

Abstract
Background
Plasma cell leukemia (PCL) is a rare and aggressive plasma cell neoplasm. In PCL, clonal plasma cells comprise ≥20% of the peripheral blood (PB) leukocytes and/or the absolute clonal PB plasma cell count is ≥2×109/L. Primary PCL (PPCL) originates de novo, whereas, secondary PCL (SPCL) evolves from pre-existing multiple myeloma.
Methods
Clinicohematological features, immunophenotypic profile, and survival of PCL patients were analyzed retrospectively.
Results
Between January 2007 and December 2014, ten PPCL and four SPCL patients were investigated (8 PPCLs and 3 SPCLs had complete clinical data). All were North Indians, sharing common geography and ethnicity. Our cohort showed less frequent renal failure, more frequent hepatomegaly, and non-secretory type disease. In contrast to western literature, flow cytometric immunophenotyping of our cohort revealed altered expression of CD138 (67%), CD56 (33%), and CD20 (0%). With novel therapeutic agents, these PPCL patients had a median overall survival of 15 months.
Conclusion
We highlight that our PPCL patients from North India had distinct clinicohematological and immunophenotypic profiles. The significance of our findings must be tested in a larger patient cohort and must be supported by molecular and cytogenetic investigations to unmask possible significant effects on pathogenesis.
Keywords: Clinicohematological profile, North India, Plasma cell leukemia, Immunophenotype, Survival
INTRODUCTION
Plasma cell leukemia (PCL) is a rare and aggressive plasma cell neoplasm characterized by clonal plasma cells comprising more than 20% of the leukocytes in the circulation and/or an absolute clonal plasma cell count of more than 2×109/L [1,2]. According to the diagnostic criteria defined by Kyle et al. in 1974 [1], PCL accounts for 2 to 4% of plasma cell dyscrasias [3]. In 2013, the International Myeloma Working Group (IMWG) had proposed modified diagnostic criteria for PCL as having PB plasma cells ≥5% and/or absolute plasma cell count ≥0.5×109/L. If this criteria is implied, the prevalence of PCL would increase further [3]. PCL is more common in African Americans compared to Caucasians [3]. PCL arising de novo without prior evidence of multiple myeloma (MM) is termed primary PCL (PPCL), and PCL evolving from a pre-existing MM is termed secondary PCL (SPCL). According to literature, 60–70% of PCLs are primary, and 30–40% are secondary [3,4]. The chances of refractory and relapsed MM progressing to SPCL is 1–2% [3,5]. PCL is a rare disease and the precise information regarding its incidence, clinical presentation, and pathological features are unfolding gradually [6].
Since SPCL is a leukemic evolution of MM, there are no major differences in the clinical profiles of MM and SPCL. On the contrary, PPCL is usually presented a decade earlier (median age, 52–65 yr), and PPCL patients have been documented to show a higher prevalence of hepatosplenomegaly, lymphadenopathy, pleural effusion, neurological deficits, anemia, thrombocytopenia, hypercalcemia, and extramedullary plasmacytomas, while osteolytic lesions are relatively few [3]. In addition, PPCL is associated with significant leukocytosis, markedly elevated lactate dehydrogenase levels, high beta-2-microglobulin (B2M) levels, and a higher incidence of tumor lysis syndrome [3]. All these clinical and laboratory features underline the critical differences in the natural history of PPCL and SPCL [3]. Till date, there is no curative treatment for PCL and the median survival is merely 7–13 months for PPCL and 2–7 months for SPCL [4,7].
There have been few case reports and case series in the literature, both from Western countries [1,3,7,8,9,10,11,12,13,14,15] and India [5,6,16,17,18,19,20,21] describing the clinical presentations, morphologic variations, immunophenotype, and treatment modalities of PCL. We describe a cohort of PCL patients originating from a single geographic locality (Punjab and Haryana) in Northern India. Their relatively unique clinicohematological and immunophenotypic profiles prompted us to report this series.
