P)7 to P14 on behavior in adult rats. We compared these effects to those produced by phenobarbital (as a positive control) and saline (as a negative control). Motor behavior was assessed using the open field and rotarod, anxiety-like behavior by the open field, elevated plus maze, and light-dark transition task, and learning/memory by the passive avoidance task; social interactions were assessed in same-treatment pairs and nociceptive sensitivity was assessed via the tail-flick assay. Motor behavior was unaltered by exposure to either drug. We found that retigabine and phenobarbital exposure both induced increased anxiety-like behavior in adult animals. Phenobarbital, but not retigabine exposure impaired learning and memory. These drugs also differed in their effects on social behavior, with retigabine-exposed animals displaying greater social interaction than phenobarbital-exposed animals. These results indicate that neonatal retigabine induces a subset of behavioral alterations previously described for other anticonvulsant drugs, and extend our knowledge of drug-induced behavioral teratogenesis to a new mechanism of anticonvulsant action.
Keywords: development, neonatal, epilepsy, antiepileptic drug, anxiety, memory, anticonvulsant, pregnancy, rat
1. Introduction
Exposure to neuroactive drugs during critical periods of brain development may have long-lasting consequences. This is of particular concern for drugs used to treat epilepsy [1], one of the most common neurological conditions of infancy [2]. Similarly, the treatment of pregnant women with epilepsy results in an appreciable population of infants who are exposed in utero to anticonvulsant drugs [3]. A now substantial body of clinical [4–11] and preclinical literature [12–21] shows that these exposures can have long-lasting effects on brain structure and function.
Acute exposure to phenobarbital, one of the most commonly utilized anticonvulsants world-wide and the most common treatment for neonatal seizures, induces a profound increase in the number of apoptotic neurons in the developing (postnatal day [P]7) rat brain [12,15,21]. These effects are not limited to phenobarbital: phenytoin, the prototypical voltage-gated sodium channel blocker [12,15,20]; lamotrigine, a newer generation sodium channel blocker [21]; benzodiazepines [12], and anesthetic agents [22–24] have all been reported to trigger apoptosis under the right conditions/doses.
In addition to the excessive pruning of neurons, phenobarbital, phenytoin, and lamotrigine trigger a lasting derangement of synaptogenesis in the striatum, with a failure of both excitatory and inhibitory synaptic transmission to develop appropriately after even a single (acute) exposure [14]. Moreover, phenobarbital has been reported to cause long-term alterations in the cortical proteome [18]. Perhaps most importantly, many anticonvulsant drugs (phenobarbital, phenytoin, lamotrigine, clonazepam) have been shown to cause short and/or long-term alterations in a variety of behavioral domains with exposures as brief as one day [14,16,25–29].
Other anticonvulsant drugs, such as levetiracetam, topiramate, and carbamazepine, which have a benign profile with respect to neuronal apoptosis [15,20,30], remain to be evaluated for behavioral teratogenesis. Here, we turn our attention to retigabine. Retigabine is currently labeled as an adjunctive therapy in adults who have failed on several alternative treatments. This a first-in-class anticonvulsant acts as a positive allosteric modulator of KCNQ channels [31,32]. These channels mediate the M-type potassium current, resulting in neuronal hyperpolarization. Retigabine shifts the activation voltage of these channels towards more negative membrane potentials. Several KCNQ channel mutations have been associated with benign familial neonatal convulsions,[33] raising additional interest in retigabine during brain development.
We have previously reported that retigabine is an effective anticonvulsant drug in neonatal rats, acting at doses ranging from 5 to 30 mg/kg [34]. Moreover, we have reported that retigabine, when administered repeatedly over the course of 24 h triggers apoptosis in a subset of vulnerable brain regions (Brown et al., In Press). This profile, while more benign than what was seen with phenobarbital or phenytoin, for example, raises an obvious question: will retigabine induce long-lasting changes in behavioral function?
To address this question, we exposed neonatal (P7 to P14) rats to retigabine, phenobarbital (as a positive control), or vehicle (as a negative control) and examined their behavior as adults. We examined behavioral domains that we and others have previously shown to be sensitive to anticonvulsant-induced behavioral teratogenesis [13,16,25,26], including anxiety-like behavior, learning and memory, motor function, and social behavior beginning at P45.
2. Materials and Methods
2.1 Animals
Male Sprague-Dawley rats were used for these studies. Treatments were counterbalanced within and across litters. Two separate cohorts of animals were treated, spaced by several months. Pups were born to timed-pregnant dams (Harlan, Indianapolis, IN, U.S.A.) with P0 designated as the date of parturition. Animals were maintained in a temperature-controlled (21°C) room with a 12-h light cycle, with food and water available ad libitum. Pups were treated as described below, and weaned to same-treatment cages on P21. All experiments were approved by the Georgetown University Animal Care and Use Committee and conducted in accordance with the Guide for the Care and Use of Laboratory Animals [35].
2.2 Drug treatments
Drug treatments were administered intraperitoneally at a volume of 0.01 ml/g. Control rats received equivalent volumes of saline vehicle (0.01 ml/g body weight). Pups were treated on P7, 8, 9, 11, 12, and 13 as we have done previously [16]. For drug treatments, pups were briefly removed from their dam, weighed, labeled, injected and returned.
2.3 Drug solutions
Retigabine (ezogabine; ethyl N-[2-amino-4-[(4-fluorophenyl)methylamino]phenyl]carbamate) was provided by GlaxoSmithKline (Research Triangle, NC) and dissolved in sterile water containing 0.1% tween-20. Retigabine was given at a dose of 30 mg/kg. Phenobarbital (Sigma-Aldrich) was dissolved in 0.9% saline and given at a dose of 75 mg/kg.
