

Abstract
Purpose of review:
The landscape of genetic diagnostic testing has changed dramatically with the introduction of next-generation clinical exome sequencing (CES), which provides an unbiased analysis of all protein-coding sequences in the roughly 21,000 genes in the human genome. Use of this testing, however, is currently limited in clinical neurologic practice by the lack of a framework for appropriate use and payer coverage.
Recent findings:
CES can be cost-effective due to its high diagnostic yield in comparison to other genetic tests in current use and should be utilized as a routine diagnostic test in patients with heterogeneous neurologic phenotypes facing a broad genetic differential diagnosis. CES can eliminate the need for escalating sequences of conventional neurodiagnostic tests.
Summary:
This review discusses the role of clinical exome sequencing in neurologic disease, including its benefits to patients, limitations, appropriate use, and billing. We also provide a reference template policy for payer use when considering testing requests.
Clinical exome sequencing (CES) is a new state-of-the-art molecular diagnostic genetic test. It has the potential to rapidly and efficiently detect disease-causing genetic mutations within any gene in the human genome and is therefore becoming widely used in clinical practice. This template policy addresses the health benefits, limitations, and appropriate use of this type of testing in patients with neurologic disease.
The health benefits of genetic diagnosis
Recent evidence suggests that health outcomes improve as a result of accurate genetic diagnosis.1–3 Results of genetic testing in patients have led to identification, initiation, or change of symptomatic and disease-modifying treatments. Other benefits have occurred in reproductive counseling and family planning. In some instances, test findings have extended the diagnostic evaluation to discover unknown significant and actionable systemic involvements and comorbidities. These tests have also facilitated prognostication and selective participation in research or clinical treatment trials.
Many common disease phenotypes exist that could result from mutations in various different genes. This genetic overlap is particularly true in neurologic disorders, including disorders causing neuropathy, epilepsy, ataxia, special sensory impairment, myopathy, movement disorders, intellectual disability, and others, which creates a particular challenge for accurate genetic diagnosis. In addition, there are many examples where one gene can cause more than one phenotypic, and sometimes disparate, illness. Effective strategies are therefore necessary to achieve accurate genetic diagnosis efficiently within this complex landscape. Broad indiscriminate testing and incorrect interpretation result in inaccurate conclusions. For proper genetic testing, careful patient selection through expert clinical evaluation is an invariant prerequisite. Validation of testing methodology and demonstration of clinical utility are also essential. Failure to follow these sequential steps will limit the realization of the emerging health benefits of genetic testing to patient care.
Current methods for genetic diagnostic testing
Single-gene sequencing (Sanger sequencing)
At present, the most common form of genetic testing employed is single-gene sequencing, i.e., testing for a mutation in a specific gene that is known to be associated with a particular disease. In clinical practice, the clinician must determine the most probable disorder and order specific testing of the gene (or genes) that causes the illness. The technique currently employed most widely is Sanger sequencing, a method capable of sequencing portions of DNA in short (<1 kilobase) segments (table 1).4 Sanger sequencing can detect most common mutations that affect coding sequences (e.g., missense and nonsense point mutations, small insertions/deletions, and mutations involving the splice site junctions).4 This method would not detect large insertions/deletions, copy number variations, or nucleotide repeat expansions.4 This form of testing is also not applicable to the testing of all genes, because sequencing large or numerous regions of DNA is prohibitive due to the exponential increases in labor and cost incurred as the method is scaled up to evaluate larger or multiple genes. The cost of Sanger sequencing varies widely based on the amount of DNA sequenced per gene, but relative estimates of ∼$1,000 per gene have been suggested,5 although the commercial pricing of single gene testing may further increase that figure, which can rapidly limit cost-effectiveness if multiple genes must be considered for a clinically heterogeneous disorder.
Table 1.