MATERIALS AND METHODS
Retrospective analysis was performed in the Department of Hematology of a state-funded tertiary care center. Using the world health organization (WHO) 2008 criteria (clonal PB plasma cells ≥2×109/L or ≥20% of the leukocyte differential count) [2], departmental records (between January 2007 and December 2014) were searched for cases of PCL. Subclassification into PPCL and SPCL was based on the absence or presence of prior MM. The selected cases were systematically reviewed for demographic profile, presenting features, radiologic studies, biochemistry tests, blood counts (performed on LH 750 and LH 780 hematology analyzers; Beckman Coulter, Florida, USA), bone marrow (BM) findings, flow cytometric immunophenotyping (FCI), serum and urine protein electrophoresis (performed on SAS-2 electrophoresis system, Helena Biosciences, UK), serum-free light chain assay (SFLCA), abdominal fat-pad aspirate detection of amyloid, treatment details and follow-up. The overall survival (OS) was calculated from the date of diagnosis to the date of last follow-up for PPCL. Similarly, for SPCL, the period from evolution to PCL until the last follow-up was calculated for OS. Treatment response was defined as complete response (CR), stringent CR, partial response (PR), very good PR, stable disease, progressive disease, relapse, and relapse from CR, according to the IMWG uniform response criteria [22]. The findings were summarized and compared to existing literature on PCL. Statistical analysis was performed using SPSS version-17.0 (SPSS Inc., Chicago, Illinois, USA).
PB and BM samples were analyzed on May-Grünwald Giemsa stained smears. For FCI, cells were prepared by lysestain-wash technique. The panel of fluorochrome conjugated monoclonal antibodies included CD138-PE (clone Mi15), CD20-PerCP (clone L27) (L27-PerCP), CD19-PECy7 (clone SJ25C1), CD38-PerCPCy5.5 (clone HIT2), CD56-APC (clone B159), and CD45-APCH7 (clone 2D1). Using BD Cytofix/Cytoperm solution (Becton Dickinson., San Jose, USA), intracytoplasmic expression of kappa (κ) and lambda (λ) light chains were analyzed using κ-FITC (clone TB28-2), and λ-PE (clone 1-155-2). Minimum of 50,000 events were acquired per tube in BD FACS Canto II flow cytometer (Becton Dickinson, San Jose, USA) and the list mode data was analysed with BD FACS Diva software. Events with CD38bright and CD138bright/dim expression were identified as plasma cells. The immunophenotypic profile of each case was noted for comparison with existing literature.
RESULTS
Among 1,152 MM cases diagnosed during the study period, 14 were PCLs (1.2%). Of these PCLS, 10 (71%) were PPCLs, and four (29%) were SPCLs. Complete medical records were available for eight PPCLs and three SPCLs. All patients belonged to adjacent North Indian states of Punjab and Haryana. A wide variety of occupational profiles were observed among the patients, making it unlikely for a particular occupation to be associated with higher risk of PCL. The clinical and hematological parameters of patients are summarized in Table 1.
Table 1. Clinical and hematological parameters of PCL patients.

Note: Age and gender were available for ten PPCLs and four SPCLs. Clinicohematological and biochemical data were available for eight PPCLs and three SPCLs. SPEP and UPEP data were available for six PPCLs and three SPCLs. Serum protein immunofixation data was available for six PPCLs and two SPCLs.
Abbreviations: PPCL, primary plasma cell leukemia; BM, bone marrow; B2M, beta-2-microglobulin; κ, kappa light chain; λ, lambda light chain; MM, multiple myeloma; PB, peripheral blood; PEP, protein electrophoresis; S. Ca, serum calcium; S. Cr, serum creatinine; TLC, total leukocyte count; NA, not applicable.
Clinical features
The median age of 10 PPCL patients at diagnosis was 59 years (range, 39–80 yr) with a male to female ratio of 1.5:1. Details regarding clinical features and investigations were available for eight patients. Hepatosplenomegaly (3/8) and isolated hepatomegaly (3/8) were both present in 37.5% of patients. Hence, enlarged liver (6/8) was noted in 75% of the patients. By imaging, 62% were confirmed to have lytic bone lesions. None of the patients had significant lymphadenopathy, neurological deficits, or bleeding diathesis.
