

SUMMARY
The RpoS transcription factor of Borrelia burgdorferi is a “gatekeeper” because it activates genes required for spirochetes to transition from tick to vertebrate hosts. However, it remains unknown how RpoS becomes repressed to allow the spirochetes to transition back from the vertebrate host to the tick vector. Here we show that a putative carbohydrate-responsive regulatory protein, designated BadR (Borrelia host adaptation Regulator), is a transcriptional repressor of rpoS. BadR levels are elevated in B. burgdorferi cultures grown under in vitro conditions mimicking unfed-ticks and badR-deficient strains are defective for growth under these same conditions. Microarray and immunoblot analyses of badR-deficient strains showed up-regulation of rpoS and other factors important for virulence in vertebrate hosts, as well as down-regulation of putative tick-specific determinants (e.g. linear plasmid 28-4 genes). DNA-binding assays revealed BadR binds to upstream regions of rpoS. Site-directed mutations in BadR and the presence of phosphorylated sugars affected BadR’s binding to the rpoS promoters. badR-deficient B. burgdorferi were unable to colonize mice. Several putative tick-specific targets have been identified. Our study identified a novel regulator, BadR, and provides a link between nutritional environmental cues utilized by spirochetes to adaptation to disparate conditions found in the tick and vertebrate hosts.
INTRODUCTION
Lyme disease is a persistent multi-system inflammatory disease and the most frequently reported arthropod-borne disease in the U.S. with over 30,000 cases reported to the Centers for Disease Control and Prevention (CDC) in 2010. Currently, there is no vaccine available and the CDC has classified Lyme disease as a re-emerging infection (CDC, 2012).
Borrelia burgdorferi, the etiologic agent of Lyme disease, is a spirochetal pathogen that exists in nature in an enzootic cycle between its tick and vertebrate hosts (Steere, 2006; Barbour et al., 1983). The spirochetes’ ability to survive in these disparate hosts is dependent on the mechanisms to rapidly adapt by sensing the surrounding environment and altering gene expression accordingly (Samuels, 2011; Radolf et al., 2012). While Escherichia coli has between 30 to 60 two-component regulatory systems involved in sensing the environment and regulating genes accordingly, B. burgdorferi’s genome encodes for only two (Fraser et al., 1997). These systems include the Hpk2-Rrp2 system that regulates RpoS, which in turn regulates many factors required for transmission and vertebrate infection; and Hpk1-Rrp1, which operates through the signaling molecule cyclic di-GMP and regulates metabolic and virulence genes (Hubner et al., 2001; Rogers et al., 2009; Yang et al., 2003; Sultan et al., 2010; Caimano et al., 2011; Samuels, 2011; Radolf et al., 2012).
B. burgdorferi relies intimately on its host for nutrients and metabolites because many biosynthetic genes are absent (Fraser et al., 1997). Many bacteria use nutrients as cues to sense the environment and evidence suggests B. burgdorferi uses different carbon sources from each host (Pappas et al., 2011). In other organisms, nutrients stimulate adaptive responses such as proliferation, motility, and virulence factor production (Poncet et al., 2009). Carbohydrate responsive regulators are crucial for relaying these nutrient signals for subsequent modulation of virulence mechanisms (Postma et al., 1993).
Only two carbohydrate responsive regulators are apparent in B. burgdorferi’s genome, genes bb0693 and bb0831, annotated as xylose operon regulatory proteins (XylR1, XylR2) respectively. XylRs are part of the ROK (repressor, ORF, kinase) family of proteins (Titgemeyer et al., 1994). In addition to XylRs, ROK repressors also include Mlc and NagC in other bacterial systems. Both Mlc and NagC are involved in regulating utilization of phosphotransferase system (PTS) sugars (Plumbridge, 2001). NagC is a dual regulator that represses nagE-nagBACD and chb operons for n-acetyl-glucosamine (GlcNAc) and chitin/chitobiose catabolism, respectively. In addition, NagC also activates glmS operons involved in the biosynthesis of GlcNAc precursors for cell wall biogenesis (Plumbridge & Pellegrini, 2004). Mlc is a global regulator of glucose metabolism and represses ptsG and ptsHI operons encoding the transporter and cytoplasmic components of the glucose PTS (PTSGlc), respectively (Hosono et al., 1995; Plumbridge, 2002; Schiefner et al., 2005). Mlc also modulates expression of the chb operon (Berg et al., 2007). Both Mlc and NagC regulate genes for bacterial virulence. For example, NagC regulates fimB recombinase involved in phase variation in Type I fimbriae in E. coli, whereas Mlc regulates Salmonella enterica hilE, which represses genes in Salmonella pathogenicity island I, important for epithelial cell invasion (Poncet et al., 2009; Sohanpal et al., 2007).
Regulation of ROKs involves interaction with their inducing signals. NagC’s operons are induced when NagC binds its inducer, GlcNAc-6-phosphate (GlcNAc-6P)(Plumbridge, 1991). In contrast, Mlc is displaced from its operators by sequestration in the inner membrane by binding to the glucose transporting protein PtsG of the PTSGlc (Tanaka et al., 2000).
Given that ROKS play a role in bacterial virulence in other systems, our study aimed to characterize the ROK regulator, XylR1, in B. burgdorferi. Our study is the first to demonstrate B. burgdorferi’s use of a putative carbohydrate responsive regulator and provides a link coordinating host-specific nutrients to host adaptation and thus virulence potential.
RESULTS
Absence of xylose utilization in B. burgdorferi
B. burgdorferi has a gene annotated as a xylulokinase (BB0545), but other genes for xylose utilization are absent (Fig. 1A). Previous studies demonstrated glucose, mannose, glycerol, GlcNAc and GlcNAc oligomers (chitin and chitobiose) could all support growth of B. burgdorferi in vitro, but not xylose (von Lackum & Stevenson, 2005). This suggests that XylR1 and XylR2 are misannotated, as there appears to be no functional need for regulating xylose utilization in B. burgdorferi.
Fig. 1.
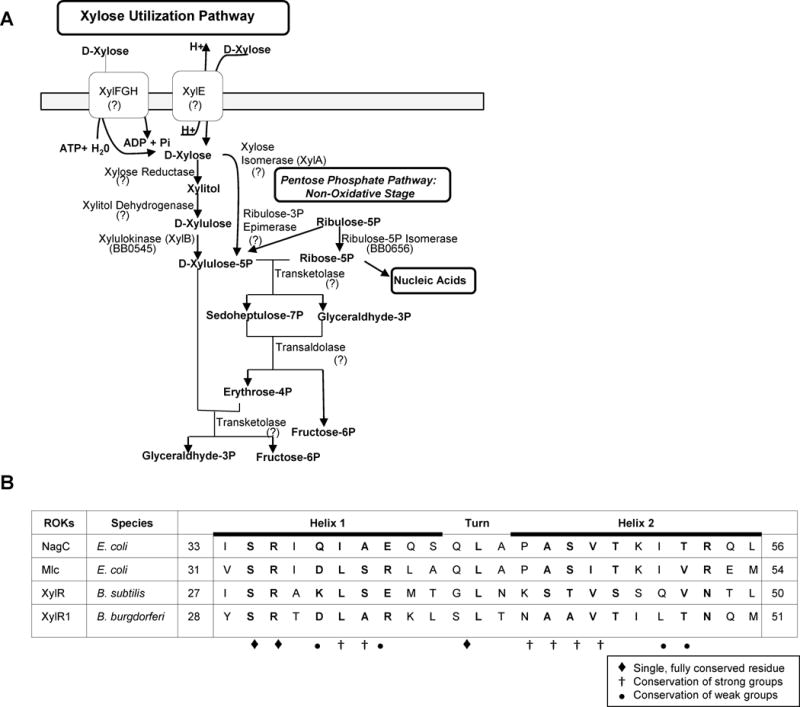
Analysis of xylose utilization of B. burgdorferi and ROK DNA-binding domains. BLAST analysis in conjunction with the borrelial genome annotations (Fraser et al., 1997) demonstrated xylulokinase (BB0545) as the only enzyme for xylose utilization in B. burgdorferi suggesting ORFs annotated as xylose operon regulatory proteins (XylR1 and XylR2) are misannotated and have alternative functions (A). Comparison of ROK DNA-binding motifs. Alignment of the HTH motifs in the N-terminal regions of NagC and Mlc from E. coli, XylR repressor from Bacillus subtilis, and XylR1 from B. burgdorferi. The locations of the motifs relative to the N-terminus of the proteins are given. Conservation stringency of residues is depicted using symbols below (B).
XylRs are included in a family of proteins known as ROKs (repressor, ORF, kinase). Specifically, ROK regulators contain an N-terminal DNA-binding domain, adaptor domain, and a C-terminal sugar-binding domain (Pennetier et al., 2008). The ROK kinases are shorter than the ROK regulators by about 80 residues due to the absence of the N-terminal DNA-binding domain (Titgemeyer et al., 1994). In silico analysis of the borrelial XylRs revealed XylR2 has features of a sugar kinase whereas XylR1 demonstrated properties of a transcriptional regulator. XylR2 is lacking the N-terminal DNA-binding domain, whereas this domain is maintained in XylR1.
XylR1 of B. burgdorferi shares homology with two other ROK regulatory proteins, NagC (BLASTp=e−16) and Mlc (BLASTp=e−14), involved in regulating GlcNAc and glucose metabolism, respectively. Comparative sequence analysis of the helix-turn-helix motif of the DNA-binding domain demonstrated that XylR1 has many conserved residues found in the other ROKs (e.g. NagC and Mlc of E. coli and XylR of Bacillus subtilis) (Fig. 1B).
Based on these in silico findings, the inability of B. burgdorferi to utilize xylose sugars, and several discoveries reported below, we renamed XylR1 as Borrelia host adaptation Regulator (BadR) and will refer to it as such throughout this study.
BadR is elevated in cultures grown under conditions mimicking tick hosts
We hypothesized BadR was a ROK regulator of glucose and/or GlcNAc utilization (e.g. Mlc and/or NagC homolog), as these are sugars utilized by B. burgdorferi. We further hypothesized that BadR expression would change in B. burgdorferi grown under inducing conditions. In previous studies using qRT-PCR, we found that badR transcript levels were negligibly changed in B. burgdorferi grown in BSK–Lite media (medium free of supplemental sugars (von Lackum & Stevenson, 2005)) supplemented with various sugars (2X glucose (0.8% w v−1), 2X GlcNAc (0.08% w v−1), 0.4% v v−1 glycerol, or 75uM chitobiose) (data not shown). In E. coli, NagC is induced only 2-fold by growth under inducing conditions, whereas NagA and NagB expression is induced 20-fold (Plumbridge, 1996). Likewise in E. coli, Mlc transcript levels changed less than 2-fold by glucose (Shin et al., 2001). Taken together, these aforementioned studies and the fact that many components in BSK-Lite media are undefined, help explain the negligible changes seen in BadR transcript levels in our sugar-supplemented growth studies.
To assess regulatory targets of BadR, BadR and putative BadR regulated proteins were over-expressed, purified, and used to generate mono-specific anti-sera. Using the antisera generated, levels of these determinants were observed by immunoblot analysis from borrelial cultures grown under in vitro conditions mimicking fed-tick (pH6.8, 37°C) and unfed-tick (pH7.6, 23°C) hosts (Fig. 2A, B). BadR levels were elevated during growth under conditions mimicking the unfed-tick host and displayed opposing trends to the levels of NagC-regulated, GlcNAc catabolic enzymes, NagA (BB0151) and NagB (BB0152), which showed elevated levels under fed-tick growth conditions. Chitobiose transporter subunit, ChbA (BBB05), regulated by both NagC and Mlc in other bacteria, was not preferentially expressed in either host condition. Levels of Mlc-regulated glucose transporter, PtsG (BB0645), were highest in cultures grown under unfed-tick conditions. As a control, we assessed the levels of outer surface protein C, OspC. OspC is a vertebrate-specific determinant, highly up-regulated in spirochetes when the ticks are acquiring a blood meal, and is a critical element for the spirochetes to transmit from the tick to the vertebrate host (Grimm et al., 2004; Pal et al., 2004). OspC levels were highest under conditions mimicking vertebrate hosts indicating proper cell signaling (Fikrig & Narasimhan, 2006). As an additional control, no variations in P66 levels were detected demonstrating equal protein loading.
Fig. 2.
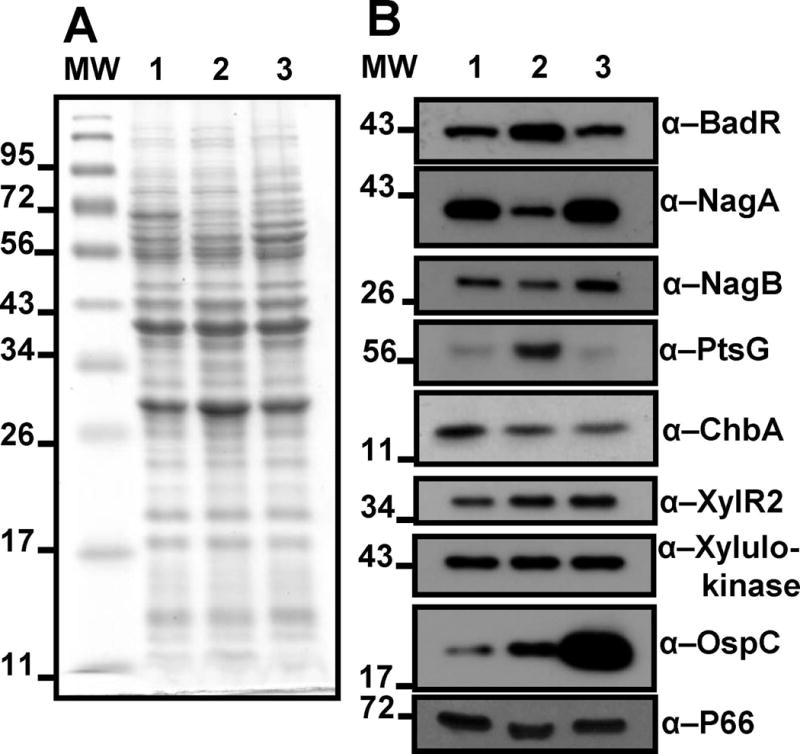
Immunoblot analysis determines regulatory contributions of BadR. Coomassie blue stained SDS-PAGE of B. burgdorferi B31-A3 lysates grown under host-mimicking conditions (A). Immunoblot analysis using depicted antibodies (B). Lanes: MW: molecular weight marker, 1: grown at pH7.6/32°C; 2: grown at pH 7.6/23°C (mimicking unfed-tick); 3: grown at pH 6.8/37°C (mimicking fed-tick).