2.4 Selection of drug doses
The dose of retigabine (30 mg/kg) was selected because it falls at the upper end of the effective anticonvulsant range in neonatal rats [34]. We selected a high dose to best test the hypothesis would induce behavioral alterations. The dose of phenobarbital was selected on the basis of prior reports from our group [36,37] and others [38] showing the efficacy of phenobarbital in P7 rats. This dose provides almost complete suppression of pentylenetetrazole-induced seizures in neonatal rats and falls above the range for complete blockade of forebrain seizures induced by methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate in P7 rats [37,38].
2.5.1 Behavioral Testing
Prior to each test day, animals were transported from the vivarium to the testing room. Animals were allowed a minimum of 30 min to acclimate to the testing environment prior to initiating testing. Test apparatus were wiped with 70% ethanol solution between each animal. Testing was conducted in the order specified below.
2.5.2 Behavioral Testing - Open Field Test
Another test measuring anxiety, the open field test measures a rat’s natural drive to explore novel environments against their propensity to remain in the periphery of a brightly lit environment [39]. This test also serves as a measure of general locomotor activity. As we have previously described [16,40], each animal was placed in a 40cm by 40cm open field box (TruScan Arena) for thirty minutes. They were filmed and their movements recorded via ANY-maze, measuring the amount of time each animal spent within 5cm of the perimeter of the open field versus in the middle (as a measure of anxiety-like behavior) and the total distance traveled in the apparatus (as a measure of overall activity).
2.5.3 Behavioral Testing - Elevated Plus Maze
The elevated plus maze is a widely used test on rodents to measure anxiety-like behavior, pitting the natural inclination of rats to explore against their aversion to open, elevated spaces [41]. Testing was conducted as we have previously described [16,25,42] Rats were placed one at a time on a standard Stoelting Co. plus maze 40cm off the ground, with 50cm long arms. Testing took place in a dark room under red 20-lux light. Two arms of the maze (opposite from each other) had walls approximately one foot high, whereas the other two arms were not covered. Each rat was placed at the center of the maze, facing an open arm, and allowed to explore freely for five minutes. Animals were filmed, and the amount of time each animal spent in the open versus closed arms of the maze was recorded and analyzed using ANY-maze software.
2.5.4 Behavioral Testing - Light/Dark Transition Task
As with the elevated plus maze, this task pits rats innate aversion to bright areas against their natural drive to explore in response to mild stressors such as a novel environment [43]. Animals were placed into a testing apparatus (Coulbourn Instruments) that was half open and half covered by a black box with an opening for animals to enter. Ambient illumination of the room was 770 lux. Animals were initially placed in the light side of the apparatus facing the door to the “dark” chamber and filmed for five minutes. Latency for each animal to initially cross into the dark side of the testing apparatus, as well as total amount of time spent in the dark part of the box and total crossovers between the light and dark sides were scored. Video was recorded via ANY-maze and hand-scored by an observer blinded to treatment status of the animals.
2.5.5 Behavioral Testing - Rotarod
This task measures the motor coordination and learning of rats by placing them on an accelerating rod and measuring the latency to fall from the rod. Testing was conducted as we have previously described using an accelerating rotarod (Accuscan Instruments) [16]. Animals were placed on the stationary rod facing the rear wall of the apparatus. When the device was turned on, the central rod began to move in the opposite direction than the animals were facing, accelerating from 0 to 45 rpm over the course of five minutes. Latency to fall was hand-recorded by observers. Each animal was tested on total of five consecutive trials with at least three minutes of rest time in between.
2.5.6 - One-trial step-through Passive Avoidance learning
This task examines hippocampal-dependent learning [44] in animals by delivering an aversive stimulus (foot shock) in one area of a chamber and measuring the latency of crossing from one area of the chamber to the other over multiple consecutive days. Passive avoidance conditioning and testing was performed as we have previously described [26] using a two-chamber passive avoidance apparatus (Coulbourn) consisting of a lit chamber and a dark chamber separated by a guillotine door. On the first day (habituation) animals were allowed to freely explore the lit chamber for 180 sec with the door to the dark side closed to prevent entry. One the second day (conditioning) animals were placed into the lit chamber and allowed to explore for 30 sec, after which the guillotine door was lifted allowing access to the dark chamber. When the animal crossed into the dark chamber, the door was lowered to prevent re-entry into the lit chamber, and a mild foot shock was delivered (0.4 mA, 2 sec duration). Animals were removed from the dark side within 30 sec of the conclusion of the conditioning trial. On the third day (Retention Probe) animals were placed in the light side of the apparatus, and after 30 sec the door was lifted allowing access to the dark side. Latency to enter the dark chamber was the dependent variable. If an animal did not enter the dark side within 300 sec of the door opening, the trial was terminated and a latency of 300 sec was assigned.
2.5.7 Behavioral Testing - Tail Flick
As a control for nociceptive sensitivity, we measured tail flick latency. This task places the tails of rats under a heat lamp and measuring the latency at which rats remove their tails from this uncomfortable stimulus. Animals were wrapped and immobilized in a cloth so that only their tails were unrestrained. Their tails were then placed under a heat lamp of a standard tail-flick apparatus and timed to how long it took for each animal to move its tail away from the heat.