There were three men and one woman among the SPCLs. The median age at diagnosis was 55 years (range, 38–79 yr). The three SPCLs, whose medical records could be traced, showed progression from MM to PCL in a median time period of 38 months. Isolated hepatomegaly was observed in one patient, while splenomegaly was absent. Lytic bone lesions were observed in all three of them with two patients showing vertebral involvement and one with skull lesions.
Laboratory investigations
In PPCL patients, the baseline median hemoglobin (Hb) was 7.7 g/dL (range, 5.6–14.7 g/dL) and severe anemia (Hb of <8.0 g/dL) was observed in 67% of the patients. With a median platelet count of 71×109/L (range, 22×109–186×109/L), 90% had thrombocytopenia at presentation. Leukocytosis was observed in 80% of the patients and the median leukocyte count was 14.6×109/L (range, 6.59×109–134×109/L) with a median proportion of circulating plasma cells of 41% (range, 21–98%). Concurrent BM examination revealed the median proportion of plasma cells to be 62% (range, 34–90%) with a proportional reduction in normal hematopoietic elements. These findings were confirmed on BM trephine biopsies.
The four SPCL patients had mean Hb and platelet counts of 6.4 g/dL (range, 5.2–6.9 g/dL) and 53×109/L (range, 22×109–176×109/L), respectively. Severe anemia was observed in all four (100%) patients and three (75%) of them had thrombocytopenia at presentation. The median leukocyte count was 10.9×109/L (range, 2×109–28.2×109/L), and the median circulating plasma cells was 68% (range, 33–90%). Concurrent bone marrow aspirates revealed 33–94% plasma cells.
None of the patients with PPCL and SPCL had deranged screening coagulation profiles. Serum protein electrophoresis (SPEP) data was available for six PPCL patients, of which four (67%) were positive for M-protein indicated by the presence of M-band. On serum immunofixation electrophoresis (SIF), these four cases were sub-classified into IgGκ (17%), IgGλ (17%), and IgAλ (33%) restricted PPCLs. Two cases (33%) were M-band negative on SPEP, and did not show bands on SIF. SFLCA of these two cases revealed λ light chain restriction (κ: λ of 0.05) in one, and κ light chain restriction (κ: λ of 116.6) in the other. Among the three M-band positive SPCLs, SIF was available for two patients, both with IgGκ restriction. Urine M-band was negative in all PCLs and Bence-Jones proteinuria was observed in one IgAλ restricted PPCL.
Among the PPCLs, the median serum creatinine (S.Cr) and serum calcium (S.Ca) were 1.0 mg/dL (range, 0.8–2.25) and 8.4 mg/dL (range, 6.1–16.1), respectively. Only one patient presented with renal insufficiency and myeloma defining hypercalcemia (Sr.Ca >11 mg/dL). Within the SPCLs, median S.Cr and S.Ca were 1.4 mg/dL (range, 0.5–2.3) and 9 mg/dL (range, 8–10), with only one patient having renal insufficiency at diagnosis. The median B2M levels among the PPCLs and SPCLs were 4.2 (range, 2.5–6.7) mg/L and 4.3 (2.0–6.7) mg/L, respectively (Table 1).