Taken together, BadR is synthesized at higher levels in the conditions mimicking unfed-ticks. Ticks can persist months between blood meals, so we hypothesize a repressor of sugar utilization genes would be up-regulated because the tick environment is a nutrient limiting state for B. burgdorferi. High PtsG levels under unfed-tick growth conditions may illustrate the importance for spirochetes to utilize glucose first, the primary sugar for most microbes, for growth in the tick midgut, which is normally a co-inhabited and thus competitive niche (Benson et al., 2004). Studies have shown that expression of the chitobiose transporter, chbC (bbb04), is up-regulated under GlcNAc starved in vitro growth conditions (Rhodes et al., 2009; Tilly et al., 2001). Other studies found chbC transcripts were higher in borrelial cultures grown at 23°C compared to cultures shifted to 34°C. This study also demonstrated that the chbA (bbb05) and chbB (bbb06) transcripts were unaffected by this temperature shift (Tilly et al., 2001). The aforementioned studies support our findings regarding ChbA levels (Fig. 2).
Generation and growth phenotype of badR-deficient B. burgdorferi B31-A3 strains
To characterize gene regulation attributed to BadR, a badR-deficient strain, badR− (StrpR), was generated in the infectious B31-A3 strain by allelic exchange and confirmed by Southern blot analysis (Fig. 3A, B, and C). In addition, PCR was performed to confirm all essential plasmids were maintained for B. burgdorferi to complete the infectious enzootic cycle (Fig. 3D). After numerous attempts to generate a badR cis-complemented strain, we decided to generate a second badR-deficient strain, badR2− (GentR), by allelic exchange using a different antibiotic cassette from badR−. The badR2− strain displayed an identical phenotype as badR− (Fig. 4). The badR-deficient strains displayed a growth defect under in vitro conditions mimicking unfed-tick hosts (Fig. 4B), which suggests a role for BadR in modulating growth kinetics of B. burgdorferi during its residence in ticks.
Fig. 3.
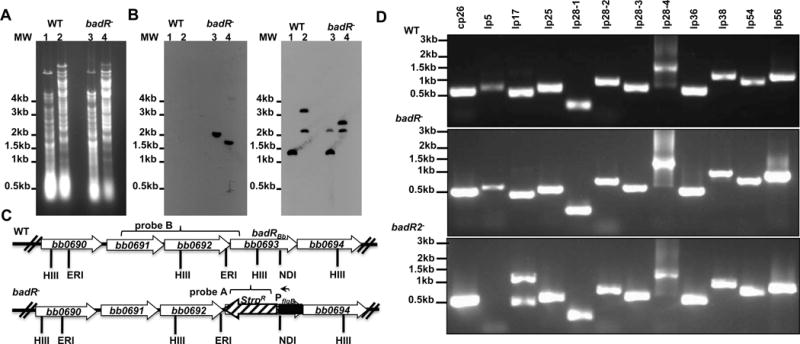
Genetic analyses of badR-deficient B. burgdorferi strains. Total genomic DNA from parental B31-A3 (WT) and badR-deficient strain (badR−) was digested with HindIII (lanes 1 and 3) or EcoRI and NdeI (lanes 2 and 4), separated on a 1% agarose gel, and transferred to nylon membranes (A, B, C). Membranes were hybridized with PCR amplified probes corresponding to the aadA gene (StrR marker) (B) or to a region upstream of the badR gene (C). Schematic representation of the badR region of the chromosome for both WT and badR−. Probes used are indicated with brackets. HIII-HindIII, ERI-EcoRI, ND1-NdeI (D). PCR confirmation to assess plasmid profile was performed on parental and badR-deficient strains. Strains and molecular weight (in base pairs) are indicated on the left (E).
Fig. 4.
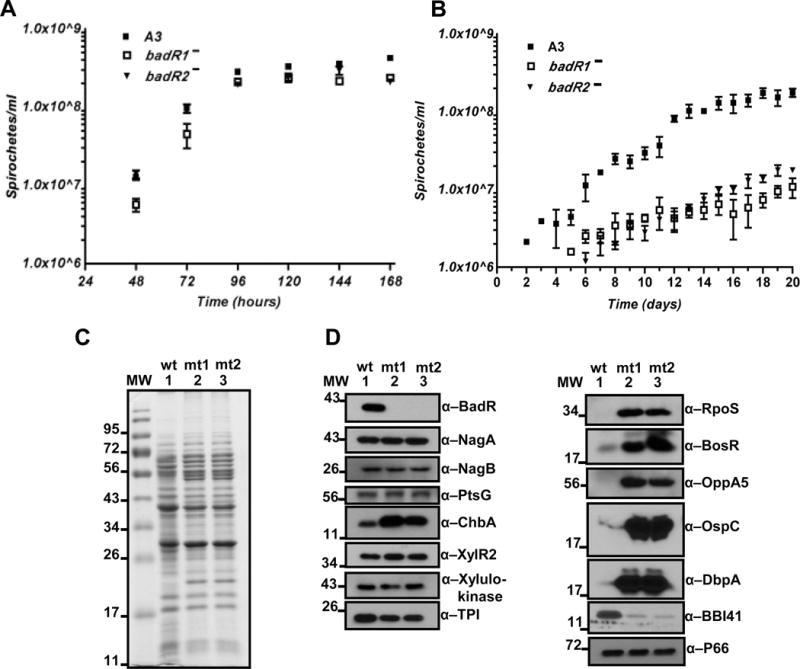
Growth analysis and protein profile of badR-deficient strains. Growth analysis of WT and badR-deficient strains grown under laboratory (pH7.6/32°C) (A) and unfed-tick (pH7.6/23°C) (B) conditions. Bacteria were diluted from stationary phase (1×108 bacteria ml−1), seeded at 5×105 bacteria ml−1, and enumerated every 24 hours using dark field microscopy. The cultures were grown in triplicate, with three independent trials. Error bars indicate standard error. Levels of significance were determined using two-way ANOVA with α=95% and there were statistical differences in growth in the badR-deficient strains from day 11 on with a P<0.01 (A, B). Protein profile of badR-deficient B. burgdorferi. Coomassie blue stained SDS-PAGE of B. burgdorferi lysates from WT (B31-A3) and badR-deficient strains grown under laboratory growth conditions (pH7.6/32°C) (C). Immunoblot analysis using depicted antibodies. Lanes: MW: molecular weight marker in kDa; 1: parental B31-A3 (wt); 2: badR− (mt1); 3: badR2− (mt2) (D).
RpoS and vertebrate host-induced factors are up-regulated in badR-deficient strains
To delineate the regulatory role of BadR, the protein profile of the badR-deficient strains grown under laboratory conditions (pH7.6, 32°C) was assessed. The badR-deficient strains showed negligible changes in NagA, NagB, and PtsG but displayed increases in the levels of chitobiose transporter protein, ChbA, (Fig. 4C, D) suggesting BadR’s role in regulating chitobiose metabolism. Intriguingly, badR-deficient strains revealed increases in levels of vertebrate-specific factors (e.g. RpoS, BosR, OspC, DbpA) implying BadR’s ability to modulate gene expression required for the spirochetes’ adaptation to the mammalian host. In addition, our immunoblot analyses demonstrated significant down-regulation of BBI41 levels, an ORF located on linear plasmid 28-4 (lp-28-4), in the badR-deficient strains and supported our microarray findings discussed below.
BadR binds to the upstream region of rpoS and binding is alleviated by phosphorylated sugars
To define whether the up-regulation of rpoS in the badR-deficient strains was a result of indirect mechanisms or by direct regulation of rpoS by BadR, DNA-binding assays were performed using recombinant BadR and two rpoS promoters. One rpoS promoter (PrpoS1) included the RpoS start codon and BosR binding site 3 (BS3) (Ouyang et al., 2011). The second rpoS promoter (PrpoS2) included the RpoN binding site, rpoS transcriptional start site, and BosR binding sites 2 and 3 (BS2, BS3) (Fig. 5A). Both promoters, when incubated with BadR, caused a mobility shift and this interaction was outcompeted when 200-fold molar excess of unlabeled promoter was added. A BadR protein lacking the N-terminal helix-turn-helix (HTH) domain (BadRΔHTH) was unable to bind these promoters (Fig. 5B).
Fig. 5.
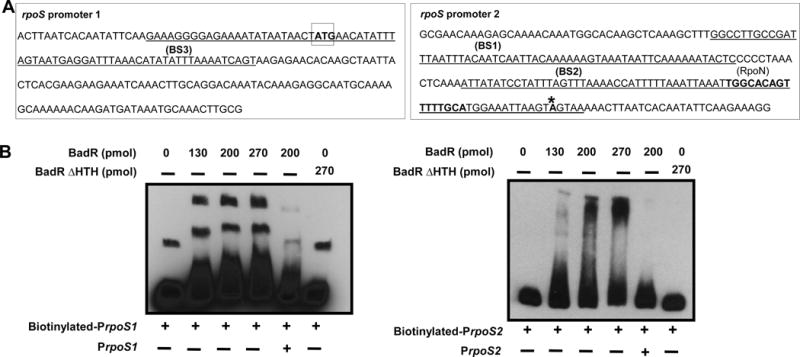
BadR binds rpoS promoters. Schematic diagram illustrating the two regions upstream of rpoS used for the EMSAs. RpoS 1 promoter (PrpoS 1(813088-813282)) includes the RpoS start codon (boxed) and BosR binding site 3 (BS3 -Underlined). RpoS2 promoter (PrpoS 2(813258-813471)) includes the RpoN binding site (bold), both the BosR binding sites 1 and 2 (BS1, BS2 -Underlined), and the rpoS transcriptional start site (bolded with asterisk) (A). 5′ biotin-labeled promoters (2 nmols) were mixed with various amounts of purified BadR(734081-735289) (130, 200, 270 pmols) in a 20μl binding reaction. Some reactions were incubated with unlabeled promoters (200-fold molar excess) for competition reactions or a N′ terminal HTH deficient BadR(734225-735289) (BadR Δ HTH) (B). The binding reactions were incubated at room temperature for 20 minutes, run on a 6% polyacrylamide gel, and transferred onto a positively charged Nylon membrane. After transfer the membrane was cross-linked by UV irradiation, blocked, incubated with streptavidin-horseradish peroxidase conjugate and luminol enhancer substrate solution allowed visualization of labeled DNA by exposure to X-ray film.
We hypothesized BadR may be analogous to NagC which is unable to bind DNA in the presence of the inducer GlcNAc-6P. DNA-binding assays were performed with increasing concentrations of GlcNAc-6P (10-200nmols). Increasing amounts of GlcNAc-6P relieved binding of BadR from PrpoS1 and PrpoS2 (Fig. 5).
To assess if other sugars were able to inhibit BadR binding, gel mobility shift reactions were performed in the presence of other sugars including: glucose-6P (Glc-6P), xylulose-5P, ribose-5P, xylose, or chitobiose (Fig. 6). Densitometry using ImageJ software assessed inhibition of binding by each sugar (Table S1) (Schneider et al., 2012). The shifted band corresponding to BadR bound to the rpoS promoters in the absence of sugars was designated as 100% bound. These studies demonstrated that all phosphorylated sugars inhibited BadR binding to the rpoS promoters in a concentration dependent manner, whereas unphosphorylated sugars, such as chitobiose, xylose, and acetate (data not shown), did not disrupt DNA binding (Fig. 6).
Fig. 6.
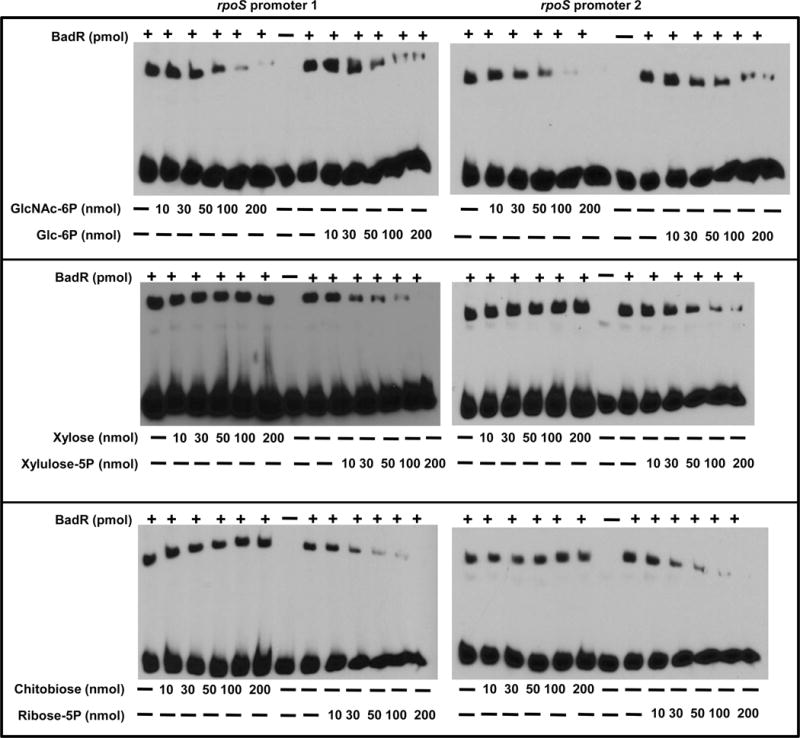
Phosphorylated sugars alleviate BadR binding to rpoS promoters. Two nmols of 5′ biotinylated rpoS promoters (PrpoS 1(A), PrpoS 2 (B)) were mixed with 130 pmols of WT BadR. The binding reactions were performed in the absence or presence of various sugars (GlcNAc-6P, glucose-6P (Glc-6P), xylulose-5P, ribose-5P, xylose, or chitobiose). Binding reactions with various sugar concentrations (10, 30, 50,100, or 200mM) determined the influence of each sugar on BadR binding to the rpoS promoters.