2.5.8 Behavioral Testing - Social Behavior
Rats are highly social animals, and spend appreciable amounts of time grooming and interacting with one another when pair housed. Social interaction between novel conspecifics has also been used as a measure of anxiety-like behavior in rodents. Here, we monitored the interactions (grooming, climbing, sniffing or otherwise touching) of same-treatment pairs of rats (housed in different cages, so they were unfamiliar). Animals were observed in a familiar environment, a 40cm by 40cm open-field (TruScan Arena), over the twenty-minute test. Social behavior was hand-scored.
2.6 - Statistical Analysis
Graphs were generated and statistical analyses were performed via GraphPad Prism (GraphPad Software, Inc.). Outlier analysis (ROUT procedure) resulted in two animals being excluded from the vehicle and retigabine group and five animals being excluded from the phenobarbital group; outliers were excluded from all analyses. It is worth noting that two of the outliers in the phenobarbital treated group were the only animals in the study to lose weight during the initial treatment, providing an additional strong justification for excluding them. Data not meeting the assumptions of normality were analyzed using Kruskal-Wallis test with Dunn’s correction for multiple comparisons. Data meeting the assumptions of normality were analyzed by analysis of variance (ANOVA) with Holm-Sidak’s correction for multiple comparisons. P values less than 0.05 (two-tailed) were considered to be statistically significant.
3. Results
3.1 Neonatal anticonvulsant exposure results in reduced weight gain during the treatment period
As shown in Figure 1, both phenobarbital and retigabine treatment during the second postnatal week suppressed weight gain by pups. Figure 1A shows weights prior to each treatment, whereas Figure 1B shows the percent body weight gained from P7 to P13. Vehicle-treated pups gained an average of 15.2 g over the course of the second postnatal week, while retigabine and phenobarbital treated animals gained an average of 9.9 and 7.4 g, respectively over the same time period. ANOVA revealed a significant main effect of postnatal day (F5,290=753.2, P<0.0001), a significant main effect of treatment (F2,58=8.7, P=0.0005) and a significant day-by-treatment interaction (F10,290=31.1, P<0.0001). Holm-Sidak post-hoc tests showed that group differences (control vs each treatment) reached the level of significance on P9 (P=0.016), P11 (Ps<0.0005) and P12-P13 (Ps<0.0001). Percent weight gain also differed significantly across groups (F2,58=58.87, P<0.0001), with both the phenobarbital and retigabine groups showing significantly (P<0.0001) less weight gain than vehicle-treated control animals.
Figure 1.

Anticonvulsant exposure results in reduced weight gain during the treatment period
(A) Mean + SEM body weight (in grams) for groups exposed to vehicle, phenobarbital (PHB) or retigabine (RTG) on P7, P8, P9, P11, P12, and P13. * indicates significantly (P<0.05) lower weight as compared to control on the same measurement day. Body weights were measured immediately prior to daily treatment. (B) Percent increase in body weight from P7 to P13. * indicates significantly (P<0.05) less weight gain as compared to vehicle treated controls.
3.2 Neonatal anticonvulsant exposure results in decreased center exploration, without altering total activity, in the open field task
As shown in Figure 2A, all groups displayed the typical pattern of habituation of activity within the open field over the course of the 30 min test session. Consistent with this, there was a main effect of time (F29,1740=29.71, P<0.001) on locomotor activity. However, there was neither a main effect of drug treatment (F2,60=0.56, P=0.57) nor a time-by-drug interaction (F58,1740=0.89, P=0.70). Thus, neonatal exposure to phenobarbital or retigabine were without effect on adult locomotor activity in the open field.
Figure 2.

Neonatal anticonvulsant exposure results in decreased center exploration, without altering total activity, in the open field task.
(A) Distance traveled in the open field as a function of time. Error bars = +/− SEM (B) Time spent in the center of the open field as a function of treatment. Bars show mean + SEM. * indicates significantly different than control (P<0.05). Decreased time in the center of the open field is consistent with increased anxiety-like behavior.
By contrast, when we examined the total time spent in the center of the arena, a commonly utilized indicator of anxiety-like behavior, we found that groups significantly differed (Figure 2B). Control animals spent a mean 109 sec in the center of the arena, while retigabine and phenobarbital exposed groups spent 73 and 44 sec, respectively. ANOVA revealed a significant effect of drug treatment (F2,60=3.8, P=0.027), with planned comparisons demonstrating that this reached the level of significance for phenobarbital, as compared to control (P=0.019). The retigabine treated group only approached the level of significance (P=0.080).
3.3 Neonatal anticonvulsant drug exposure spares motor performance and motor learning in the rotarod task
Peak performance (defined as the best performance across the five trials) did not differ as a function of treatment (Mean+SEM for VEH: 141+10.7, PB: 139+15, RTG: 133+14). Data were first analyzed by two-way ANOVA which revealed no significant effects or interactions (Ps>0.3). Following this analysis we examined peak performance (as in a prior study [16]) and also failed to find an effect of treatment (F2,57=0.08, P=0.92).
3.4. Neonatal phenobarbital but not retigabine increases adult anxiety-like behavior in the elevated plus maze
To follow up on our finding of decreased center exploration in the open field, we examined performance in the elevated plus maze. We found that phenobarbital, but not retigabine exposed animals displayed behavior consistent with increased anxiety. As shown in Figure 3A, phenobarbital-treated animals spent significantly less time in the open arms than the vehicle treated group (Kruskal Wallis: H=6.84, P=0.033; Dunn’s multiple comparison test P=0.018). While both the number of entries to the open arms, and the total arm entries were numerically lower in the phenobarbital treated groups (Figure 3B, 3C), this did not reach the level of statistical significance (Kruskal-Wallis: H=0.84, P=0.67 and H=3.8, P=0.14, respectively).