Comparison of current technologies for genetic/genomic diagnostic testing

Multigene sequencing (Sanger sequencing, panel testing)
In clinically heterogeneous neurologic disorders (e.g., intellectual disability, epilepsy, ataxia, leukodystrophy, polyneuropathy), commercial laboratories have developed genetic panels of variable size for testing multiple genes simultaneously. As discussed above, because such panels typically utilize Sanger sequencing methods, the commercial price can be expensive (e.g., $30,000 or more), with a correspondingly low diagnostic yield in many cases due to phenotyping errors or variability in phenotype across disorders, creating a considerable burden on the health care system and the individual patient.6 Furthermore, as new genes associated with these broad neurologic disorders are described, previous testing becomes quickly obsolete.
Chromosomal microarray analysis
Chromosomal microarray analysis (CMA), an array-based form of comparative genomic hybridization, is a technique for detecting clinically significant structural changes, particularly deletions or duplications in the genome, termed copy number variation (table 1).4 Based on diagnostic yield, this method of testing is considered a first-line test for patients with unexplained developmental and intellectual disabilities, autism spectrum disorder, or congenital anomalies.7 Because it cannot detect sequence variation, CMA is used complementary to CES to detect structural variation that exome sequencing cannot. For certain phenotypes or disorders with diverse genetic causes, their use would not be mutually exclusive, and in some circumstances CMA and CES may potentially be performed sequentially or even simultaneously.
Benefits of clinical exome sequencing
CES utilizes a method commonly referred to as next-generation sequencing, consisting of a massively parallel sequencing strategy enabling rapid genome-scale sequencing of DNA at a significantly reduced cost relative to the Sanger method, yet capable of detecting all the same types of mutations (e.g., point mutations, small insertions/deletions, and splice site mutations).5,8,9 Because of the associated reduction in cost and time, next-generation sequencing allows for sequencing of the entire exome, i.e., the 1%–2% of the human genome representing all protein-coding regions.9 This technology involves isolating genomic DNA from a patient, fragmenting this DNA into small nucleotide segments, coupling these fragments to artificial linkers, isolating the specific fragments corresponding to exomic sequences, and then sequencing those fragments in parallel on a slide using an in situ amplification method involving fluorescent dyes that are read by computer using a laser in conjunction with a microscope and a digital camera.9 The resulting sequence reads are next assembled as a series of overlapping fragments and aligned to a reference sequence of the human genome so differences between the patient and the reference sequences can be noted. These differences are collectively termed variants because they can (and in the majority typically do) represent benign polymorphisms (genetic variants found in a single person or in a percentage of the population but not associated with any primary disease), as opposed to pathogenic mutations. In this manner, the same classes of mutation can be identified as for Sanger sequencing (i.e., missense and nonsense point mutations, small insertions/deletions, and mutations involving splice site junctions).9 An important consideration for next-generation sequencing is depth of coverage, which refers to the average times each nucleotide is viewed and provides an estimate of certainty in how likely a mutation at a specific position would be detected, if present, reducing the risk of false-negative results. For example, a depth of coverage of 10-fold means that each nucleotide has been examined 10 times on average. In addition, there are numerous metrics available to calculate the quality of next-generation data.10 Routinely attainable minimum read depths and quality levels can be determined where the false-positive rate of next-generation sequencing is less than the false-negative rate of Sanger sequencing, suggesting that this method will likely become the next gold standard in diagnostic testing.10 The commercial price is approximately $5,000 for sequencing and bioinformatic variant analysis (which includes personnel and computing costs) of the entire exome, approximately 21,000 genes, translating into roughly $0.24 per gene. Because of this significant pricing advantage, CES is rapidly becoming the new standard for sequencing in genetic diagnostics, as all 21,000 genes in the human genome can be evaluated in an essentially unbiased manner.11–13 This limits the effect of phenotyping errors or variability as testing is not solely limited to genes associated with one particular symptom or phenotype.