Site-directed mutations in BadR affect its binding to rpoS
To further characterize the ROK regulator, site-directed mutants of BadR were generated for use in mobility shift assays using the rpoS promoters. E. coli Mlc and NagC ROK regulators share residues thought to be important for binding their inducer, which include: H194, D195, E244, H247, and E266 (Mlc numbering; Fig. 7A, B) (Pennetier et al., 2008). Of these residues, BadR maintains only H247, which corresponds to H243 in Figure 7B. Consistent with the phenotype observed with E. coli NagC and Mlc H247 mutants, BadR with a H243A substitution was unable to bind to the rpoS promoter, which suggests H243 may also be critical for DNA binding of BadR (Fig. 7C, lane 5). Mutations in H194, E244, E266, and G246 in the E. coli NagC resulted in proteins insensitive to their inducer, GlcNAc-6P (Pennetier et al., 2008). Since BadR does not maintain any of these conserved residues, we focused on BadR-unique residues. In silico analyses utilizing the UniProtKB/Swiss-Prot database, suggested acetate could serve as a putative ligand of BadR. Acetate’s conversion to acetyl-P plays a crucial role in triggering vertebrate specific adaptation (Fig. 8C) (Raju et al., 2011; Van Laar et al., 2012; Sze & Li, 2011). Therefore, we decided to mutate N253 and P256 residues in BadR’s inducer binding domain (IBD) because previous studies identified N91 and P232 as crucial residues in mediating acetate kinase’s ability to bind acetate in Methanosarcina thermophila (Ingram-Smith et al., 2005). N253A/P256A BadR mutant bound to the rpoS promoter (Fig. 7C, lane 7) and responded to various sugars similarly as the WT (Fig. S1 and Table S2). The last 18 amino acids of E. coli Mlc are critical in orienting the HTH domain to facilitate DNA binding (Schiefner et al., 2005). Removal of the entire IBD C-terminal domain yielded a mutant BadR unable to bind the rpoS promoter (Fig. 7C, lane 4). Mutations in the putative dimerization/metal binding residues (C254, C257) also prevented BadR from binding to the rpoS promoter (Fig. 7C, lane 6) and parallels results seen in equivalent Mlc mutants in E. coli. Ultimately, this demonstrated that C254 and C257 may also play a structural role and contribute to orientation of the HTH domain in BadR (Schiefner et al., 2005). Taken together, BadR has several residues important for function that are shared with both Mlc and NagC.
Fig. 7.
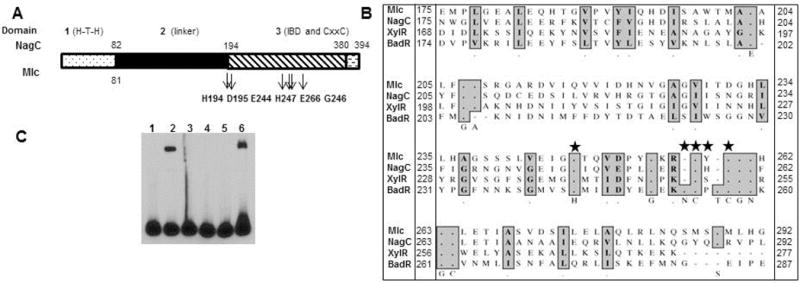
Characterization of BadR: Site-directed mutations and domain-deficient BadRs. Schematic representation of BadR domains (A) and comparisons of residues in ROK inducer-binding domains (IBD). Mutated residues are indicated (star) (B). 5′ biotinylated rpoS promoter 1 (2 nmols) was incubated with WT or mutant BadR proteins (130pmols) as previously described. Lanes: binding reaction absent of BadR (1), WT BadR(734081-735289) (2), BadR Δ HTH (BadR(734225-735289)) (3), BadR deficient of inducer binding domain BadR(734081-734659) (BadRΔIBD) (4), BadR with a site directed mutation in the equivalent residue of the ROK conserved residue, H247, (H243A) (5), BadR with site-directed mutations in the putative ROK CxxC metal binding/dimerization residues, (C254A/C257A) (6), BadR with site directed mutations in unique residues in BadR’s putative inducer binding domain (N253A/P255A) (7) (C).
Fig. 8.
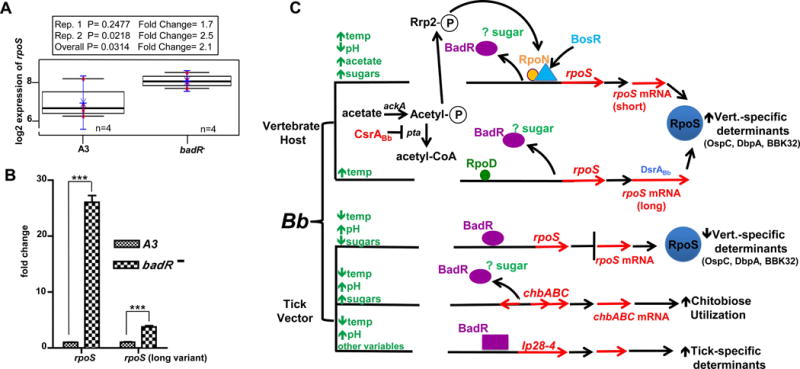
BadR represses rpoS to regulate host-specific adaptation in B. burgdorferi. The rpoS gene is upregulated in a badR-deficient B. burgdorferi strain. Log2 fold change of rpoS derived from two microarrays from (pH7.6/32°C) grown cultures. P values from pooled (overall) and individual arrays (Rep.1, Rep.2) are indicated using n=4 for both A3 (WT) and badR− (A). Quantitative RT-PCR fold changes of rpoS are indicated with P≤0.01. Error bars indicate s.e.m. (B). Predicted BadR regulatory network. Many activators of rpoS have been identified (e.g. DsrABb, BosR, RpoN, RpoD, CsrABb). Our study is the first to demonstrate BadR as a repressor of rpoS and thus may facilitate the spirochete’s transition back into ticks. A model for BadR regulation entails the following: 1.) BadR binds upstream of rpoS to repress rpoS in unfed ticks; 2.) Since ROK regulators are nutrient-responsive, BadR regulation may be modulated directly or indirectly by nutrient inducers; 3.) Nutrient surges from the blood meal may provide the inducer to allow BadR’s derepression of rpoS; and 4.) BadR regulates chitobiose utilization genes and may activate genes to enhance the spirochetes maintenance in ticks (lp28-4 genes). square= activation, oval=repression by BadR, respectively (C).
Up-regulation of vertebrate-specific determinants and down-regulation of tick-specific determinants in badR-deficient strains
To identify, further, BadR’s regulatory network, badR− and parental WT were compared using microarray analyses of these cultures grown under laboratory growth conditions (pH7.6, 32°C). Two independent but identically designed microarrays were performed and results were consistent with our immunoblot findings (e.g. up-regulation of chitobiose utilization genes in badR−) (Table 1, and accession GSE38827). To validate our microarray, quantitative RT-PCR was performed on a handful of transcripts (Table 2 and Fig. 8). Table 1 demonstrates data derived from both microarrays and specifically displays genes with at least a 2-fold difference in gene expression when comparing badR− to WT strains. Approximately one-third (39%) of the genes displayed in Table 1 were down-regulated in badR− compared to WT. Among those down-regulated genes, a large proportion (43% and 29%) was present on the chromosome and lp28-4, respectively. The majority of these down-regulated genes (63%) are annotated as hypothetical proteins. Approximately two-thirds (62%) of the genes displayed in Table 1 were up-regulated in badR− compared to WT. Among those up-regulated genes, most (38% and 17%) were present on circular plasmids 32 (cp32 1-4) and the chromosome, respectively. The majority (65%) of these up-regulated genes are annotated as hypothetical proteins. Intriguingly, badR− displayed up-regulation of rpoS, as well as genes regulated by the Rrp2-RpoN-RpoS pathway, such as ospC, dbpA, dbpB, bba07, bba64, and bb844. Other factors important for virulence in vertebrate hosts including antigens recognized by Lyme disease patient sera (e.g. BBP28 (mlp), BBI42, BBQ47 (erpX), BBS41 (erpG), BBO39 (erpL), BBG23, and BBQ03) were also up-regulated in badR− (Xu et al., 2008) (Fig. 8, Table 1, and Table 2).
Table 1.
Genes differentially expressed in the badR-deficient strain.
| Gene ID | Function (Annotation) | Replicon | FCOverall | 95%CILow | 95%CIHigh | POverall | FDR |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Up-regulated in badR− | |||||||
| BBB19 | outer surface protein C (ospC) | cp26 | 75.21 | 54.07 | 104.61 | 2.84E-08 | 4.74E-06 |
| BBA36 | lipoprotein | lp54 | 64.27 | 52.31 | 78.97 | 6.62E-10 | 1.02E-06 |
| BBA37 | hypothetical protein | lp54 | 61.94 | 48.21 | 79.58 | 3.93E-09 | 1.53E-06 |
| BBA65 | hypothetical protein | lp54 | 54.41 | 39.08 | 75.74 | 5.66E-08 | 6.69E-06 |
| BBA73 | antigen, P35, putative | lp54 | 36.52 | 27.23 | 48.96 | 4.97E-08 | 6.37E-06 |
| BBD07 | hypothetical protein | lp17 | 33.11 | 13.25 | 82.78 | 5.37E-04 | 8.42E-03 |
| BBA25 | decorin binding protein B (dbpB) | lp54 | 32.85 | 17.48 | 61.71 | 3.50E-05 | 1.15E-03 |
| BBA72 | hypothetical protein | lp54 | 30.38 | 20.93 | 44.10 | 5.91E-07 | 3.63E-05 |
| BBB05 | PTS system, chitobiose-specific IIA component (chbC) | cp26 | 24.06 | 17.73 | 32.64 | 1.99E-07 | 1.60E-05 |
| BBJ23 | hypothetical protein | lp38 | 22.83 | 13.95 | 37.36 | 1.19E-05 | 4.59E-04 |
| BBA66 | antigen, P35, putative | lp54 | 19.76 | 14.63 | 26.69 | 3.00E-07 | 2.03E-05 |
| BBA07 | chpAI protein, putative | lp54 | 17.67 | 12.25 | 25.49 | 2.15E-06 | 1.00E-04 |
| BBA71 | hypothetical protein | lp54 | 17.48 | 13.52 | 22.59 | 1.12E-07 | 1.23E-05 |
| BBH41 | hypothetical protein | lp28-3 | 17.23 | 10.13 | 29.29 | 4.48E-05 | 1.41E-03 |
| BBB06 | PTS system, chitobiose-specific IIB component (chbA) | cp26 | 17.07 | 12.03 | 24.23 | 1.64E-06 | 8.19E-05 |
| BBA05 | antigen, S1 | lp54 | 16.00 | 9.34 | 27.42 | 6.13E-05 | 1.66E-03 |
| BB0844 | hypothetical protein | chromosome | 15.82 | 9.65 | 25.91 | 3.25E-05 | 1.11E-03 |
| BBA33 | hypothetical protein | lp54 | 13.89 | 10.18 | 18.95 | 1.14E-06 | 6.49E-05 |
| BBP27 | rev protein | cp32-1 | 12.50 | 10.33 | 15.14 | 2.65E-08 | 4.74E-06 |
| BBQ37 | hypothetical protein | lp56 | 10.48 | 6.48 | 16.95 | 9.13E-05 | 2.24E-03 |
| BBM29 | hypothetical protein | cp32-6 | 10.26 | 6.41 | 16.44 | 8.37E-05 | 2.21E-03 |
| BBP28 | lipoprotein | cp32-1 | 9.98 | 7.51 | 13.28 | 1.70E-06 | 8.19E-05 |
| BBM38 | ErpK | cp32-6 | 8.91 | 6.56 | 12.10 | 4.65E-06 | 1.99E-04 |
| BBB07 | outer surface protein, putative | cp26 | 8.62 | 7.30 | 10.17 | 3.08E-08 | 4.74E-06 |
| BBO39 | ErpL | cp32-7 | 8.50 | 5.66 | 12.77 | 5.17E-05 | 1.53E-03 |
| BBA34 | oligopeptide ABC transporter, periplasmic oligopeptide-binding protein (oppAV) | lp54 | 7.83 | 5.10 | 12.02 | 1.04E-04 | 2.43E-03 |
| BBA24 | decorin binding protein A (dbpA) | lp54 | 7.65 | 6.28 | 9.32 | 2.18E-07 | 1.60E-05 |
| BBK32 | immunogenic protein P35 | lp36 | 7.36 | 4.57 | 11.86 | 2.82E-04 | 5.33E-03 |
| BBA04 | antigen, S2 | lp54 | 7.06 | 5.54 | 8.99 | 1.68E-06 | 8.19E-05 |
| BBB04 | PTS system, chitobiose-specific IIC component (chbB) | cp26 | 6.71 | 5.34 | 8.44 | 1.33E-06 | 7.04E-05 |
| BBJ01 | hypothetical protein | lp38 | 5.66 | 4.60 | 6.96 | 1.24E-06 | 6.81E-05 |
| BBI42 | hypothetical protein | lp28-4 | 4.96 | 3.67 | 6.71 | 4.87E-05 | 1.47E-03 |