Figure 3.

Neonatal phenobarbital, but not retigabine, increases adult anxiety-like behavior in the elevated plus maze.
(A) Mean + SEM time spent in the open arms of the elevated plus maze. (B) Mean + SEM number of entries to the open arms. (C) Mean + SEM total number of arm entries. * indicates significantly different than vehicle (P<0.05). Decreased time in the open arms is consistent with increased anxiety-like behavior.
3.5 Neonatal anticonvulsant treatment increases anxiety-like behavior in adults tested in the light-dark transition task
While both phenobarbital- and retigabine-treated animals showed increased anxiety-like behavior in both the open field and elevated plus maze tasks, retigabine-exposed animals only displayed this profile in the open field task. To provide a third measure of anxiety-like behavior, we next evaluated animals on the light-dark transition task. As shown in Figure 4A and B, neither the latency to enter the dark chamber, nor the total number of transitions differed between groups (F2,58=1.06, P=0.35; H=4.8, P=0.09, respectively), although the latter measure approached the level of statistical significance. A measure that takes into account both latency and transitions is “dwell time”, or average duration of a visit to the dark chamber. As shown in Figure 4C, average visit duration to the dark chamber was increased in the phenobarbital and retigabine-exposed groups (F2,58=3.8, P=0.028; Holm-Sidak multiple comparison tests, Ps<0.05).
Figure 4.

Neonatal anticonvulsant treatment increases anxiety-like behavior in adults tested in the light-dark transition task.
(A) Mean + SEM of latency to enter the dark chamber. (B) Mean + SEM of number of transitions between the light and dark chambers. (C) Mean + SEM of average duration of a visit to the dark zone. * indicates significantly different than vehicle treated controls (P<0.05). Increased duration of dark-zone visit is consistent with increased anxiety-like behavior.
3.6. Neonatal phenobarbital, but not retigabine, exposure impairs adult learning/memory
To determine if neonatal exposure to phenobarbital or retigabine would alter adult learning and memory, we examined performance on the step-through passive avoidance task. As shown in Figure 5, latency to enter the dark chamber did not differ between groups on the pre-conditioning test. On the post-conditioning test, all groups showed a significant (Ps<0.0001 for vehicle and retigabine, P=0.03 for phenobarbital) increase in latency to enter the dark (shock-paired) chamber. However, the magnitude of this increase was significantly lower for phenobarbital treated animals as compared to vehicle treated animals (P<0.0012). These effects were revealed by ANOVA which showed main effects of treatment (F2,58=4.9, P=0.01), stage (F1,58=80.80, P<0.0001), and a treatment-by-stage interaction (F2,58=4.1, P=0.02). The planned comparison results reported above (treatment across stage and treatment within stage) were corrected using the Holm-Sidak approach. To rule out differential nociceptive sensitivity as a contributor to this effect, we also examined tail flick latency and found no difference across groups (H=0.08, P=0.96).
Figure 5.
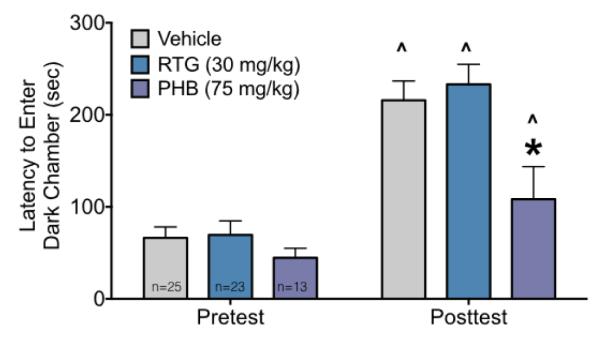
Neonatal phenobarbital, but not retigabine, exposure impairs adult learning/memory.
Latencies to enter the dark chamber during the pretest (left) and post conditioning session (right) as a function of treatment. Bars show mean + SEM. ^ indicates significantly increased latency as compared to the pretest session, * indicates significantly shorter latency than vehicle treated controls.
3.7 Social Interaction
Finally, we examined social interactions in same-treatment pairs of rats. We found a high rate of social interaction in all dyads tested as shown in Figure 6. We found an effect of treatment (F2,32=4.03, P=0.027), however, the only group difference we detected was between retigabine and phenobarbital (P=0.02). Numerically, retigabine-exposed dyads spent more time interacting than vehicle, whereas phenobarbital exposed dyads spent less time interacting than vehicle. This test was less powered than the others in this manuscript as each dyad was treated as an experimental unit.
Figure 6.
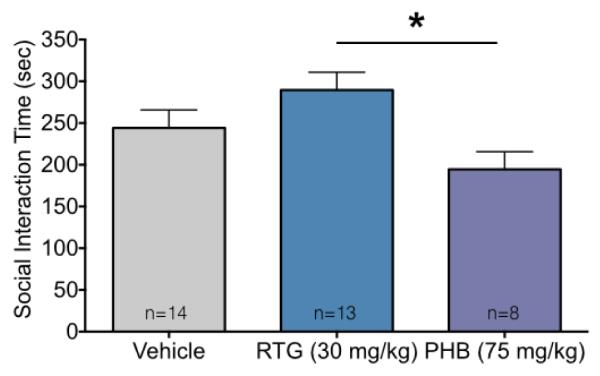
Adult social interactions in dyads after early life anticonvulsant drug exposure.