Appropriate use
Clinical prioritization is a prerequisite before undertaking any test, especially expensive, invasive ones. For instance, in the United States, more than 1 million patients undergo a $9,000 per procedure coronary angiography yearly.14 Not all of these procedures lead to appropriate revascularization interventions,15,16 and it has been suggested that proper patient selection is the key to reducing inappropriate percutaneous coronary intervention procedures. Similarly, careful patient selection is even more important for exome sequencing (figure). At present, it is imprudent to consider exome sequencing as a primary or first-order endeavor towards establishing a diagnosis when any genetic disorder is suspected. Thorough clinical evaluation by appropriately trained personnel is the essential first step. The decision to proceed should rest on the choice of tests available with established analytic and clinical validity, and with demonstrated clinical utility for the phenotype in question. At this stage, the testing recommendations of a treating health care provider merit serious coverage consideration. Even if an actionable curative decision may not be imminent, there are downstream long-term health benefits.17,18 For instance, in situations of diagnostic uncertainty when phenotype alone is unhelpful, an escalating series of diagnostic tests (neuroimaging, biopsies, invasive chemistries, CSF analysis) is frequently employed. Before undertaking these expensive and time-consuming tests, exome sequencing could abbreviate or stop the diagnostic testing cascade by establishing the specific rare diagnosis genetically. A Pediatric Genomic Medicine group found that next-generation sequencing has the potential to truncate the diagnostic odyssey by 5 years.19 The average price of testing was over $19,000 prior to establishing a sequencing-based diagnosis, of which 23% were actionable.19 Exome sequencing can also improve preventive care by alerting physicians to unanticipated comorbidities. An important example of this is in the preventive benefit for family members. Exome sequencing results can alleviate family member concerns regarding potential risk of transmission or identify individuals in need of more specialized care or surveillance to reduce future disease complications.