| BBS41 | ErpG | cp32-3 | 4.84 | 3.79 | 6.18 | 1.04E-05 | 4.11E-04 |
| BBQ05 | antigen, P35, putative | lp56 | 4.74 | 3.36 | 6.69 | 1.65E-04 | 3.58E-03 |
| BBF01 | erpD protein, putative | lp28-1 | 4.56 | 3.34 | 6.23 | 9.18E-05 | 2.24E-03 |
| BBE31 | antigen, P35, putative | lp-25 | 4.53 | 2.98 | 6.89 | 7.91E-04 | 1.16E-02 |
| BBQ03 | outer membrane protein, putative | lp56 | 4.50 | 3.38 | 6.00 | 5.33E-05 | 1.55E-03 |
| BBK12 | hypothetical protein | lp36 | 4.43 | 2.46 | 7.96 | 6.81E-03 | 4.99E-02 |
| BBK03 | hypothetical protein | lp36 | 4.14 | 2.85 | 6.01 | 5.38E-04 | 8.42E-03 |
| BBJ48 | hypothetical protein | lp38 | 4.12 | 2.73 | 6.22 | 1.11E-03 | 1.52E-02 |
| BBK53 | outer membrane protein | lp36 | 4.07 | 2.97 | 5.59 | 1.88E-04 | 3.80E-03 |
| BBK07 | hypothetical protein | lp36 | 4.00 | 2.94 | 5.44 | 1.69E-04 | 3.58E-03 |
| BBA64 | antigen, P35 | lp54 | 3.95 | 2.54 | 6.14 | 2.06E-03 | 2.33E-02 |
| BBQ89 | hypothetical protein | lp56 | 3.94 | 3.11 | 5.00 | 2.57E-05 | 9.40E-04 |
| BBR28 | lipoprotein | cp32-4 | 3.71 | 2.64 | 5.21 | 5.04E-04 | 8.08E-03 |
| BBD24 | hypothetical protein | lp17 | 3.69 | 2.42 | 5.60 | 2.04E-03 | 2.33E-02 |
| BBK52 | protein p23 | lp36 | 3.56 | 2.35 | 5.41 | 2.36E-03 | 2.42E-02 |
| BBM39 | hypothetical protein | cp32-6 | 3.40 | 2.74 | 4.22 | 2.83E-05 | 9.90E-04 |
| BBQ38 | hypothetical protein | lp56 | 3.19 | 1.94 | 5.25 | 1.06E-02 | 6.65E-02 |
| BBP03 | hypothetical protein | cp32-1 | 3.17 | 2.54 | 3.96 | 5.72E-05 | 1.63E-03 |
| BBJ24 | hypothetical protein | lp38 | 3.16 | 1.91 | 5.21 | 1.14E-02 | 6.92E-02 |
| BBA57 | hypothetical protein | lp54 | 3.14 | 2.53 | 3.90 | 4.84E-05 | 1.47E-03 |
| BBA06 | hypothetical protein | lp54 | 3.04 | 2.07 | 4.45 | 3.08E-03 | 2.93E-02 |
| BBA01 | hypothetical protein | lp54 | 2.92 | 2.15 | 3.97 | 9.83E-04 | 1.37E-02 |
| BBL30 | hypothetical protein | cp32-8 | 2.86 | 1.84 | 4.44 | 9.66E-03 | 6.27E-02 |
| BBB10 | hypothetical protein | cp26 | 2.85 | 1.73 | 4.70 | 1.77E-02 | 8.98E-02 |
| BBJ46 | hypothetical protein | lp38 | 2.79 | 1.99 | 3.90 | 2.34E-03 | 2.42E-02 |
| BBR29 | hypothetical protein | cp32-4 | 2.78 | 1.94 | 3.98 | 3.55E-03 | 3.19E-02 |
| BBP38 | ErpA | cp32-1 | 2.78 | 1.87 | 4.13 | 6.33E-03 | 4.77E-02 |
| BBO40 | ErpM | cp32-7 | 2.76 | 1.68 | 4.53 | 2.03E-02 | 9.65E-02 |
| BBJ03 | hypothetical protein | lp38 | 2.74 | 1.55 | 4.84 | 3.71E-02 | 1.39E-01 |
| BBO02 | hypothetical protein | cp32-7 | 2.72 | 2.30 | 3.22 | 2.02E-05 | 7.59E-04 |
| BBS31 | hypothetical protein | cp32-3 | 2.69 | 1.77 | 4.10 | 1.01E-02 | 6.41E-02 |
| BBQ47 | ErpX | lp56 | 2.69 | 1.81 | 4.00 | 7.54E-03 | 5.34E-02 |
| BBS42 | BapA | cp32-3 | 2.66 | 1.86 | 3.82 | 4.52E-03 | 3.78E-02 |
| BBQ82 | hypothetical protein | lp56 | 2.65 | 2.07 | 3.41 | 4.55E-04 | 7.45E-03 |
| BBR40 | ErpH | cp32-4 | 2.62 | 1.91 | 3.59 | 2.34E-03 | 2.42E-02 |
| BBP29 | hypothetical protein | cp32-1 | 2.60 | 1.73 | 3.93 | 1.07E-02 | 6.65E-02 |
| BBJ02 | hypothetical protein | lp38 | 2.59 | 2.09 | 3.20 | 1.77E-04 | 3.68E-03 |
| BB0845 | hypothetical protein | chromosome | 2.55 | 1.69 | 3.86 | 1.20E-02 | 7.11E-02 |
| BBR31 | hypothetical protein | cp32-4 | 2.54 | 1.57 | 4.13 | 2.62E-02 | 1.14E-01 |
| BBA32 | hypothetical protein | lp54 | 2.54 | 1.91 | 3.36 | 1.45E-03 | 1.82E-02 |
| BBP02 | hypothetical protein | cp32-1 | 2.51 | 2.10 | 2.99 | 5.82E-05 | 1.63E-03 |
| BBO29 | hypothetical protein | cp32-7 | 2.49 | 1.63 | 3.81 | 1.62E-02 | 8.48E-02 |
| BBJ25 | hypothetical protein | lp38 | 2.47 | 1.89 | 3.24 | 1.29E-03 | 1.66E-02 |
| BBK50 | immunogenic protein P37 | lp36 | 2.44 | 1.48 | 4.02 | 3.65E-02 | 1.39E-01 |
| BBP35 | BppA | cp32-1 | 2.42 | 1.84 | 3.18 | 1.59E-03 | 1.96E-02 |
| BBQ52 | hypothetical protein | lp56 | 2.42 | 2.04 | 2.87 | 6.14E-05 | 1.66E-03 |
| BBP01 | hypothetical protein | cp32-1 | 2.40 | 1.96 | 2.94 | 2.27E-04 | 4.53E-03 |
| BBJ47 | hypothetical protein | lp38 | 2.38 | 1.72 | 3.29 | 5.11E-03 | 4.16E-02 |
| BBS38 | BppA | cp32-3 | 2.38 | 1.81 | 3.12 | 1.83E-03 | 2.16E-02 |
| BBM02 | hypothetical protein | cp32-6 | 2.37 | 1.95 | 2.88 | 1.86E-04 | 3.80E-03 |
| BBS02 | hypothetical protein | cp32-3 | 2.36 | 1.98 | 2.82 | 9.46E-05 | 2.24E-03 |
| BBA23 | hypothetical protein | lp54 | 2.36 | 1.65 | 3.37 | 9.13E-03 | 6.02E-02 |
| BBM01 | antigen, P35, putative | cp32-6 | 2.35 | 1.95 | 2.84 | 1.55E-04 | 3.45E-03 |
| BBR01 | antigen, P35, putative | cp32-4 | 2.35 | 1.95 | 2.83 | 1.39E-04 | 3.15E-03 |
| BBM35 | BppA | cp32-6 | 2.32 | 1.81 | 2.99 | 1.27E-03 | 1.66E-02 |
| BBN38 | ErpP | cp32-9 | 2.32 | 1.77 | 3.05 | 2.08E-03 | 2.33E-02 |
| BBE04 | hypothetical protein | lp-25 | 2.31 | 1.53 | 3.51 | 2.12E-02 | 9.94E-02 |
| BBS01 | hypothetical protein | cp32-3 | 2.29 | 1.84 | 2.85 | 5.48E-04 | 8.44E-03 |
| BBA35 | hypothetical protein | lp54 | 2.27 | 1.78 | 2.89 | 1.24E-03 | 1.64E-02 |
| BBK48 | immunogenic protein P37, putative | lp36 | 2.26 | 1.56 | 3.26 | 1.41E-02 | 7.94E-02 |
| BBP10 | hypothetical protein | cp32-1 | 2.25 | 1.84 | 2.75 | 3.89E-04 | 6.65E-03 |
| BBQ88 | hypothetical protein | lp56 | 2.23 | 1.47 | 3.39 | 2.59E-02 | 1.14E-01 |
| BBJ19 | hypothetical protein | lp38 | 2.23 | 1.71 | 2.90 | 2.35E-03 | 2.42E-02 |
| BBG22 | hypothetical protein | lp28-2 | 2.23 | 1.73 | 2.87 | 1.84E-03 | 2.16E-02 |
| BBH36 | hypothetical protein | lp28-3 | 2.20 | 1.72 | 2.82 | 1.76E-03 | 2.12E-02 |
| BBQ43 | BppA | lp56 | 2.20 | 1.80 | 2.69 | 4.47E-04 | 7.40E-03 |
| BBM37 | BppC | cp32-6 | 2.20 | 1.73 | 2.78 | 1.35E-03 | 1.72E-02 |
| BBR10 | hypothetical protein | cp32-4 | 2.20 | 1.75 | 2.75 | 9.81E-04 | 1.37E-02 |
| BBO30 | hypothetical protein | cp32-7 | 2.19 | 1.72 | 2.80 | 1.69E-03 | 2.06E-02 |
| BBO36 | BppA | cp32-7 | 2.19 | 1.80 | 2.65 | 3.63E-04 | 6.28E-03 |
| BBK11 | hypothetical protein | lp36 | 2.18 | 1.80 | 2.63 | 3.24E-04 | 5.87E-03 |
| BBN10 | hypothetical protein | cp32-9 | 2.17 | 1.77 | 2.67 | 5.42E-04 | 8.42E-03 |
| BBQ06 | hypothetical protein | lp56 | 2.16 | 1.57 | 2.98 | 9.18E-03 | 6.03E-02 |
| BBQ10 | hypothetical protein | lp56 | 2.16 | 1.79 | 2.60 | 3.02E-04 | 5.53E-03 |
| BBJ26 | ABC transporter, ATP-binding protein | lp38 | 2.16 | 1.55 | 3.00 | 1.07E-02 | 6.65E-02 |
| BBQ45 | BppC | lp56 | 2.15 | 1.63 | 2.83 | 4.17E-03 | 3.59E-02 |
| BBL36 | BppA | cp32-8 | 2.14 | 1.77 | 2.58 | 3.52E-04 | 6.23E-03 |
| BBN30 | hypothetical protein | cp32-9 | 2.13 | 1.63 | 2.77 | 3.56E-03 | 3.19E-02 |
| BBO01 | hypothetical protein | cp32-7 | 2.12 | 1.78 | 2.53 | 2.46E-04 | 4.79E-03 |
| BBQ29 | hypothetical protein | lp56 | 2.11 | 1.64 | 2.71 | 2.87E-03 | 2.80E-02 |
| BBH02 | hypothetical protein | lp28-3 | 2.10 | 1.49 | 2.95 | 1.49E-02 | 8.11E-02 |
| BBA76 | thymidylate synthase | lp54 | 2.07 | 1.56 | 2.73 | 6.00E-03 | 4.58E-02 |
| BBN35 | BppA | cp32-9 | 2.07 | 1.63 | 2.62 | 2.36E-03 | 2.42E-02 |
| BB0771 | RNA polymerase sigma factor (rpoS) | chromosome | 2.06 | 1.39 | 3.05 | 3.14E-02 | 1.28E-01 |
| BBS04 | hypothetical protein | cp32-3 | 2.06 | 1.71 | 2.48 | 4.94E-04 | 7.99E-03 |
| BBG27 | hypothetical protein | lp28-2 | 2.05 | 1.73 | 2.44 | 2.84E-04 | 5.33E-03 |
| BBF28 | hypothetical protein | lp28-1 | 2.05 | 1.38 | 3.03 | 3.25E-02 | 1.30E-01 |
| BBL26 | hypothetical protein | cp32-8 | 2.04 | 1.55 | 2.69 | 6.12E-03 | 4.63E-02 |
| BBR26 | hypothetical protein | cp32-4 | 2.04 | 1.52 | 2.73 | 8.39E-03 | 5.66E-02 |
| BBE05 | hypothetical protein | lp-25 | 2.02 | 1.41 | 2.90 | 2.40E-02 | 1.07E-01 |
| BB0519 | grpE protein (grpE) | chromosome | 2.02 | 1.46 | 2.81 | 1.61E-02 | 8.47E-02 |
| BBP26 | hypothetical protein | cp32-1 | 2.02 | 1.53 | 2.67 | 6.71E-03 | 4.94E-02 |
| BBN01 | hypothetical protein | cp32-9 | 2.02 | 1.70 | 2.40 | 3.52E-04 | 6.23E-03 |
| BBE21 | hypothetical protein | lp-25 | 2.01 | 1.46 | 2.76 | 1.41E-02 | 7.94E-02 |
| BBP04 | hypothetical protein | cp32-1 | 2.01 | 1.65 | 2.45 | 9.54E-04 | 1.36E-02 |
|
| |||||||
| Down-regulated in badR− | |||||||
|
| |||||||
| BBI29 | hypothetical protein | lp28-4 | −90.58 | −128.92 | −63.65 | 3.52E-08 | 4.93E-06 |
| BBU05 | plasmid partition protein, putative | lp21 | −51.70 | −75.70 | −35.31 | 2.12E-07 | 1.60E-05 |
| BBI24 | hypothetical protein | lp28-4 | −40.19 | −68.62 | −23.54 | 6.09E-06 | 2.53E-04 |
| BBI11 | hypothetical protein | lp28-4 | −35.56 | −50.99 | −24.80 | 3.04E-07 | 2.03E-05 |
| BBI12 | hypothetical protein | lp28-4 | −30.74 | −39.39 | −24.00 | 1.78E-08 | 4.56E-06 |
| BBI21 | hypothetical protein | lp28-4 | −27.35 | −33.44 | −22.36 | 3.99E-09 | 1.53E-06 |
| BBI22 | conserved hypothetical protein, | lp28-4 | −21.83 | −27.66 | −17.23 | 2.96E-08 | 4.74E-06 |
| BBI23 | hypothetical protein | lp28-4 | −19.25 | −25.23 | −14.69 | 1.32E-07 | 1.35E-05 |
| BBI14 | hypothetical protein | lp28-4 | −19.18 | −25.51 | −14.43 | 2.09E-07 | 1.60E-05 |
| BBI03 | hypothetical protein | lp28-4 | −17.79 | −29.88 | −10.59 | 3.44E-05 | 1.15E-03 |
| BBI41 | hypothetical protein | lp28-4 | −14.57 | −16.86 | −12.60 | 1.51E-09 | 1.16E-06 |
| BBI28 | hypothetical protein | lp28-4 | −9.99 | −16.01 | −6.24 | 9.19E-05 | 2.24E-03 |
| BBI17 | hypothetical protein | lp28-4 | −9.68 | −13.28 | −7.06 | 4.44E-06 | 1.95E-04 |
| BBU07 | hypothetical protein | lp21 | −9.03 | −11.11 | −7.33 | 1.72E-07 | 1.60E-05 |
| BBI20 | hypothetical protein | lp28-4 | −7.99 | −10.45 | −6.11 | 2.42E-06 | 1.09E-04 |
| BBI16 | hypothetical protein | lp28-4 | −7.90 | −9.97 | −6.27 | 7.45E-07 | 4.41E-05 |
| BBU04 | hypothetical protein | lp21 | −7.67 | −11.60 | −5.07 | 8.58E-05 | 2.21E-03 |
| BBI13 | hypothetical protein | lp28-4 | −6.92 | −8.33 | −5.74 | 2.04E-07 | 1.60E-05 |
| BBI19 | hypothetical protein | lp28-4 | −6.37 | −9.94 | −4.09 | 2.89E-04 | 5.36E-03 |
| BBI06 | pfs protein (pfs) | lp28-4 | −6.04 | −9.81 | −3.72 | 6.64E-04 | 1.00E-02 |
| BBI31 | hypothetical protein | lp28-4 | −5.02 | −6.42 | −3.92 | 9.37E-06 | 3.79E-04 |