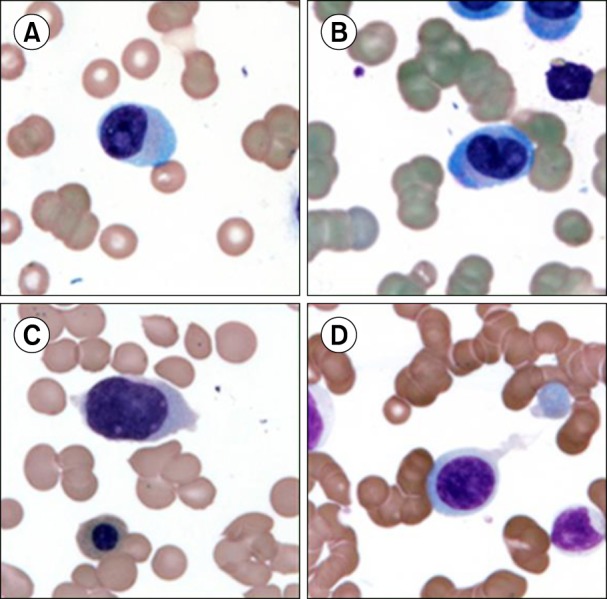
PB films showed both mature plasma cells and lymphoplasmacytoid cells in four PPCLs and four SPCLs (Fig. 1A, B). In the remaining six PPCLs, the plasma cells were very immature and had blastoid morphologies with prominent nucleoli; rendering a definitive morphological diagnosis of plasma cell neoplasm difficult (Fig. 1C, D).
Fig. 1. (A, B) Circulating plasma cells with lymphoplasmacytoid morphology, (C, D) circulating plasma cells with blast like morphology (Magnification ×1,000, May-Grünwald-Giemsa staining).
Immunophenotyping by flow cytometry was attempted in these six PPCLs with blastoid morphology. Table 2 summarizes the immunophenotypic profile of these cases. All six cases showed surface expression of CD38 and two cases had aberrant dim expression of CD138. CD56 was expressed in two cases and none of these cases were CD20 positive. Among ten PPCLs, one was lost to follow-up, and two opted against treatment. All seven patients were treated with novel therapeutic agents and fared a median OS of 15 months.
Table 2. Immunophenotypic profiles of six PPCL cases by multicolor flow cytometry.

Abbreviations: Pos, positive; Neg, negative; Equ, equivocal; Cyto κ, cytoplasmic kappa light chain; Cyto λ, cytoplasmic lambda light chain.
Among SPCLs, two patients opted against treatment and one was given palliative therapy (OS of 5 mo), while only one was treated appropriately (OS of 30 mo). Table 3 and 4 summarize the combination chemotherapies used in these patients. Of the autologous stem cell transplant eligible patients, only one underwent the procedure and others opted out. Interestingly, a 52 year-old woman was diagnosed with PPCL characterized by a baseline plasma cell immunophenotype of CD38+CD138+CD56-. She was lost to follow-up after eight cycles of velcade-thalidomide-dexamethasone (VTD). Thirteen months later, she returned with a left breast mass. The lesion was diagnosed as a plasmacytoma by fine needle aspiration. Flow cytometry of the breast mass revealed an altered immunophenotypic profile with loss of CD138 and gain of CD56. She was restarted on VTD and lost to follow-up after two months.
Table 3. Summary of treatment in seven PPCL and two SPCL patients.

a)Not affordable for bortezomib.
Note: Among ten PPCLs, one lost follow-up before start of therapy and two opted against therapy. Out of four SPCLs, two opted against therapy.
Abbreviations: PPCL, primary plasma cell leukemia; SPCL, secondary plasma cell leukemia; CR, complete response; PR, partial response; sCR, stringent CR.
Table 4. Dosage and schedule of the treatment regimen.

a)Started with 100 mg, maximum 200 mg. Dose modifications: In case of peripheral neuropathy, thalidomide dose was reduced by 50% and bortezomib was reduced to 1 mg/m2. In case of renal impairment, melphalan dose was reduced by 25% if Cr clearance was <30 mL/min. For lenolidomide, the dose adjustments were: 10 mg for Cr clearance of <30 mL/min, 15 mg for Cr clearance <30–60 mL/min and 5 mg for patients on hemodialysis. The treatment response criteria was as defined by IMWG uniform response criteria [22].
Abbreviations: PO, per os (by oral); SC, sub-cutaneous; HS, hora somni (bed-time); IV, intra venous.