Dyadic social interaction time (mean + SEM). * indicates significantly difference between retigabine (RTG) and phenobarbital (PHB). Despite differences between these drug-exposed groups, neither group differed significantly from the vehicle control group.
4. Discussion
Here we have found that neonatal exposure to the new-generation anticonvulsant drug, retigabine, induced a long-lasting alteration in anxiety-like behavior in rats. This effect mirrored that seen with phenobarbital. Retigabine, as compared to phenobarbital, spared other behavioral domains, showing no effect on learning/memory function. Neither drug produced alterations in overall activity or motor learning. Finally, these drugs produced opposite effects on social behavior, with retigabine-exposed animals displaying increased social interactions as compared to phenobarbital-exposed animals. These data fit in with a growing literature regarding long-term behavioral teratogenesis after early life anticonvulsant drug exposure [14,16,25,28,29].
Our results with phenobarbital with respect to anxiety-like behavior were somewhat surprising to us; we had previously reported increased exploration of the open arms of the elevated plus maze after one week of phenobarbital exposure [16], a phenotype consistent with decreased anxiety-like behavior. In another report [25], a single dose of phenobarbital was without effect on anxiety-like behavior. How can we reconcile these findings? One salient difference between the present experiment and our previous experiments was the fact that for the present study, pups arrived at our animal facility on P5-6, rather than being born in the facility. It is possible that the pups and/or dams were subject to additional stress from shipping and that this interacted with drug treatment. Indeed, early life stress and/or disruption in maternal care during critical periods have been associated with increased anxiety-like behavior in adulthood.[45,46] Our finding that retigabine increased anxiety-like behavior in two of the three tasks (open field and light-dark, but not the elevated plus maze) suggests a subtler effect of early-life retigabine on conflict-exploratory behavior and/or anxiety state as compared to that of phenobarbital.
We found that phenobarbital, but not retigabine, induced deficits in learning/memory in the passive avoidance task. This effect of phenobarbital is consistent with another recent study from our group [26]. This task, which classically is thought to probe hippocampal-dependent memory processes also engages frontal cortex. Indeed, deficits in either frontal or hippocampal function can result in impaired performance on this task [44,47]. Because both KCNQ2 and KCNQ3 channels are expressed in the developing hippocampus [48,49] and transcript and protein are present in both frontal cortex and hippocampus of rodents and humans [50,51], we had hypothesized that retigabine exposure might impact behavior mediated by these circuits. However, despite the widespread expression of KCNQ2/3 in both frontal and temporal structures, neonatal retigabine exposure was without effect on passive avoidance behavior.
We have recently reported that retigabine exposure (15 mg/kg, 3 times over 24 h) induces neuronal apoptosis in P7 rat pups [52]. This profile was similar to that seen (and previously reported) for phenobarbital [12], but impacted only a subset of vulnerable regions, including dorsolateral thalamus, cingulate cortex, motor cortex, retrosplenial cortex, and somatosensory cortex. While the degree to which enhanced neuronal apoptosis caused by anticonvulsant compounds is predictive of later-in-life behavioral changes, it is worth noting that early life damage to the thalamus has been associated with increased anxiety-like behaviors in rats [53]. While induction of apoptosis in limbic targets has not been examined for retigabine, we have previously reported that phenobarbital induced profound neuronal apoptosis in the amygdala, another region closely associated with the regulation of anxiety-like behavior in rodents [15]. Similarly, hippocampal apoptosis has been reported after phenobarbital exposure [12,21] (but is unexamined after retigabine exposure), which may contribute to the phenobarbital-induced deficits in passive avoidance we reported here. Additional alterations in hippocampus may also contribute to the behavioral deficits reported, indeed, rodents exposed neonatally to phenobarbital show persistent decreases in hippocampal volume [54], decreased cholinesterase activity [55], increased muscarinic receptor expression [56], decreased levels of norepinephrine [57], and decreased dendritic complexity [57,58].
One limitation to the direct comparison of phenobarbital and retigabine in the present study is their differing pharmacokinetic profiles. Phenobarbital has been reported to have a half-life of ~16 h in neonatal mice [59], and a range of 9 to 20 h has been reported in adult rats [60]. By contrast, retigabine has a half-life in the range of 2 h; thus levels of phenobarbital in the present study likely remained elevated substantially longer than those of retigabine. It is interesting to note, that despite the rather large difference in half-life, both groups of treated animals displayed increased anxiety behavior in adulthood. Moreover, both groups also displayed reduced weight gain during the period of treatment. Thus, while the duration of exposure to phenobarbital may have been effectively longer than that of retigabine, our data indicate that even the once-daily dosing with retigabine was sufficient to impact nervous system development. The degree to which multiple-time per day dosing of retigabine (designed to match the half life of phenobarbital) would result in more profound behavioral changes remains to be explored.
5. Conclusions
Both retigabine and phenobarbital produced long-lasting effects on behavior after a confined period of exposure during the second postnatal week. While the pattern of behavioral changes in some cases differed between these drugs, both induced an increased profile of anxiety-like behavior in adult rats after neonatal treatment. Neonatal phenobarbital, but not retigabine impaired adult learning and memory function. While recent concerns regarding abnormal pigmentation following retigabine use have limited its clinical use [61], these data show that in some respects (e.g., learning/memory function) it has a more benign profile than phenobarbital, whereas in other respects (e.g., anxiety-like behavior) it has a similar profile of behavioral teratogenesis. In addition, the present findings extend our knowledge of behavioral teratogenesis after anticonvulsant drugs to a new class, KCNQ activators, which may be relevant if and when additional drugs of this class reach the market.