| BBI26 | multidrug-efflux transporter | lp28-4 | −4.59 | −7.23 | −2.91 | 1.31E-03 | 1.67E-02 |
| BBI02 | hypothetical protein | lp28-4 | −4.35 | −5.87 | −3.21 | 9.31E-05 | 2.24E-03 |
| BB0692 | hypothetical protein | chromosome | −3.97 | −8.92 | −1.77 | 4.33E-02 | 1.56E-01 |
| BBU09 | hypothetical protein | lp21 | −3.57 | −6.43 | −1.98 | 1.55E-02 | 8.29E-02 |
| BB0019 | hypothetical protein | chromosome | −3.44 | −4.42 | −2.68 | 8.63E-05 | 2.21E-03 |
| BBI15 | hypothetical protein | lp28-4 | −3.40 | −4.46 | −2.59 | 1.66E-04 | 3.58E-03 |
| BBA20 | hypothetical protein | lp54 | −2.87 | −5.28 | −1.57 | 3.99E-02 | 1.47E-01 |
| BB0548 | DNA polymerase I (polA) | chromosome | −2.84 | −4.19 | −1.93 | 4.83E-03 | 3.99E-02 |
| BBA75 | hypothetical protein | lp54 | −2.84 | −4.78 | −1.69 | 2.17E-02 | 1.01E-01 |
| BB0310 | hypothetical protein | chromosome | −2.82 | −5.11 | −1.56 | 3.93E-02 | 1.46E-01 |
| BBU06 | hypothetical protein | lp21 | −2.79 | −3.37 | −2.32 | 3.65E-05 | 1.17E-03 |
| BBJ10 | hypothetical protein | lp38 | −2.78 | −4.10 | −1.88 | 5.62E-03 | 4.48E-02 |
| BBA19 | hypothetical protein | lp54 | −2.74 | −3.85 | −1.94 | 2.92E-03 | 2.83E-02 |
| BB0077 | hypothetical protein | chromosome | −2.73 | −3.74 | −2.00 | 1.72E-03 | 2.08E-02 |
| BBU01 | hypothetical protein | lp21 | −2.67 | −3.67 | −1.94 | 2.16E-03 | 2.35E-02 |
| BBA21 | hypothetical protein | lp54 | −2.63 | −3.41 | −2.03 | 6.44E-04 | 9.81E-03 |
| BB0790 | hypothetical protein | chromosome | −2.63 | −3.88 | −1.78 | 7.65E-03 | 5.37E-02 |
| BB0210 | surface-located membrane protein 1 (lmp1) | chromosome | −2.57 | −3.50 | −1.89 | 2.21E-03 | 2.38E-02 |
| BB0828 | DNA topoisomerase I (topA) | chromosome | −2.57 | −3.48 | −1.90 | 2.09E-03 | 2.33E-02 |
| BB0198 | guanosine-3′,5′-bis(di-P) 3′-pyrophosphohydrolase (RelBbu) | chromosome | −2.56 | −3.14 | -2.09 | 1.37E-04 | 3.14E-03 |
| BBI01 | hypothetical protein | lp28-4 | −2.55 | −3.57 | −1.82 | 4.18E-03 | 3.59E-02 |
| BB0200 | D-alanine–D-alanine ligase (ddlA) | chromosome | −2.52 | −3.26 | −1.95 | 8.06E-04 | 1.17E-02 |
| BBI33 | hypothetical protein | lp28-4 | −2.49 | −3.13 | −1.98 | 4.05E-04 | 6.85E-03 |
| BBU08 | hypothetical protein | lp21 | −2.45 | −2.99 | −2.01 | 1.70E-04 | 3.58E-03 |
| BBG31 | hypothetical protein | lp28-2 | −2.42 | −2.83 | −2.08 | 2.69E-05 | 9.61E-04 |
| BB0197 | protoporphyrinogen oxidase, putative | chromosome | −2.38 | −3.15 | −1.80 | 2.17E-03 | 2.35E-02 |
| BB0673 | hypothetical protein | chromosome | −2.32 | −3.25 | −1.66 | 7.28E-03 | 5.21E-02 |
| BB0595 | hypothetical protein | chromosome | −2.31 | −2.82 | −1.90 | 2.70E-04 | 5.20E-03 |
| BB0251 | leucyl-tRNA synthetase (leuS) | chromosome | −2.31 | −3.04 | −1.76 | 2.31E-03 | 2.42E-02 |
| BB0012 | pseudouridylate synthase I (hisT) | chromosome | −2.30 | −3.16 | −1.67 | 5.95E-03 | 4.58E-02 |
| BB0528 | aldose reductase, putative | chromosome | −2.29 | −3.15 | −1.67 | 5.93E-03 | 4.58E-02 |
| BB0027 | hypothetical protein | chromosome | −2.28 | −2.98 | −1.75 | 2.17E-03 | 2.35E-02 |
| BBU02 | hypothetical protein | lp21 | −2.27 | −3.53 | −1.46 | 3.01E-02 | 1.25E-01 |
| BBN33 | hypothetical protein | cp32-9 | −2.24 | −3.32 | −1.51 | 1.97E-02 | 9.49E-02 |
| BBS35 | plasmid partition protein, putative | cp32-3 | −2.24 | −3.34 | −1.50 | 2.18E-02 | 1.01E-01 |
| BB0184 | carbon storage regulator (csrA) | chromosome | −2.20 | −3.16 | −1.54 | 1.42E-02 | 7.96E-02 |
| BB0013 | hypothetical protein | chromosome | −2.20 | −2.82 | −1.71 | 1.90E-03 | 2.21E-02 |
| BB0363 | periplasmic protein | chromosome | −2.19 | −2.83 | −1.69 | 2.40E-03 | 2.44E-02 |
| BB0758 | hypothetical protein | chromosome | −2.15 | −3.30 | −1.40 | 3.61E-02 | 1.38E-01 |
| BB0571 | uridylate kinase | chromosome | -2.14 | -2.83 | -1.61 | 4.89E-03 | 4.01E-02 |
| BB0782 | hypothetical protein | chromosome | −2.10 | −2.70 | −1.63 | 3.02E-03 | 2.90E-02 |
| BBB11 | hypothetical protein | cp26 | −2.09 | −2.62 | −1.67 | 1.48E-03 | 1.84E-02 |
| BB0768 | pyridoxal kinase (pdxK) | chromosome | −2.09 | −2.60 | −1.68 | 1.22E-03 | 1.63E-02 |
| BBB01 | hypothetical protein | cp26 | −2.08 | −2.76 | −1.58 | 5.61E-03 | 4.48E-02 |
| BB0754 | ABC transporter, ATP-binding protein | chromosome | −2.08 | −2.87 | −1.51 | 1.19E-02 | 7.11E-02 |
| BBH27 | hypothetical protein | lp28-3 | −2.08 | −2.96 | −1.45 | 1.96E-02 | 9.49E-02 |
| BB0305 | hypothetical protein | chromosome | −2.08 | −3.16 | −1.36 | 3.98E-02 | 1.47E-01 |
| BBB29 | PTS system, maltose and glucose-specific IIABC component (malX) | cp26 | −2.05 | −2.63 | −1.60 | 3.15E-03 | 2.97E-02 |
| BBA42 | hypothetical protein | lp54 | −2.05 | −3.17 | −1.33 | 4.83E-02 | 1.69E-01 |
| BB0080 | ABC transporter, ATP-binding protein | chromosome | −2.05 | −2.82 | −1.49 | 1.29E-02 | 7.48E-02 |
| BB0587 | methionyl-tRNA synthetase | chromosome | −2.05 | −2.86 | −1.47 | 1.58E-02 | 8.41E-02 |
| BB0078 | hypothetical protein | chromosome | −2.04 | −3.12 | −1.33 | 4.54E-02 | 1.62E-01 |
| BB0749 | hypothetical protein | chromosome | −2.03 | −2.73 | −1.52 | 8.88E-03 | 5.91E-02 |
| BB0009 | hypothetical protein | chromosome | −2.03 | −3.08 | −1.34 | 4.31E-02 | 1.56E-01 |
| BB0308 | hypothetical protein | chromosome | −2.02 | −2.89 | −1.42 | 2.29E-02 | 1.03E-01 |
| BB0249 | phosphatidyltransferase | chromosome | −2.02 | −2.56 | −1.60 | 2.47E-03 | 2.49E-02 |
| BBS36 | hypothetical protein | cp32-3 | −2.02 | −2.62 | −1.56 | 4.61E-03 | 3.83E-02 |
| BB0838 | hypothetical protein | chromosome | −2.02 | −3.09 | −1.32 | 4.85E-02 | 1.69E-01 |
Function (Annotations) are based on JCVI/CMR B. burgdorferi strain B31-and in (Fraser et al., 1997).
FCOverall= fold changes are between B31-A3 (WT) and badR− and derived by pooling both independent microarrays.
POverall= P value derived by pooling both independent microarrays.
Genes ≥2 fold differences in expression and POverall ≤0.05 are reported.
95%CILow-High =95% confidence intervals indicating range of fold change expression.
FDR = adjusted Benjamini-Hochberg False Discovery Rates are all statistically significant (≤0.2).
Genes validated by qRT-PCR and/or western blotting are bolded.
Table 2.
Quantitative RT-PCR of select genes.
| Gene ID | Function (Annotation) | Array | qRT-PCR |
|---|---|---|---|
| BB0771 | RNA polymerase sigma factor (rpoS) | 2.1 | 26.06 |
| RNA polymerase sigma factor (rpoS) – long transcript | N/A | 3.75 | |
| BBB19 | outer surface protein C (ospC) | 75.2 | 55.38 |
| BBA24 | decorin binding protein A (dbpA) | 7.7 | 30.91 |
| BBA34 | oligopeptide ABC transporter, periplasmic oligopeptide-binding protein (oppAV) | 7.8 | 211.37 |
| BBB05 | PTS system, chitobiose-specific IIA component (chbC) | 24.1 | 54.21 |
| BB0240 | glycerol uptake facilitator (glpF) | 1.8 | 7.04 |
| BBI42 | hypothetical protein | 5.0 | 7.07 |
| BBA07 | chpAI protein, putative | 17.7 | 362.04 |
| BBP27 | rev protein | 12.5 | 35.59 |
| BBP28 | lipoprotein | 10.0 | 16.95 |
| BBB29 | PTS system, maltose and glucose-specific IIABC component (malX) | −2.1 | −3.07 |
| BBI16 | hypothetical protein | −7.9 | Undetectable |
| BB0019 | hypothetical protein | −3.4 | −1.44 |
| BB0198 | guanosine-3′,5′-bis(di-P) 3′-pyrophosphohydrolase (RelBbu) | −2.6 | −1.19 |
| BBU05 | plasmid partition protein, putative | −51.7 | Undetectable |
Function (Annotations) are based on JCVI/CMR B. burgdorferi strain B31-and in (Fraser et al., 1997).
Exception of rpoS long transcript; qRT-PCR primers were used (Table S3) to amplify only the long rpoS transcript variant.
Values expressed as a ratio of expression for the gene in B31-A3 WT relative to badR− and derived by pooling both independent microarrays.
BadR binds to the same promoter as BosR in our DNA-binding assays. Ouyang et al. predicted additional putative BosR-regulated promoters (Ouyang et al., 2011). Interestingly, 25% of the putative BosR-regulated genes displayed significant expression changes in badR− including: bb0020 (pfpB), bb0055 (tpi), and bbb29 (malX), a PtsG homolog (Table 1, and accession GSE38827).
Collectively, these results suggest BadR functions as a versatile ROK regulator, regulating both chitobiose utilization and genes required for host adaptation (e.g. rpoS). BadR may also interplay with BosR at various gene targets. Our results demonstrate a regulatory network unique to B. burgdorferi pathogenesis, because a significant proportion of differentially expressed genes in badR− encode hypothetical proteins of unknown function and located on replicon, lp28-4; a plasmid shown to be important for the spirochete’s survival in ticks (Strother et al., 2005; Elias et al., 2002).
BadR is required for mammalian infectivity
To determine if BadR is critical for B. burgdorferi to infect the mammalian host, C3H/HeN mice were infected with 105 B. burgdorferi strains by needle inoculation. Fourteen days post infection the badR-deficient strains were not isolated from any of the infected tissues, while the wild-type parental strain, B31-A3, could be isolated from all the tissues tested (Table 4). Since badR-deficient strains display increased expression of outer surface antigens (e.g. OspC and DbpA) which might enhance bacterial clearance, immunodeficient SCID mice were also infected with 105 spirochetes. Similarly, badR-deficient B. burgdorferi could not be propagated from the infected tissues of SCID mice 14 days post infection. Since the innate immune system of SCID mice is still intact, it is possible that key components of innate immunity (e.g. complement) play a role in the clearance of the badR-deficient strains. Moreover, other mechanisms responsible for the unsuccessful colonization, dissemination, and survival of these mutant strains in mammalian hosts cannot be ruled out. Taken together, BadR is critical for B. burgdorferi to colonize mammalian hosts.