DISCUSSION
Ever since the recognition of first PCL by Gluzinski and Reichenstein in 1906 [23], the disease still remains largely unexplored due to its rarity. Even if multiple cases and some case series of PCL have been reported in the literature, there are no formal studies describing the incidence of PCL in the general population [24].
In our study, consistent with existing literature, PPCLs (71%) were much higher than SPCLs (29%). The male preponderance observed among our PPCL cohort (M:F ratio of 1.5:1) is consistent with existing literature [14], although some studies showed equal sex incidence [8] and female preponderance [21]. Consistent with previous Western and Indian publications, the current cohort had a median age of 59 years (range, 39–80 yr) at presentation [3,6,16,17,18,21,25]. Table 5 compares the clinical and laboratory data of the current study with previously published large case series.
Table 5. Comparison between the clinical and laboratory data of PPCL cases in the current study and other large case series.

a)Median value, b)Median not available, c)98% had ≥40% PC, d)≥2 mg/dL in 44%, e)≥6 mg/mL in 65%.
Abbreviations: κ, kappa light chain; λ, lambda light chain; NA, Not available; PC, plasma cells; PB, peripheral blood; BM, bone marrow; B2M, beta-2-microglobulin; Hb, hemoglobin; S.Cr, serum creatinine; S.Ca, serum calcium.
The frequency of osteolytic lesions and splenomegaly among our PPCL cases were comparable to previous reports, but the incidence of hepatomegaly (75%) was higher and lymphadenopathy was absent in our cohort. Thrombocytopenia was noted in 80% of PPCLs at presentation; however, none of them had bleeding diathesis. The degree of anemia, leukocytosis, and PB plasmacytosis were comparable to the data described in other case series, but the median BM plasma cells (62%) in our series was the same as that described by Peijing et al. in 2009 [26]. Even if renal failure is described frequently in PCL [27], in our cohort, S.Ca >11 mg/dL, S.Cr >2 mg/dL, and renal failure were observed in only one patient (12.5%). On SIF, the frequency for all of IgG, IgA, and non-secretor phenotype of our PPCL cohort was 33.3%. This is in sharp contrast with the most recent literature including the IMWG 2013 data, where light chain disease is considered to be more frequent [27], with the frequencies of IgG, IgA, light chain, and non-secretory PCL in the ranges 28–58%, 4–17%, 23–44%, and 0–12%, respectively [3,6,9,10,11,24,26,28,29]. In addition, similar to Toma et al. [30] and Majumdar et al. [6], our PCL cohort also had higher λ light chain restriction than κ light chain restriction.
In comparison to MM, the immunophenotypic information on PPCL is limited. It is described in the literature that in addition to bright CD38 and CD138 expression, leukemic plasma cells of PPCL display low frequency of CD56 positivity and higher frequencies of CD20, CD45, and CD19 positivity [27]. Flow cytometric analysis of our cohort showed that only CD38 expression (100% of cases) was consistent with existing literature [6,11,16]. Our cohort showed that the frequencies of CD138 (67%) and CD56 (33%) were less [6,11,16], and CD20 was not expressed. A comparable immunophenotypic profile of 10 PCL cases was described by Tembhare et al. [16]; however, in our study, the frequencies of CD138 and CD56 positivity were even less. This atypical immunophenotypic profile (Table 6) might suggest a possible genetic heterogeneity among North Indian PCL patients. A definite conclusion may also not be possible due to the lack of conventional cytogenetic/FISH analysis in the present study. A collaborative effort to analyze the gene expression profile of Indian and Western PCL patients might provide concrete evidence for disease heterogeneity in the future.
Table 6. Comparison of marker expression profile with immunophenotyping by flow cytometry performed in six PPCL patients.

Abbreviations: cKappa, cytoplasmic kappa light chain restriction; cLambda, cytoplasmic lambda light chain restriction.