Figure. Clinical exome sequencing workflow.
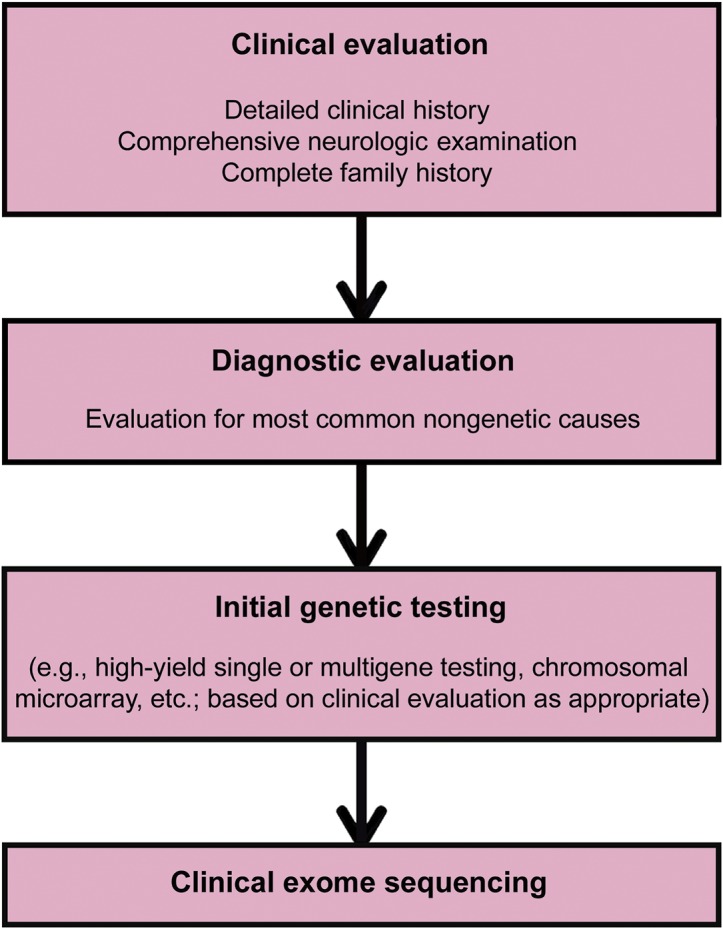
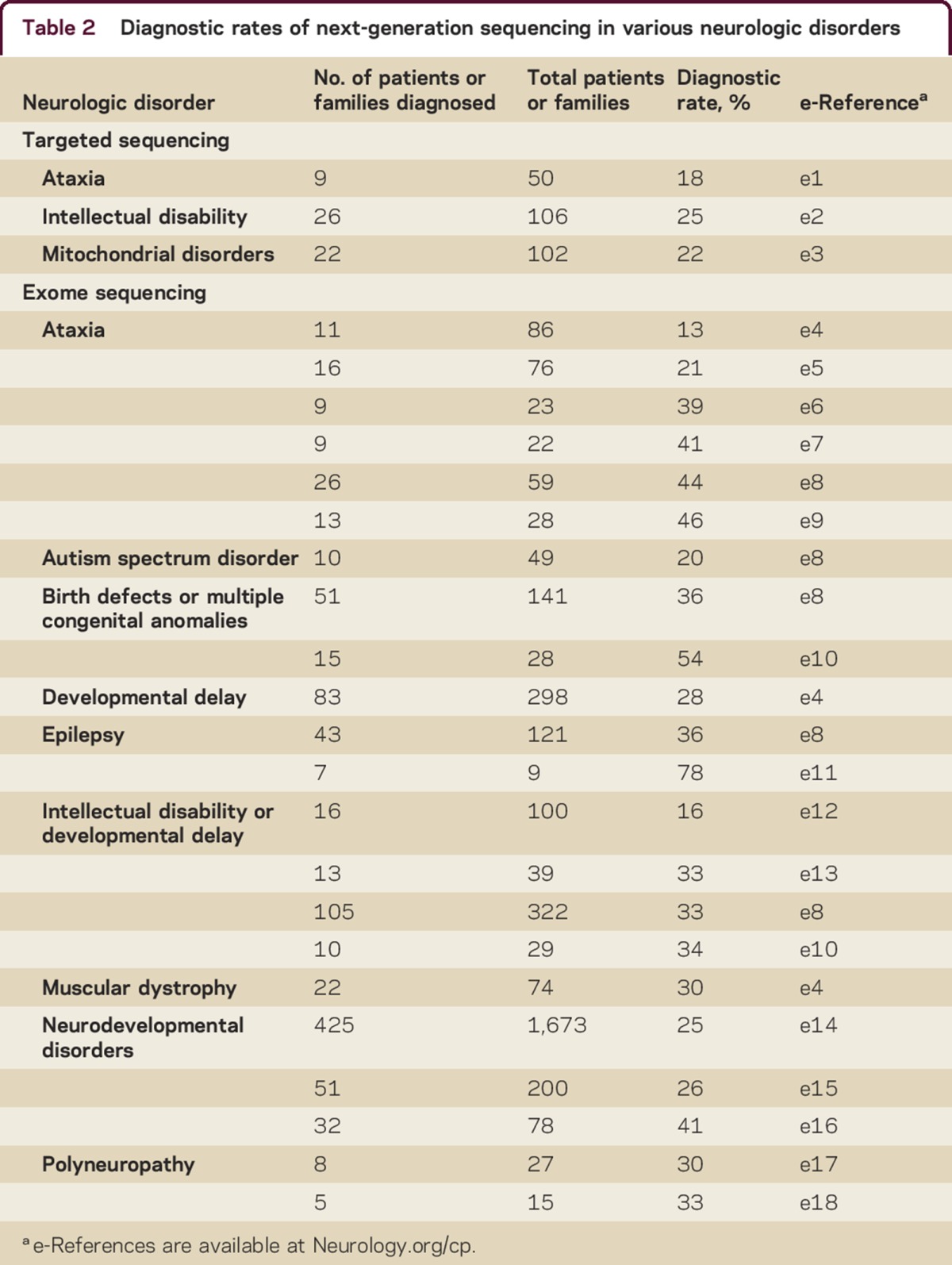
As described above, CES is not expected to be utilized as a first-line or screening test (figure). Patients would be expected to undergo an initial typical clinical evaluation for common acquired etiologies and high-yield genetic testing (e.g., single-gene or multigene testing, chromosomal microarray) if indicated. For example, it would be more cost-effective for a patient with a specific suspected genetic disorder and the classically described phenotype to receive single-gene testing initially for only that disorder. This would likely be the case for the majority of the most common genetic diseases seen in a typical neurology practice. However, if a genetic etiology with a broad differential basis is suspected, then exome sequencing may be the most high-yield and cost-effective evaluation strategy to pursue. This has been reflected in numerous clinical studies to date. Although much of this early analysis has been performed in pediatric populations, CES is not limited by patient age. It is applicable to both children and adults, and can yield clinically meaningful findings in patients without a clear family history of disease (reflecting either de novo mutation or recessive inheritance). The effectiveness of exome sequencing is also not limited to specific categories of disease but is widespread across phenotypes resulting from disparate genetic etiologies. There are numerous examples in the literature of such effectiveness with diagnosis rates ranging from ∼20%–80% for various phenotypes tested. CES and targeted next-generation sequencing focused on large numbers of genes associated with specific phenotypes have shown high diagnostic rates for various disorders (table 2). Clinical experience and best practices will dictate the need for single gene testing vs clinical exome for a specific phenotype, but the above data clearly demonstrate the effectiveness of exome sequencing in clinically heterogeneous conditions.