Highlights.
-
-
Neonatal phenobarbital exposure increased adult anxiety-like behavior in rats
-
-
Neonatal retigabine exposure increased adult anxiety-like behavior in rats
-
-
Neonatal phenobarbital, but not retigabine, exposure impaired adult learning/memory
-
-
Neonatal phenobarbital decreases and retigabine increases adult social behavior
Acknowledgements
This work was supported by a research grant from GlaxoSmithKline to AK and PAF. PAF also received support from HD046388. We thank Colin Soper and Isabelle Orozco for assistance with treating and testing animals, respectively.
Footnotes
Author contributions
PAF and AK designed the study
SF, NM, SG, CK performed experiments
PAF and SF analyzed data
PAF and SF wrote the manuscript, which was edited by NM, SG, CK, and AK.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. References
- [1].Turski CA, Ikonomidou C. Neuropathological sequelae of developmental exposure to antiepileptic and anesthetic drugs. Front Neur. 2012;3:120. doi: 10.3389/fneur.2012.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hauser WA. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35(Suppl 2):S1–6. doi: 10.1111/j.1528-1157.1994.tb05932.x. [DOI] [PubMed] [Google Scholar]
- [3].Gedzelman E, Meador K. Neurological and psychiatric sequelae of developmental exposure to antiepileptic drugs. Front Neur. 2012;3:182. doi: 10.3389/fneur.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ikonomidou C, Scheer I, Wilhelm T, Juengling FD, Titze K, Stöver B, et al. Brain morphology alterations in the basal ganglia and the hypothalamus following prenatal exposure to antiepileptic drugs. Eur J Paediatr Neurol. 2007;11:297–301. doi: 10.1016/j.ejpn.2007.02.006. [DOI] [PubMed] [Google Scholar]
- [5].Meador KJ, Baker GA, Browning N, Cohen MJ, Clayton-Smith J, Kalayjian LA, et al. Foetal antiepileptic drug exposure and verbal versus non-verbal abilities at three years of age. Brain. 2011;134:396–404. doi: 10.1093/brain/awq352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Meador KJ, Baker GA, Browning N, Clayton-Smith J, Combs-Cantrell DT, Cohen M, et al. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N Engl J Med. 2009;360:1597–605. doi: 10.1056/NEJMoa0803531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Meador KJ, Penovich P, Baker GA, Pennell PB, Bromfield E, Pack A, et al. Antiepileptic drug use in women of childbearing age. Epilepsy Behav. 2009;15:339–43. doi: 10.1016/j.yebeh.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McVearry KM, Gaillard WD, VanMeter J, Meador KJ. A prospective study of cognitive fluency and originality in children exposed in utero to carbamazepine, lamotrigine, or valproate monotherapy. Epilepsy Behav. 2009;16:609–16. doi: 10.1016/j.yebeh.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pennell PB, Klein AM, Browning N, Baker GA, Clayton-Smith J, Kalayjian LA, et al. Differential effects of antiepileptic drugs on neonatal outcomes. Epilepsy Behav. 2012;24:449–56. doi: 10.1016/j.yebeh.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Farwell JR, Lee YJ, Hirtz DG, Sulzbacher SI, Ellenberg JH, Nelson KB. Phenobarbital for febrile seizures--effects on intelligence and on seizure recurrence. N Engl J Med. 1990;322:364–9. doi: 10.1056/NEJM199002083220604. [DOI] [PubMed] [Google Scholar]
- [11].Sulzbacher S, Farwell JR, Temkin N, Lu AS, Hirtz DG. Late cognitive effects of early treatment with phenobarbital. Clin Pediatr (Phila) 1999;38:387–94. doi: 10.1177/000992289903800702. [DOI] [PubMed] [Google Scholar]
- [12].Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U S A. 2002;99:15089–94. doi: 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Forcelli PA, Janssen MJ, Stamps LA, Sweeney C, Vicini S, Gale K. Therapeutic strategies to avoid long-term adverse outcomes of neonatal antiepileptic drug exposure. Epilepsia. 2010;51(Suppl 3):18–23. doi: 10.1111/j.1528-1167.2010.02603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Forcelli PA, Janssen MJ, Vicini S, Gale K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann Neurol. 2012;72:363–72. doi: 10.1002/ana.23600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Forcelli PA, Kim J, Kondratyev A, Gale K. Pattern of antiepileptic drug-induced cell death in limbic regions of the neonatal rat brain. Epilepsia. 2011;52:e207–11. doi: 10.1111/j.1528-1167.2011.03297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Forcelli PA, Kozlowski R, Snyder C, Kondratyev A, Gale K. Effects of neonatal antiepileptic drug exposure on cognitive, emotional, and motor function in adult rats. J Pharmacol Exp Ther. 2012;340:558–66. doi: 10.1124/jpet.111.188862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bittigau P, Sifringer M, Ikonomidou C. Antiepileptic drugs and apoptosis in the developing brain. Ann N Y Acad Sci. 2003;993:103–14. doi: 10.1111/j.1749-6632.2003.tb07517.x. discussion 123–4. [DOI] [PubMed] [Google Scholar]