Using novel therapeutic agents, our PPCL cohort fared an OS of 15 months (refer to Table 5), which is similar to that reported by Intergroupe Fracophone du Myélome on PCL patients treated with novel drugs [25,27]. Only two of our SPCL patients opted for treatment, which makes the cohort too small for satisfactory survival analysis.
In conclusion, the present study highlights the clinicohematological and immunophenotypic profile of PCL patients belonging to a distinct North Indian population. These patients had a low frequency of renal insufficiency and higher frequencies of hepatomegaly and non-secretory phenotype of PCL. With the administration of novel therapeutic agents, the patients had 15 months OS.
A smaller cohort size and relatively short follow-up limits a definitive conclusion. The significance of our findings must be tested in a larger patient cohort, and must include molecular and cytogenetic investigations to unmask any significant mechanism of pathogenesis.
ACKNOWLEDGMENTS
We thank Mrs. Parveen Bose, senior technician, for her help in flow cytometric sample processing.
Footnotes
Authors' Disclosures of Potential Conflicts of Interest: No potential conflicts of interest relevant to this article were reported.
References
- 1.Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia. Report on 17 cases. Arch Intern Med. 1974;133:813–818. doi: 10.1001/archinte.133.5.813. [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC Press; 2008. [Google Scholar]
- 3.Fernández de Larrea C, Kyle RA, Durie BG, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013;27:780–791. doi: 10.1038/leu.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cifola I, Lionetti M, Pinatel E, et al. Whole-exome sequencing of primary plasma cell leukemia discloses heterogeneous mutational patterns. Oncotarget. 2015;6:17543–17558. doi: 10.18632/oncotarget.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raj RS, Najeeb S, Aruna R, Pavithran K, Thomas M. Primary plasma cell leukemia occuring in the young. Indian J Cancer. 2003;40:116–117. [PubMed] [Google Scholar]
- 6.Majumdar N, Kumar R, Anand M, et al. Plasma cell leukemia-a study of 28 cases from India. Hematology. 2009;14:198–203. doi: 10.1179/102453309X426191. [DOI] [PubMed] [Google Scholar]
- 7.Johnson MR, Del Carpio-Jayo D, Lin P, et al. Primary plasma cell leukemia: morphologic, immunophenotypic, and cytogenetic features of 4 cases treated with chemotherapy and stem cell transplantation. Ann Diagn Pathol. 2006;10:263–268. doi: 10.1016/j.anndiagpath.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Woodruff RK, Malpas JS, Paxton AM, Lister TA. Plasma cell leukemia (PCL): A report on 15 patients. Blood. 1978;52:839–845. [PubMed] [Google Scholar]
- 9.Noel P, Kyle RA. Plasma cell leukemia: an evaluation of response to therapy. Am J Med. 1987;83:1062–1068. doi: 10.1016/0002-9343(87)90942-9. [DOI] [PubMed] [Google Scholar]
- 10.Dimopoulos MA, Palumbo A, Delasalle KB, Alexanian R. Primary plasma cell leukaemia. Br J Haematol. 1994;88:754–759. doi: 10.1111/j.1365-2141.1994.tb05114.x. [DOI] [PubMed] [Google Scholar]
- 11.García-Sanz R, Orfão A, Gonzalez M, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics. Blood. 1999;93:1032–1037. [PubMed] [Google Scholar]
- 12.Saccaro S, Fonseca R, Veillon DM, et al. Primary plasma cell leukemia: report of 17 new cases treated with autologous or allogeneic stem-cell transplantation and review of the literature. Am J Hematol. 2005;78:288–294. doi: 10.1002/ajh.20272. [DOI] [PubMed] [Google Scholar]
- 13.Musto P, Rossini F, Gay F, et al. Efficacy and safety of bortezomib in patients with plasma cell leukemia. Cancer. 2007;109:2285–2290. doi: 10.1002/cncr.22700. [DOI] [PubMed] [Google Scholar]