Table 4.
Mammalian infectivity analysis of badR-deficient strains.
| No. of cultures positive /total No. | No. of mice positive/total No. of mice | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Mouse strain | Strain | Skin | Lymph | Spleen | Bladder | Heart | Joint | All sites | |
|
| |||||||||
| C3H/HeN | A3 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 30/30 | 5/5 |
| badR1− | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/30 | 0/5 | |
| badR2− | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/30 | 0/5 | |
|
| |||||||||
| BALB/c | A3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 18/18 | 3/3 |
| badR1− | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/18 | 0/3 | |
| badR2− | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/18 | 0/3 | |
|
| |||||||||
| SCID | A3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 18/18 | 3/3 |
| badR1− | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/18 | 0/3 | |
| badR2− | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/18 | 0/3 | |
DISCUSSION
BadR repression of RpoS
For the host-propagated pathogen B. burgdorferi, adaptive phenotypic changes required to switch between its two hosts is energetically expensive and resembles an all-or-none response. At some point, there is a crucial signal that triggers the spirochetes to commit, spend energy, and undergo these adaptive changes. The transcription factor RpoS has been termed the “gatekeeper” because it promotes rapid adaptation as it expresses the genes required for B. burgdorferi to transmit to and infect vertebrate hosts (Caimano et al., 2007; Dunham-Ems et al., 2012; Ouyang et al., 2012). Understandably, expression of rpoS is a committed step that must be appropriately regulated. A slew of studies have identified factors that activate rpoS, but there is a dearth of information on how rpoS is repressed to down-regulate vertebrate-specific adaptations, to allow B. burgdorferi’s transition back into ticks. While a reduction in intensity of external signals that lead to rpoS activation is one possibility, our study found that the transcriptional regulator BadR mediates rpoS repression by binding upstream regions of rpoS directly (Fig. 8C).
Intriguingly, the phenotype of badR-deficient strains is an increase in expression and level of rpoS and RpoS-regulated genes. Generally, expression of rpoS is mediated by the alternative σ factor RpoN. However RpoN-independent rpoS activation has also been suggested to occur through σ70 factor, RpoD (Samuels, 2011). Increased temperatures from blood meals promotes a small RNA, DsrABb, to unfold and promote RpoS translation from a longer rpoS mRNA, transcribed with the aid of RpoD (Lybecker & Samuels, 2007). Previous studies have shown acetyl phosphate (elevated due to inhibitory effects of Carbon Storage Regulator A (CsrABb) on phosphate acetyl-transferase (Pta)), phosphorylates and activates Rrp2, an enhancer binding protein which activates RpoN dependent transcription of rpoS (Karna et al., 2011; Sze et al., 2011). Borrelia oxidative stress regulator, BosR, also binds to upstream regions of rpoS and enhances rpoS expression (Ouyang et al., 2011; Hyde et al., 2006). It is plausible that the high RpoS levels and transcripts in a badR-deficient strain could be a consequence of modulating any one of these processes; however, our findings support the hypothesis that BadR is a direct transcriptional repressor of rpoS.
First, our microarray analyses did not reflect significant increases in expression of rrp2, rpoN, rpoD, dsrABb, csrABb, bosR, or acetate metabolic enzymes, pta and ackA, in badR−, suggesting increases in rpoS could not be attributed to BadR’s transcriptional regulation of these factors. To further support that BadR is a direct transcriptional repressor of rpoS, we found elevated levels of BadR in A3 WT grown under unfed-tick conditions where there is little or no RpoS levels. Furthermore, we found increases in both rpoS transcripts and RpoS levels in the badR-deficient strains compared to WT. We do understand, however, that these are in vitro conditions and the repression of rpoS by BadR needs to be addressed in future in vivo studies. Finally, to further support that BadR is a direct transcriptional repressor of rpoS, BadR bound to two regions upstream of rpoS in vitro.
Taken together, BadR appears to repress rpoS expression presumably by means of excluding the binding of rpoS activators, e.g. BosR, RpoN, and/or RpoD. Additionally, since BadR is a ROK family member comprised of regulators that are responsive to carbohydrates and typically bind sugars to modulate their regulatory function, we predict that BadR’s repression of rpoS is alleviated through its direct or indirect interaction with an inducing signal, probably a sugar or byproduct linked to nutrient metabolism (Fig. 8C).
ROK Regulators: Modulators of Virulence Potential
badR-deficient B. burgdorferi strains were completely unable to establish infection in both immunocompetent (C3H/HeN, BALB/c) and immunodeficient mice (SCID), indicating BadR is an important regulator of B. burgdorferi virulence. Microarray analyses revealed that BadR regulates the transcription of virulence genes. In the badR-deficient strains, there was an up-regulation of rpoS, the genes regulated by the Rrp2-RpoN-RpoS pathway, as well as other vertebrate specific factors important for virulence (e.g. antigens recognized by the sera of Lyme disease patients).
B. burgdorferi strains carry up to ten cp32 plasmids and numerous questions remain unanswered regarding cp32 genes and the role of their encoded proteins in pathogenesis (Stevenson et al., 2000; Kenedy et al., 2012). Because approximately one-third of genes up-regulated were from cp32 plasmids and approximately two-thirds of differentially expressed genes, both up- and down-regulated, were annotated as hypothetical proteins of unknown function in badR−, we suggest a number of these genes are likely critical for B. burgdorferi pathogenesis and virulence.
ROKs can function as both repressors and activators (Plumbridge, 1995). Our results imply BadR is an activator of tick-specific factors because badR− had significant decreases in expression of genes encoded on lp28-4. Previous studies found that B. burgdorferi strains absent of lp28-4 were unable to be transmitted from ticks to naïve mice and overall had reduced infectivity of tick midguts (Strother et al., 2005; Elias et al., 2002). Therefore, it has been hypothesized that lp28-4 is important for B. burgdorferi’s residence in the tick, specifically flat ticks and hence nutrient depleted environments (Tokarz et al., 2004). Consistent with this observation, our microarray showed down-regulation of guanosine-3′, 5′-bis (diphosphate) 3′-pyrophosphohydrolase (RelBbu) in the badR− strain. RelBbu is up-regulated during starvation and important for B. burgdorferi to adapt to stationary phase (Bugrysheva et al., 2005). Collectively, BadR positively regulates genes most likely important for the survival of spirochetes in the nutrient-limiting environments within the tick (Fig. 8C). Therefore, we hypothesize that badR-deficient strains will also have reduced infectivity in ticks and we will assess this in future in vivo studies.
The ability of BadR to repress vertebrate-specific virulence factors while simultaneously activating the expression of tick-specific factors demonstrates a central role of this regulator in the lifecycle of B. burgdorferi. We suggest that the dysregulation of these host-specific factors within the badR− mutant is the cause of the high level of attenuation seen in this strain in mice.
ROK Regulators: Modulated by Nutrients
Many studies highlight food availability as the principle factor for microbes to adapt and survive, as well as provide the evolutionary push for the development of pathogenic characteristics (Poncet et al., 2009). We investigated whether host-specific nutrients contribute to B. burgdorferi’s pathogenesis. Our study aimed to characterize the only apparent carbohydrate responsive regulatory protein, a gene we have renamed BadR. BadR is a ROK family member, which includes repressors such as NagC, Mlc, and XylR involved in regulating GlcNAc, glucose, and xylose metabolism, respectively. B. burgdorferi does not utilize xylose but does utilize GlcNAc and glucose sugars. Many bacteria have multiple ROK regulators, specific for each sugar utilized (Titgemeyer et al., 1994). Interestingly, this is not the case with B. burgdorferi as BadR is the only ROK regulator identified in the genome. Since the B. burgdorferi genome has few transcriptional regulators, this suggests that these regulators are versatile and lead us to hypothesize BadR is a unique ROK regulator, unlike NagCs and Mlcs in other systems, and to which our findings support.
First, BadR’s original annotation as a XylR1 would imply its regulation is through the binding of xylose, but xylose had no effect on the binding of BadR to the rpoS promoters. Secondly, the badR-deficient strains displayed a significant up-regulation of chitobiose utilization genes, suggesting BadR functions similar to NagC and Mlc, because both modulate expression of these genes in other bacteria (Berg et al., 2007; Plumbridge & Pellegrini, 2004). BadR is unlike a NagC, however, because levels of catabolic enzymes, NagA and NagB, were not affected in badR-deficient strains; whereas a nagC-deficient E. coli demonstrates a 20-fold induction of nagB (Plumbridge, 1991). On the other hand, BadR is like a NagC in that GlcNAc-6P disrupted BadR’s binding to the rpoS promoters. However, our studies demonstrated that several phosphorylated sugars (e.g. GlcNAc-6P, Glc-6P, ribose-5P) disrupted the binding of BadR to the rpoS promoter.
Site-directed mutants of BadR were generated in the putative inducer/sugar binding domain (IBD) to generate a mutant BadR unresponsive to its inducer, which would aid in identifying any specific interactions with the sugars and BadR. Previous studies identified residues in both NagC and Mlc proteins in E. coli that when mutated result in regulators insensitive to their inducer (Pennetier et al., 2008). However, BadR does not maintain any of these conserved residues, further illustrating its unique qualities. A couple of residues that were mutated in the IBD domain of BadR had no effect on BadR binding to the rpoS promoter in the presence of various sugars. Removal of the entire IBD domain yielded a BadR protein unable to bind to the rpoS promoters and is consistent with E. coli Mlc mutants lacking the last 18 amino acids (Schiefner et al., 2005).
BadR’s regulatory function may not be modulated like NagCs and XylRs, which are directly bound by their specific sugar/inducer. It is feasible that BadR is similarly regulated as an Mlc, specifically being sequestered in the inner membrane through its binding of the PTS transporter protein, PtsG. Mlc-deficient strains of E. coli display a modest 2-fold induction of ptsG (Plumbridge, 2001; Kimata et al., 1998). Even though our study did not address spatial location of BadR in the presence or absence of these sugars, badR− did not show a significant induction of ptsG transcript, suggesting that BadR is unlike an Mlc.
Further structure/function studies of mutant BadR proteins gave more insight into the characteristics of BadR. We analyzed the effects of site-specific changes in residues conserved in BadR that have been shown to be important for the function of both Mlc and NagC in E. coli. First, C254A/C257A (putative metal binding residues) and H243A BadR mutants were unable to bind the rpoS promoter and parallels results seen in equivalent Mlc and NagC mutants in E. coli (Pennetier et al., 2008).
Taken together, BadR has many characteristics similar to Mlc and NagC but our results suggest BadR is a unique ROK regulator. Our study did not identify a specific inducer of BadR, however, it cannot be ruled out that phosphorylated sugars may serve as inducing signals for BadR in vivo. In general, B. burgdorferi has a limited genome with limited metabolic capabilities, so perhaps BadR has evolved to recognize multiple phospho-sugars because it functions in place of multiple regulators found in other bacteria. Nevertheless, we postulate that under conditions encountered within different host environments, BadR modifies its regulatory function at its various gene targets through its interaction with host-specific nutrients or by collaboration with other proteins or regulatory elements (Jutras et al., 2012).
ROK Regulators: Modulators of Metabolism
BadR negatively regulates chitobiose transporter genes (chbA, chbB, chbC) indicated by the up-regulation of these genes in the badR-deficient strains. Chitobiose, a GlcNAc dimer, constitutes the subunit of chitin, which is an abundant component found only in ticks and not in mammals. Evidence supports that chitin and chitobiose (GlcNAc dimer) alone can provide the GlcNAc source to support growth of B. burgdorferi in vitro (Tilly et al., 2001). Interestingly, a chbC-deficient borrelial strain demonstrated ChbC was not essential for any stage of the mouse-tick-mouse infection model (Tilly et al., 2004; Tilly et al., 2001).
Nevertheless, it has been hypothesized that expression of these genes may enhance bacterial survival in nutritionally stressful circumstances (Tilly et al., 2001; Tilly et al., 2004; Rhodes et al., 2010; Rhodes et al., 2009). However, studies suggest that the presence of exogenous chitobiose is not sufficient to induce the expression of the chitobiose transport genes. chbC transcripts are not induced from B. burgdorferi cultures grown in media supplemented with chitobiose (Tilly et al., 2001), and ChbC levels remain low in the presence of free GlcNAc (Rhodes et al., 2009; Tilly et al., 2001). We postulate that the presence of an inducer molecule for BadR may be only one requirement necessary to facilitate expression of the chitobiose utilization genes, similar to E. coli, in which expression of chbC is regulated by NagC as well as two additional regulatory proteins, ChbR and CAP (Plumbridge & Pellegrini, 2004).
Conclusion
Our findings suggest that the transcriptional regulator, BadR, is a key component in the lifecycle of B. burgdorferi because it simultaneously activates genes important for the tick-specific lifestyle while repressing genes critical for vertebrate-specific infection. Its repressive effects on mammalian-specific virulence factors could be traced to regulation of the “gatekeeper,” rpoS; BadR binds to the rpoS promoter and represses its transcription. Since ROK regulators, including BadR, are typically modulated through their direct interactions with carbohydrates or byproducts of metabolism, influx of vertebrate-specific sugars may be a trigger that induces the RpoS-dependent expression of virulence factors upon entry of B. burgdorferi into the mammalian host. Our results also imply that BadR is an activator of tick determinants such as chitobiose utilization genes and genes of lp28-4, which are thought to be involved in B. burgdorferi’s maintenance in ticks. Thus, BadR is a critical regulator that most likely couples nutrient availability to niche-specific gene expression, allowing appropriate adaptation to both the tick and vertebrate environments. This study is the first to identify and characterize this novel regulator in B. burgdorferi, and reveals novel targets to reduce the transmission and incidence of Lyme disease (Fig. 8).
EXPERIMENTAL PROCEDURES
Recombinant proteins and antibody production
Recombinant proteins and monospecific sera were generated as previously described (Karna et al., 2011; Esteve-Gassent et al., 2009; Van Laar et al., 2012). Briefly, total genomic DNA obtained from B. burgdorferi clonal isolate MSK5 was used as a template to PCR amplify genes previously discussed and using primers listed in Table S3. Amplified genes were cloned into the inducible protein expression vector pET23a (Novagen). E. coli expression host Rosetta™ (Novagen) were transformed with the expression vector and induced with 1mM IPTG. Recombinant proteins were gel purified using the ELUTRAP System (Whatman). Six to eight weeks-old female BALB/c mice (n=5) were immunized subcutaneously with 50–100 μg of protein with equal proportion of adjuvant, TiterMax™ (Sigma) in phosphate saline buffer. Boosters were given at day 14 and 21 and immune serum from blood was collected on day 28.