- [18].Kaindl AM, Koppelstaetter A, Nebrich G, Stuwe J, Sifringer M, Zabel C, et al. Brief alteration of NMDA or GABAA receptor-mediated neurotransmission has long term effects on the developing cerebral cortex. Mol Cell Proteomics. 2008;7:2293–310. doi: 10.1074/mcp.M800030-MCP200. [DOI] [PubMed] [Google Scholar]
- [19].Kim J, Kondratyev A, Tomita Y, Gale K. Neurodevelopmental impact of antiepileptic drugs and seizures in the immature brain. Epilepsia. 2007;48(Suppl 5):19–26. doi: 10.1111/j.1528-1167.2007.01285.x. [DOI] [PubMed] [Google Scholar]
- [20].Kim J, Kondratyev A, Gale K. Antiepileptic drug-induced neuronal cell death in the immature brain: effects of carbamazepine, topiramate, and levetiracetam as monotherapy versus polytherapy. J Pharmacol Exp Ther. 2007;323:165–73. doi: 10.1124/jpet.107.126250. [DOI] [PubMed] [Google Scholar]
- [21].Katz I, Kim J, Gale K, Kondratyev A. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322:494–500. doi: 10.1124/jpet.107.123133. [DOI] [PubMed] [Google Scholar]
- [22].Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- [23].Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Young C, Jevtovic-Todorovic V, Qin Y-Q, Tenkova T, Wang H, Labruyere J, et al. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146:189–97. doi: 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bhardwaj SK, Forcelli PA, Palchik G, Gale K, Srivastava LK, Kondratyev A. Neonatal exposure to phenobarbital potentiates schizophrenia-like behavioral outcomes in the rat. Neuropharmacology. 2012;62:2337–45. doi: 10.1016/j.neuropharm.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gutherz SB, Kulick CV, Soper C, Kondratyev A, Gale K, Forcelli PA. Brief postnatal exposure to phenobarbital impairs passive avoidance learning and sensorimotor gating in rats. Epilepsy Behav. 2014;37:265–9. doi: 10.1016/j.yebeh.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Stefovska VG, Uckermann O, Czuczwar M, Smitka M, Czuczwar P, Kis J, et al. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann Neurol. 2008;64:434–45. doi: 10.1002/ana.21463. [DOI] [PubMed] [Google Scholar]
- [28].Mikulecká A, Subrt M, Pařízková M, Mareš P, Kubová H. Consequences of early postnatal benzodiazepines exposure in rats. II. Social behavior. Front Behav Neurosci. 2014;8:169. doi: 10.3389/fnbeh.2014.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mikulecká A, Subrt M, Stuchlík A, Kubová H. Consequences of early postnatal benzodiazepines exposure in rats. I. Cognitive-like behavior. Front Behav Neurosci. 2014;8:101. doi: 10.3389/fnbeh.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Glier C, Dzietko M, Bittigau P, Jarosz B, Korobowicz E, Ikonomidou C. Therapeutic doses of topiramate are not toxic to the developing rat brain. Exp Neurol. 2004;187:403–9. doi: 10.1016/j.expneurol.2004.01.025. [DOI] [PubMed] [Google Scholar]
- [31].Rundfeldt C. The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur J Pharmacol. 1997;336:243–9. doi: 10.1016/s0014-2999(97)01249-1. [DOI] [PubMed] [Google Scholar]
- [32].Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–45. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–9. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- [34].Forcelli PA, Soper C, Lakhkar A, Gale K, Kondratyev A. Anticonvulsant effect of retigabine during postnatal development in rats. Epilepsy Res. 2012;101:135–40. doi: 10.1016/j.eplepsyres.2012.03.006. [DOI] [PubMed] [Google Scholar]
- [35].National Research Council (U.S.) Institute for Laboratory Animal Research (U.S.) National Academies Press (U.S.) Guide for the care and use of laboratory animals. 8th ed. National Academies Press; Washington, D.C: 2011. [PubMed] [Google Scholar]
- [36].Forcelli PA, Soper C, Duckles A, Gale K, Kondratyev A. Melatonin potentiates the anticonvulsant action of phenobarbital in neonatal rats. Epilepsy Res. 2013;107:217–23. doi: 10.1016/j.eplepsyres.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kulick CV, Gutherz SB, Beck VC, Medvedeva N, Soper C, Forcelli PA. Profile of anticonvulsant action of levetiracetam, tiagabine and phenobarbital against seizures evoked by DMCM (methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate) in neonatal rats. Eur J Pharmacol. 2014;743:63–8. doi: 10.1016/j.ejphar.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kubova H, Mares P. Anticonvulsant effects of phenobarbital and primidone during ontogenesis in rats. Epilepsy Res. 1991;10:148–55. doi: 10.1016/0920-1211(91)90007-3. [DOI] [PubMed] [Google Scholar]
- [39].Crawley JN. Exploratory behavior models of anxiety in mice. Neurosci Biobehav Rev. 1985;9:37–44. doi: 10.1016/0149-7634(85)90030-2. [DOI] [PubMed] [Google Scholar]