- 14.Albarracin F, Fonseca R. Plasma cell leukemia. Blood Rev. 2011;25:107–112. doi: 10.1016/j.blre.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gogia A, Raina V, Gupta R. Massive ascites as an presenting feature of plasma cell leukemia. Indian J Cancer. 2013;50:301. doi: 10.4103/0019-509X.123632. [DOI] [PubMed] [Google Scholar]
- 16.Tembhare PR, Subramanian PG, Sehgal K, et al. Immunophenotypic profile of plasma cell leukemia: a retrospective study in a reference cancer center in India and review of literature. Indian J Pathol Microbiol. 2011;54:294–298. doi: 10.4103/0377-4929.81603. [DOI] [PubMed] [Google Scholar]
- 17.Naseem S, Kaur S, Gupta R, Kashyap R, Nityanand S. Plasma cell leukemia: case series from a tertiary center with review of literature. Indian J Hematol Blood Transfus. 2012;28:10–14. doi: 10.1007/s12288-011-0097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kar R, Priyadarshini SG, Niraimathi M, Basu D, Badhe BA. Clinico-pathological spectrum of primary plasma cell leukemia diagnosed at a tertiary care centre in South India over 5 year period. Indian J Hematol Blood Transfus. 2012;28:170–174. doi: 10.1007/s12288-011-0133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majhi U, Murhekar K, Sundersingh S, Rajalekshmi KR. Primary plasma cell leukaemia with unusual presentations: a case series. Indian J Hematol Blood Transfus. 2014;30(Suppl 1):390–393. doi: 10.1007/s12288-014-0430-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal AD, Brahmbhatt BS, Parikh BP, Shah MJ. Plasma cell leukemia: a retrospective study of 10 cases. Indian J Cancer. 2014;51:18–19. doi: 10.4103/0019-509X.134605. [DOI] [PubMed] [Google Scholar]
- 21.Rajeswari G, Paul TR, Uppin MS, et al. Plasma cell leukemia: A case series from South India with emphasis on rarer variants. Indian J Med Paediatr Oncol. 2014;35:211–214. doi: 10.4103/0971-5851.142037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–1473. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 23.Gluzinski A, Reichenstein M. Myeloma und leucaemia lymphatica plasmocellularis. Wien Klin Wochenschr. 1906;12:336–339. [Google Scholar]
- 24.Pagano L, Valentini CG, De Stefano V, et al. Primary plasma cell leukemia: a retrospective multicenter study of 73 patients. Ann Oncol. 2011;22:1628–1635. doi: 10.1093/annonc/mdq646. [DOI] [PubMed] [Google Scholar]
- 25.Royer B, Magrangeas F, Lioure B, et al. First large prospective study for patients with primary plasma cell leukemia: Bortezomib-doxorubicine-dexamethasone/bortezomib-cyclophosphamidedexamethasone regimens as induction before stem cell transplantation followed by consolidation with lenalidomide-bortezomib-dex or allograft. (a study of the IFM group) Blood (ASH Annual Meeting Abstracts) 2013;122(Suppl):761. [Google Scholar]
- 26.Peijing Q, Yan X, Yafei W, et al. A retrospective analysis of thirty-one cases of plasma cell leukemia from a single center in China. Acta Haematol. 2009;121:47–51. doi: 10.1159/000210555. [DOI] [PubMed] [Google Scholar]
- 27.van de Donk NW, Lokhorst HM, Anderson KC, Richardson PG. How I treat plasma cell leukemia. Blood. 2012;120:2376–2389. doi: 10.1182/blood-2012-05-408682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colović M, Janković G, Suvajdzić N, Milić N, Dordević V, Janković S. Thirty patients with primary plasma cell leukemia: a single center experience. Med Oncol. 2008;25:154–160. doi: 10.1007/s12032-007-9011-5. [DOI] [PubMed] [Google Scholar]
- 29.Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044–1052. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toma VA, Retief FP, Potgieter GM, Anderson JD. Plasma cell leukaemia. Diagnostic problems in our experience with 11 cases. Acta Haematol. 1980;63:136–145. doi: 10.1159/000207385. [DOI] [PubMed] [Google Scholar]