For recombinant proteins used in the EMSAs, amplified products of full-length badR and a badR amplicon lacking the N-terminal helix-turn-helix domain or the C-terminal putative sugar/inducer binding domain were subsequently cloned into the pMAL-c2X expression vector (New England Biolabs). This vector facilitates the generation of recombinant proteins fused to an N-terminal maltose binding protein. Site directed mutagenesis of the BadR protein was performed to replace conserved (H243, and C254/C257 simultaneously-putative dimerization domain) and unique residues of BadR (N253/P256 simultaneously) to alanines. The pMAL-c2X expression vector containing the WT badR gene was used as the template using Quick Change Site-Directed Mutagenesis Kit (Stratagene) and the designated primers (Table S3). Plasmids were screened by restriction enzymes and sequenced. Rosetta™ (Novagen) cells were transformed and allowed to reach logarithmic phase at 37°C. Cells were allowed to induce overnight at 16°C (1mM IPTG), collected by centrifugation, and resuspended in 30ml of low salt buffer (LSB; 10mM phosphate, 30mM NaCl, 10mM β-ME, 1mM EGTA, pH 7.0). The cells were lysed by French press (1000 psi), centrifuged, and supernatants were loaded onto an amylose column for purification using the Biologic Duo-Flow FPLC (Bio-Rad) as per manufactures instructions. Proteins were stored at −80°C and concentrations were determined using Bio-Rad protein assay with BSA as a standard (Bio-Rad).
Bacterial strains and growth conditions
Strains utilized in this study are listed in Table 3. Briefly, infectious B. burgdorferi strain, B31-A3, was cultured in Barbour-Stoenner-Kelly (BSK)-II medium with 6% rabbit serum (Pel-Freez) at 32°C or 37°C in a CO2 (1%) incubator or at 23°C and grown to 5×107 bacteria ml−1 as previously described (Van Laar et al., 2012; Karna et al., 2011; Esteve-Gassent et al., 2009). For growth curves, spirochetes were diluted from stationary phase (1×108 bacteria ml−1) and seeded at 5×105 bacteria ml−1 into 10 ml cultures in BSK-II medium supplemented with 6% NRS (for all strains), 100μg ml−1 of streptomycin (for badR−) and 40μg ml−1 of gentamycin (badR2−). Cells were enumerated every 24 hours for cultures grown at 32°C and 23°C using dark field microscopy. The cultures were grown in triplicate, with three independent trials. Levels of significance were determined using GRAPHPAD PRISM v.4.0 (Graph Pad Software) and two-way ANOVA with α=95%.
Table 3.
Plasmids and strains used in this study.
| Plasmids | Description | Source or reference |
|---|---|---|
| pCR®2.1-TOPO | PCR cloning vector | Invitrogen |
| pET23a | Expression vector with a C-terminal 6-His tag | Novagen |
| pMALc2x | Protein expression vector | NEB |
| pCLM11 | BB0693 cloned into pET23a | This study |
| pCLM25 | BB0693 cloned into pMALc2x | This study |
| pCLM40 | BB0693NoHTH cloned into pMALc2x | This study |
| pCLM55 | BB0693NoIBD cloned into pMALc2x | This study |
| pCLM67 | BB0693H243A cloned into pMALc2x | This study |
| pCLM66 | BB0693C254A/C257A cloned into pMALc2x | This study |
| pCLM65 | BB0693N253A/P255A cloned into pMALc2x | This study |
| pMP2 | BB0151 cloned into pET23a | This study |
| pMP3 | BB0152 cloned into pET23a | This study |
| pCLM22 | BB0645 cloned into pET23a | This study |
| pTS2 | BBB05 cloned into pET23a | This study |
| pBVSR2713 | BB0831 cloned into pET23a | This study |
| pMP4 | BB0545 cloned into pET23a | This study |
| pCLM26 | BB0055 cloned into pET23a | This study |
| pBVSR3212 | BB0694 cloned into pET23a | This study |
| pCLM12 | upstream (732988–734094) and downstream (735259–736264) flanking Strr cassette in pCR2.1 for BB0693 deletion 733108–735289 upstream and BB0693 and 735290–736288 downstream |
This study |
| pCLM24 | flanking Gentr cassette for BB0693 cis complementation |
This study |
|
| ||
| B. burgdorferi strains | Description | Source or reference |
|
| ||
| A3 | B31, low passage parental infectious isolate (WT) | (Elias et. al 2002) |
| badR− | B31 isolate, badR (BB0693)deficient, Strr | This study |
| badR2− | B31 isolate, badR (BB0693) deficient, Gentr | This study |
Strr, streptomycin resistance; Gentr, kanamycin resistance.
Generation of badR-deficient B31-A3 strain
A suicide vector was generated, as previously described (Karna et al., 2011), which allowed for deletion of the badR gene through allelic exchange with an antibiotic cassette, PflgBStrR or PflgBGentR, for the first and second badR-deficient strain, respectively. Primers used are listed in Table S3. The antibiotic cassettes were driven by a constituently expressed borrelial promoter, PflgB. Briefly, ~1-kb regions of the borrelial chromosome flanking the badR gene were PCR amplified from total genomic DNA from infectious, clonal isolate of B. burgdorferi B31-A3. In the second step, a ~2-kb amplicon was PCR amplified by LATaq polymerase (Takara) using the PCR products from the first step as DNA templates. The amplicon was cloned into pCR2.1 cloning vector (Invitrogen) and transformed into TOP 10 E. coli. A plasmid with an appropriate insert was identified following blue/white screening, and the antibiotic cassette was cloned into this plasmid using the SalI sites present on the primers used. The final plasmid used for the generation of the mutant in the infectious strain of B. burgdorferi B31-A3 is illustrated in Fig. 3 (Samuels, 1995).
The transformants were plated on BSK-II agarose overlays and counter selected in the presence of streptomycin or gentamycin (100μg ml−1 or 40μg ml−1, respectively). Plates were incubated at 32°C in 1% CO2 until individual colonies were visible, at which then they were isolated aseptically into BSK-II liquid medium and incubated at 32°C until the spirochetes reached a density of 5 × 107 ml−1. Total genomic DNA was extracted from this culture and the presence of the antibiotic cassettes was determined by PCR. A positive clone was further confirmed for the loss of badR by Southern blot and immunoblot analysis.
Southern blot analysis
As described previously (Raju et al., 2011), total genomic DNA was extracted from WT and badR− strains, digested with various restricted enzymes, separated on a 1% agarose gel, and transferred onto a Nylon membrane (Amersham Hybond-N+;GE Healthcare). PCR amplified probes corresponding to the aadA gene (StrR maker) or to a region upstream of the badR gene were labeled using the Enhanced Chemiluminescence Labeling and Detection System (GE Healthcare). Membranes were hybridized with the probes overnight at 42°C, washed, and developed according the manufacturer’s protocol.
SDS-PAGE and immunoblot analyses
Whole-cell B. burgdorferi lysates were prepared, separated on SDS-PAGE, and visualized either by Coomassie brilliant blue staining or transferred onto PVDF membranes for immunoblot analysis, as previously described (Van Laar et al., 2012; Sanjuan et al., 2009). Equivalent loading of protein was estimated with Coomassie blue staining and immunoblot analysis with mouse anti-P66 serum. The PVDF membranes were blocked with 1% non-fat milk, and probed with mouse, rat, or rabbit antibodies against aforementioned antigens. Subsequent probing with appropriate dilutions of horse radish peroxidase-conjugated anti-mouse, anti-rat, or anti-rabbit serum allowed for visualization using ECL Western blotting reagents (GE Healthcare).
RNA extraction and quantitative RT-PCR
Procedures were as previously described (Karna et al., 2011). WT parental A3 strain and the badR deficient mutant were grown in BSK-II at 32°C until cultures reached mid-logarithmic phase corresponding to 5×107 cells ml−1. RNA was harvested using RNA-Bee (Tel-Test, Inc.) and phenol/chloroform followed by isopropanol precipitation and washes with 75%ethanol. Genomic DNA was removed by treatment with DNAse I. RNA was quantified using the Nanodrop spectrophotometer (Invitrogen) and absence of DNA contamination was confirmed using PCR primers specific for the recA gene in B. burgdorferi. The RNA was reverse transcribed to cDNA using TaqMan reverse transcription reagents (Applied Biosystems). Quantitative real-time PCRs was performed using SYBR green master mix (Ambion) with specified primers (Table S3) and analysis by ABI Prism 7300 system (Applied Biosystems) with relative changes using recA for normalization and fold difference with 2−ΔΔCt method. Unpaired student’s t-test and P<0.05 (Prism) were implemented.
Microarray analyses
RNA from cultures grown under pH7.6/32°C was harvested as previously described, (Karna et al., 2011), and sent to Roche Nimblegen for cDNA synthesis and microarray hybridization. Two separate but identically designed microarrays were performed. Microarray data deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE38827 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38827). Hierarchical clustering and correlation analysis was used to verify quality of the normalized array data. The expression data from biological replicates 1 and 2 were both pooled and analyzed individually. We used LIMMA R package to test contrasts between WT and badR-deficient strains (Smyth, 2004). We reported raw and log2 fold change (rpoS; Fig. 8), and computed 95% confidence intervals, p-value, and adjusted Benjamini-Hochberg False Discovery Rates (FDR). Genes ≥2 fold differences in expression and overall P≤0.05 were reported (Table 1).
EMSAs
Primers used are listed (Table S3). 5′ biotinylated promoters were produced by Nucleic Acids Core Facility (University of Texas Health Science Center, SA, TX) and purified (SV gel-PCR cleanup-Promega). LightShift Chemiluminescent EMSA kit was utilized according to manufacturer’s recommendations (Pierce Biotechnology). Briefly, labeled promoters (1.9 nmols) and BadR (135, 205, or 270 pmols) were mixed in 20μl buffer (10mM Tris (pH7.5), 50mM KCl, 1mM DTT, and 2ug poly(dI-dC) for 20 min at 23°C, run on 6% polyacrylamide gel, transferred onto Nylon membranes (GE Healthcare), UV-cross-linked, and visualized with LightShift reagents. Some reactions included GlcNAc-6P, glucose-6P (Glc-6P), xylulose-5P, ribose-5P, xylose, or chitobiose (10, 30, 50, 100, or 200mM; Sigma-Aldrich) and unlabeled promoters (200-fold molar excess), for induction and competition studies, respectively. Densitometry to assess inhibition of binding by sugars was performed using ImageJ software (Table S1 and S2) (Schneider et al., 2012). A representative experiment was shown. The shifted band corresponding to BadR’s binding to the rpoS promoters in the absence of sugars was designated as a 100% bound.
Infectivity Analysis
Six-week old female C3H/HeN, SCID, or BALB/c mice were inoculated intradermally with 105 spirochetes per mouse with the following borrelial strains: wild-type (B31-A3), badR-deficient strain 1 (badR1−) and badR-deficient strain 2 (badR2−). Fourteen days after inoculation, the spleen, left-tibiotarsal joint, left inguinal lymph node, heart, bladder and a piece of abdominal skin were collected, and cultured in BSK-II growth medium to facilitate isolation of spirochetes as previously described. The cultures were scored for growth of B. burgdorferi after 2 to 3 weeks using dark field microscopy (Table 4). All animal procedures were performed in accordance with the animal use protocol approved by Institutional Animal Care and Use Committee (IACUC) at the University of Texas at San Antonio.
Supplementary Material
Acknowledgments
We thank Patti Rosa for B. burgdorferi strain B31-A3. The research was partly funded by Public Health Service Grant SC1-AI-078559 from the NIH-NIAID, the Army Research Office of DoD under Contract No: W911NF-11-1-0136, pre-doctoral fellowships from CEIG (CLM) and STCEID (SLRK). We thank Dr. J.A.L. Gelfond, UTHSC, Epidemiology & Biostatistics, San Antonio, TX, for his expertise and statistical analyses of our microarray studies. We thank Karl E. Klose and M. Neal Guentzel for critically reading the manuscript.