- [40].Forcelli PA, Turner JR, Lee BG, Olson TT, Xie T, Xiao Y, et al. Anxiolytic- and antidepressant-like effects of the methadone metabolite 2-ethyl-5-methyl-3,3-diphenyl-1-pyrroline (EMDP) Neuropharmacology. 2015;101:46–56. doi: 10.1016/j.neuropharm.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pellow S, Chopin P, File SE, Briley M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods. 1985;14:149–67. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- [42].Forcelli PA, Heinrichs SC. Teratogenic effects of maternal antidepressant exposure on neural substrates of drug-seeking behavior in offspring. Addict Biol. 2008;13:52–62. doi: 10.1111/j.1369-1600.2007.00078.x. [DOI] [PubMed] [Google Scholar]
- [43].Crawley J, Goodwin FK. Preliminary report of a simple animal behavior model for the anxiolytic effects of benzodiazepines. Pharmacol Biochem Behav. 1980;13:167–70. doi: 10.1016/0091-3057(80)90067-2. [DOI] [PubMed] [Google Scholar]
- [44].Isaacson RL, Wickelgren WO. Hippocampal ablation and passive avoidance. Science. 1962;138:1104–6. doi: 10.1126/science.138.3545.1104. [DOI] [PubMed] [Google Scholar]
- [45].Wang X-D, Labermaier C, Holsboer F, Wurst W, Deussing JM, Müller MB, et al. Early-life stress-induced anxiety-related behavior in adult mice partially requires forebrain corticotropin-releasing hormone receptor 1. Eur J Neurosci. 2012;36:2360–7. doi: 10.1111/j.1460-9568.2012.08148.x. [DOI] [PubMed] [Google Scholar]
- [46].Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–8. doi: 10.1126/science.286.5442.1155. [DOI] [PubMed] [Google Scholar]
- [47].Brennan JF, Wisniewski C. The efficacy of response prevention on avoidance behavior in young and adult rats with prefrontal cortical injury. Behav Brain Res. 1982;4:117–31. doi: 10.1016/0166-4328(82)90068-7. [DOI] [PubMed] [Google Scholar]
- [48].Weber YG, Geiger J, Kämpchen K, Landwehrmeyer B, Sommer C, Lerche H. Immunohistochemical analysis of KCNQ2 potassium channels in adult and developing mouse brain. Brain Res. 2006;1077:1–6. doi: 10.1016/j.brainres.2006.01.023. [DOI] [PubMed] [Google Scholar]
- [49].Geiger J, Weber YG, Landwehrmeyer B, Sommer C, Lerche H. Immunohistochemical analysis of KCNQ3 potassium channels in mouse brain. Neurosci Lett. 2006;400:101–4. doi: 10.1016/j.neulet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- [50].Tinel N, Lauritzen I, Chouabe C, Lazdunski M, Borsotto M. The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett. 1998;438:171–6. doi: 10.1016/s0014-5793(98)01296-4. [DOI] [PubMed] [Google Scholar]
- [51].Kanaumi T, Takashima S, Iwasaki H, Itoh M, Mitsudome A, Hirose S. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 2008;30:362–9. doi: 10.1016/j.braindev.2007.11.003. [DOI] [PubMed] [Google Scholar]
- [52].Brown L, Gutherz S, Kulick C, Soper C, Kondratyev A, Forcelli P. Profile of retigabine-induced neuronal apoptosis in the developing rat brain. Epilepsia. doi: 10.1111/epi.13335. n.d.;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ouhaz Z, Ba-M’hamed S, Mitchell AS, Elidrissi A, Bennis M. Behavioral and cognitive changes after early postnatal lesions of the rat mediodorsal thalamus. Behav Brain Res. 2015;292:219–32. doi: 10.1016/j.bbr.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bergman A, Feigenbaum JJ, Yanai J. Neuronal losses in mice following both prenatal and neonatal exposure to phenobarbital. Acta Anat (Basel) 1982;114:185–92. doi: 10.1159/000145590. [DOI] [PubMed] [Google Scholar]
- [55].Kleinberger N, Yanai J. Early phenobarbital-induced alterations in hippocampal acetylcholinesterase activity and behavior. Brain Res. 1985;354:113–23. doi: 10.1016/0165-3806(85)90074-4. [DOI] [PubMed] [Google Scholar]
- [56].Rogel-Fuchs Y, Newman ME, Trombka D, Zahalka EA, Yanai J. Hippocampal cholinergic alterations and related behavioral deficits after early exposure to phenobarbital. Brain Res Bull. 1992;29:1–6. doi: 10.1016/0361-9230(92)90002-f. [DOI] [PubMed] [Google Scholar]
- [57].Yanai J, Fares F, Gavish M, Greenfeld Z, Katz Y, Marcovici G, et al. Neural and behavioral alterations after early exposure to phenobarbital. Neurotoxicology. 1989;10:543–54. [PubMed] [Google Scholar]
- [58].Jacobson CD, Antolick LL, Scholey R, Uemura E. The influence of prenatal phenobarbital exposure on the growth of dendrites in the rat hippocampus. Brain Res Dev Brain Res. 1988;44:233–9. doi: 10.1016/0165-3806(88)90221-0. [DOI] [PubMed] [Google Scholar]
- [59].Markowitz GJ, Kadam SD, Boothe DM, Irving ND, Comi AM. The pharmacokinetics of commonly used antiepileptic drugs in immature CD1 mice. Neuroreport. 2010;21:452–6. doi: 10.1097/wnr.0b013e328338ba18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Löscher W. The pharmacokinetics of antiepileptic drugs in rats: consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy. Epilepsia. 2007;48:1245–58. doi: 10.1111/j.1528-1167.2007.01093.x. [DOI] [PubMed] [Google Scholar]
- [61].Center for Drug Evaluation and Research [accessed November 23, 2015];Drug Safety and Availability - FDA Drug Safety Communication: FDA approves label changes for anti-seizure drug Potiga (ezogabine) describing risk of retinal abnormalities, potential vision loss, and skin discoloration. 2013 http://www.fda.gov/Drugs/DrugSafety/ucm372774.htm.