References
- Barbour AG, Burgdorfer W, Grunwaldt E, Steere AC. Antibodies of patients with Lyme disease to components of the Ixodes dammini spirochete. J Clin Invest. 1983;72:504–515. doi: 10.1172/JCI110998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MJ, Gawronski JD, Eveleigh DE, Benson DR. Intracellular symbionts and other bacteria associated with deer ticks (Ixodes scapularis) from Nantucket and Wellfleet, Cape Cod, Massachusetts. Appl Environ Microbiol. 2004;70:616–620. doi: 10.1128/AEM.70.1.616-620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Schild S, Reidl J. Regulation of the chitobiose-phosphotransferase system in Vibrio cholerae. Arch Microbiol. 2007;187:433–439. doi: 10.1007/s00203-006-0207-4. [DOI] [PubMed] [Google Scholar]
- Bugrysheva JV, Bryksin AV, Godfrey HP, Cabello FC. Borrelia burgdorferi rel is responsible for generation of guanosine-3′-diphosphate-5′-triphosphate and growth control. Infect Immun. 2005;73:4972–4981. doi: 10.1128/IAI.73.8.4972-4981.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimano MJ, Iyer R, Eggers CH, Gonzalez C, Morton EA, Gilbert MA, et al. Analysis of the RpoS regulon in Borrelia burgdorferi in response to mammalian host signals provides insight into RpoS function during the enzootic cycle. Mol Microbiol. 2007;65:1193–1217. doi: 10.1111/j.1365-2958.2007.05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimano MJ, Kenedy MR, Kairu T, Desrosiers DC, Harman M, Dunham-Ems S, et al. The hybrid histidine kinase Hk1 is part of a two-component system that is essential for survival of Borrelia burgdorferi in feeding Ixodes scapularis ticks. Infect Immun. 2011;79:3117–3130. doi: 10.1128/IAI.05136-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. Summary of notifiable diseases–United States,2010. MMWR Morb Mortal Wkly Rep. 2012;59:1–111. [PubMed] [Google Scholar]
- Dunham-Ems SM, Caimano MJ, Eggers CH, Radolf JD. Borrelia burgdorferi requires the alternative sigma factor RpoS for dissemination within the vector during tick-to-mammal transmission. PLoS Pathog. 2012;8:e1002532. doi: 10.1371/journal.ppat.1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias AF, Schmutzhard J, Stewart PE, Schwan TG, Rosa P. Population dynamics of a heterogeneous Borrelia burgdorferi B31 strain in an experimental mouse-tick infectious cycle. Wien Klin Wochenschr. 2002;114:557–561. [PubMed] [Google Scholar]
- Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, et al. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun. 2002;70:2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve-Gassent MD, Elliott NL, Seshu J. sodA is essential for virulence of Borrelia burgdorferi in the murine model of Lyme disease. Mol Microbiol. 2009;71:594–612. doi: 10.1111/j.1365-2958.2008.06549.x. [DOI] [PubMed] [Google Scholar]
- Fikrig E, Narasimhan S. Borrelia burgdorferi–traveling incognito? Microbes Infect. 2006;8:1390–1399. doi: 10.1016/j.micinf.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, et al. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci U S A. 2004;101:3142–3147. doi: 10.1073/pnas.0306845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono K, Kakuda H, Ichihara S. Decreasing accumulation of acetate in a rich medium by Escherichia coli on introduction of genes on a multicopy plasmid. Biosci Biotechnol Biochem. 1995;59:256–261. doi: 10.1271/bbb.59.256. [DOI] [PubMed] [Google Scholar]
- Hubner A, Yang X, Nolen DM, Popova TG, Cabello FC, Norgard MV. Expression of Borrelia burgdorferi OspC and DbpA is controlled by a RpoN-RpoS regulatory pathway. Proc Natl Acad Sci U S A. 2001;98:12724–12729. doi: 10.1073/pnas.231442498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde JA, Seshu J, Skare JT. Transcriptional profiling of Borrelia burgdorferi containing a unique bosR allele identifies a putative oxidative stress regulon. Microbiology. 2006;152:2599–2609. doi: 10.1099/mic.0.28996-0. [DOI] [PubMed] [Google Scholar]
- Ingram-Smith C, Gorrell A, Lawrence SH, Iyer P, Smith K, Ferry JG. Characterization of the acetate binding pocket in the Methanosarcina thermophila acetate kinase. J Bacteriol. 2005;187:2386–2394. doi: 10.1128/JB.187.7.2386-2394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutras BL, Bowman A, Brissette CA, Adams CA, Verma A, Chenail AM, et al. EbfC (YbaB) is a new type of bacterial nucleoid-associated protein, and a global regulator of gene expression in the Lyme disease spirochete. J Bacteriol. 2012 doi: 10.1128/JB.00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karna SL, Sanjuan E, Esteve-Gassent MD, Miller CL, Maruskova M, Seshu J. CsrA modulates levels of lipoproteins and key regulators of gene expression critical for pathogenic mechanisms of Borrelia burgdorferi. Infect Immun. 2011;79:732–744. doi: 10.1128/IAI.00882-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenedy MR, Lenhart TR, Akins DR. The role of Borrelia burgdorferi outer surface proteins. FEMS Immunol Med Microbiol. 2012;66:1–19. doi: 10.1111/j.1574-695X.2012.00980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata K, Inada T, Tagami H, Aiba H. A global repressor (Mlc) is involved in glucose induction of the ptsG gene encoding major glucose transporter in Escherichia coli. Mol Microbiol. 1998;29:1509–1519. doi: 10.1046/j.1365-2958.1998.01035.x. [DOI] [PubMed] [Google Scholar]
- Lybecker MC, Samuels DS. Temperature-induced regulation of RpoS by a small RNA in Borrelia burgdorferi. Mol Microbiol. 2007;64:1075–1089. doi: 10.1111/j.1365-2958.2007.05716.x. [DOI] [PubMed] [Google Scholar]
- Ouyang Z, Deka RK, Norgard MV. BosR (BB0647) controls the RpoN-RpoS regulatory pathway and virulence expression in Borrelia burgdorferi by a novel DNA-binding mechanism. PLoS Pathog. 2011;7:e1001272. doi: 10.1371/journal.ppat.1001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Narasimhan S, Neelakanta G, Kumar M, Pal U, Fikrig E, et al. Activation of the RpoN-RpoS regulatory pathway during the enzootic life cycle of Borrelia burgdorferi. BMC Microbiol. 2012;12:44. doi: 10.1186/1471-2180-12-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal U, Yang X, Chen M, Bockenstedt LK, Anderson JF, Flavell RA, et al. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J Clin Invest. 2004;113:220–230. doi: 10.1172/JCI19894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas CJ, Iyer R, Petzke MM, Caimano MJ, Radolf JD, Schwartz I. Borrelia burgdorferi requires glycerol for maximum fitness during the tick phase of the enzootic cycle. PLoS Pathog. 2011;7:e1002102. doi: 10.1371/journal.ppat.1002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennetier C, Dominguez-Ramirez L, Plumbridge J. Different regions of Mlc and NagC, homologous transcriptional repressors controlling expression of the glucose and N-acetylglucosamine phosphotransferase systems in Escherichia coli, are required for inducer signal recognition. Mol Microbiol. 2008;67:364–377. doi: 10.1111/j.1365-2958.2007.06041.x. [DOI] [PubMed] [Google Scholar]
- Plumbridge J. Co-ordinated regulation of amino sugar biosynthesis and degradation: the NagC repressor acts as both an activator and a repressor for the transcription of the glmUS operon and requires two separated NagC binding sites. EMBO J. 1995;14:3958–3965. doi: 10.1002/j.1460-2075.1995.tb00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge J. How to achieve constitutive expression of a gene within an inducible operon: the example of the nagC gene of Escherichia coli. J Bacteriol. 1996;178:2629–2636. doi: 10.1128/jb.178.9.2629-2636.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge J. Regulation of PTS gene expression by the homologous transcriptional regulators, Mlc and NagC, in Escherichia coli (or how two similar repressors can behave differently) J Mol Microbiol Biotechnol. 2001;3:371–380. [PubMed] [Google Scholar]
- Plumbridge J. Regulation of gene expression in the PTS in Escherichia coli: the role and interactions of Mlc. Curr Opin Microbiol. 2002;5:187–193. doi: 10.1016/s1369-5274(02)00296-5. [DOI] [PubMed] [Google Scholar]
- Plumbridge J, Pellegrini O. Expression of the chitobiose operon of Escherichia coli is regulated by three transcription factors: NagC, ChbR and CAP. Mol Microbiol. 2004;52:437–449. doi: 10.1111/j.1365-2958.2004.03986.x. [DOI] [PubMed] [Google Scholar]
- Plumbridge JA. Repression and induction of the nag regulon of Escherichia coli K-12: the roles of nagC and nagA in maintenance of the uninduced state. Mol Microbiol. 1991;5:2053–2062. doi: 10.1111/j.1365-2958.1991.tb00828.x. [DOI] [PubMed] [Google Scholar]
- Poncet S, Milohanic E, Maze A, Nait Abdallah J, Ake F, Larribe M, et al. Correlations between carbon metabolism and virulence in bacteria. Contrib Microbiol. 2009;16:88–102. doi: 10.1159/000219374. [DOI] [PubMed] [Google Scholar]
- Postma PW, Lengeler JW, Jacobson GR. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev. 1993;57:543–594. doi: 10.1128/mr.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radolf JD, Caimano MJ, Stevenson B, Hu LT. Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol. 2012;10:87–99. doi: 10.1038/nrmicro2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju BV, Esteve-Gassent MD, Karna SL, Miller CL, Van Laar TA, Seshu J. Oligopeptide permease A5 modulates vertebrate host-specific adaptation of Borrelia burgdorferi. Infect Immun. 2011;79:3407–3420. doi: 10.1128/IAI.05234-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes RG, Atoyan JA, Nelson DR. The chitobiose transporter, chbC, is required for chitin utilization in Borrelia burgdorferi. BMC Microbiol. 2010;10:21. doi: 10.1186/1471-2180-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes RG, Coy W, Nelson DR. Chitobiose utilization in Borrelia burgdorferi is dually regulated by RpoD and RpoS. BMC Microbiol. 2009;9:108. doi: 10.1186/1471-2180-9-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers EA, Terekhova D, Zhang HM, Hovis KM, Schwartz I, Marconi RT. Rrp1, a cyclic-di-GMP-producing response regulator, is an important regulator of Borrelia burgdorferi core cellular functions. Mol Microbiol. 2009;71:1551–1573. doi: 10.1111/j.1365-2958.2009.06621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS. Electrotransformation of the spirochete Borrelia burgdorferi. Methods Mol Biol. 1995;47:253–259. doi: 10.1385/0-89603-310-4:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS. Gene regulation in Borrelia burgdorferi. Annu Rev Microbiol. 2011;65:479–499. doi: 10.1146/annurev.micro.112408.134040. [DOI] [PubMed] [Google Scholar]
- Sanjuan E, Esteve-Gassent MD, Maruskova M, Seshu J. Overexpression of CsrA (BB0184) alters the morphology and antigen profiles of Borrelia burgdorferi. Infect Immun. 2009;77:5149–5162. doi: 10.1128/IAI.00673-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefner A, Gerber K, Seitz S, Welte W, Diederichs K, Boos W. The crystal structure of Mlc, a global regulator of sugar metabolism in Escherichia coli. J Biol Chem. 2005;280:29073–29079. doi: 10.1074/jbc.M504215200. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Lim S, Seok YJ, Ryu S. Heat shock RNA polymerase (E sigma(32)) is involved in the transcription of mlc and crucial for induction of the Mlc regulon by glucose in Escherichia coli. J Biol Chem. 2001;276:25871–25875. doi: 10.1074/jbc.M101757200. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Sohanpal BK, Friar S, Roobol J, Plumbridge JA, Blomfield IC. Multiple co-regulatory elements and IHF are necessary for the control of fimB expression in response to sialic acid and N-acetylglucosamine in Escherichia coli K-12. Mol Microbiol. 2007;63:1223–1236. doi: 10.1111/j.1365-2958.2006.05583.x. [DOI] [PubMed] [Google Scholar]
- Steere AC. Lyme borreliosis in 2005, 30 years after initial observations in Lyme Connecticut. Wien Klin Wochenschr. 2006;118:625–633. doi: 10.1007/s00508-006-0687-x. [DOI] [PubMed] [Google Scholar]
- Stevenson B, Zuckert WR, Akins DR. Repetition, conservation, and variation: the multiple cp32 plasmids of Borrelia species. J Mol Microbiol Biotechnol. 2000;2:411–422. [PubMed] [Google Scholar]
- Strother KO, Broadwater A, De Silva A. Plasmid requirements for infection of ticks by Borrelia burgdorferi. Vector Borne Zoonotic Dis. 2005;5:237–245. doi: 10.1089/vbz.2005.5.237. [DOI] [PubMed] [Google Scholar]
- Sultan SZ, Pitzer JE, Miller MR, Motaleb MA. Analysis of a Borrelia burgdorferi phosphodiesterase demonstrates a role for cyclic-di-guanosine monophosphate in motility and virulence. Mol Microbiol. 2010;77:128–142. doi: 10.1111/j.1365-2958.2010.07191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CW, Li C. Inactivation of bb0184, which encodes carbon storage regulator A, represses the infectivity of Borrelia burgdorferi. Infect Immun. 2011;79:1270–1279. doi: 10.1128/IAI.00871-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CW, Morado DR, Liu J, Charon NW, Xu H, Li C. Carbon storage regulator A (CsrA(Bb)) is a repressor of Borrelia burgdorferi flagellin protein FlaB. Mol Microbiol. 2011;82:851–864. doi: 10.1111/j.1365-2958.2011.07853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kimata K, Aiba H. A novel regulatory role of glucose transporter of Escherichia coli: membrane sequestration of a global repressor Mlc. EMBO J. 2000;19:5344–5352. doi: 10.1093/emboj/19.20.5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly K, Elias AF, Errett J, Fischer E, Iyer R, Schwartz I, et al. Genetics and regulation of chitobiose utilization in Borrelia burgdorferi. J Bacteriol. 2001;183:5544–5553. doi: 10.1128/JB.183.19.5544-5553.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly K, Grimm D, Bueschel DM, Krum JG, Rosa P. Infectious cycle analysis of a Borrelia burgdorferi mutant defective in transport of chitobiose, a tick cuticle component. Vector Borne Zoonotic Dis. 2004;4:159–168. doi: 10.1089/1530366041210738. [DOI] [PubMed] [Google Scholar]
- Titgemeyer F, Reizer J, Reizer A, Saier MH., Jr Evolutionary relationships between sugar kinases and transcriptional repressors in bacteria. Microbiology. 1994;140(Pt 9):2349–2354. doi: 10.1099/13500872-140-9-2349. [DOI] [PubMed] [Google Scholar]
- Tokarz R, Anderton JM, Katona LI, Benach JL. Combined effects of blood and temperature shift on Borrelia burgdorferi gene expression as determined by whole genome DNA array. Infect Immun. 2004;72:5419–5432. doi: 10.1128/IAI.72.9.5419-5432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar TA, Lin YH, Miller CL, Karna SL, Chambers JP, Seshu J. Effect of Levels of Acetate on the Mevalonate Pathway of Borrelia burgdorferi. PLoS One. 2012;7:e38171. doi: 10.1371/journal.pone.0038171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lackum K, Stevenson B. Carbohydrate utilization by the Lyme borreliosis spirochete, Borrelia burgdorferi. FEMS Microbiol Lett. 2005;243:173–179. doi: 10.1016/j.femsle.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bruno JF, Luft BJ. Profiling the humoral immune response to Borrelia burgdorferi infection with protein microarrays. Microb Pathog. 2008;45:403–407. doi: 10.1016/j.micpath.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Yang XF, Alani SM, Norgard MV. The response regulator Rrp2 is essential for the expression of major membrane lipoproteins in Borrelia burgdorferi. Proc Natl Acad Sci U S A. 2003;100:11001–11006. doi: 10.1073/pnas.1834315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.