Table 2.
Diagnostic rates of next-generation sequencing in various neurologic disorders
It must be emphasized that all patients considered for exome sequencing must receive appropriate genetic counseling either from a trained physician or a licensed genetic counselor both before and after the testing is performed. This ensures that the patient has a complete understanding regarding the nature of the test, its benefits and limitations, and the precise clinical interpretation of the results. Additional ethical considerations include variants of uncertain significance and the potential detection of incidental/secondary findings (discussed in detail below). Given the complexity of these counseling issues, we strongly advocate for the use of exome sequencing only in the clinical setting and would not endorse direct-to-consumer testing for any indication.
As of this writing, requests for CES tests will differ from traditional and more familiar diagnostic tests (e.g., neuroimaging, electrophysiology, blood chemistries, or earlier molecular tests such as single-gene tests, chromosomal microarray, or traditional karyotype) in the following manner:
They will be very selective and, comparatively, substantially less frequent.
Clear clinically reasoned indications must accompany testing requests.
The requests will be generated by expertise-based prior evaluations from clinicians familiar with genetic disease and appropriate testing methods.
The testing itself will be performed by a few nationally recognized laboratories with extensive experience in this method and correspondingly high technical standards.
While assigning monetary value to a diagnostic test that may not immediately or directly affect the health and well-being of patients is challenging, there is still a need to recognize the indirect benefits.20 Establishing a genetic diagnosis has value to the patient, the medical care team, the family, and to public health at large by eliminating prolonged diagnostic explorations, improving family counseling, facilitating reproductive decisions, and reducing patient/parental anxiety.20
Challenges and limitations of CES
There are 2 challenges to universal acceptance and policy coverage of CES. The first is more general and the second specific. The first is a conceptual debate surrounding the value of a test that does not always immediately lead to improvement in outcome. The second deals with technical issues, predominantly completeness of gene coverage, validation of results, and variant interpretation.21,22
CES is a new and evolving technology. Some payers consider the test to be experimental and investigational.18,23 However, clinicians pursuing a genetic etiology with a broad differential consider exome sequencing as the foremost high-yield and cost-effective evaluation strategy.24–26 Whereas coverage is generally available for noninvasive prenatal genetic testing and genetic evaluation of neoplastic disease, exome sequencing is not yet recognized for coverage. At the center of the coverage debate is the difficulty of establishing an immediate and conventionally measurable index of health benefit from test results. A long-established expectation is that an action, treatment, or prognostication follow closely after the results of traditional tests. Our Code of Federal Regulations (CFR) concerning health care has manualized a definition for all diagnostic tests. It requires that the ordering and treating physician use the results in the management of the beneficiary's specific medical problem (CFR §411.15[k][1]). Genetic testing, however, does not result in a proximate actionable intervention or management with such immediacy or short temporal sequence as traditional and familiar tests. The latter tests frequently lead to an action, for example, a pharmacologic treatment, biopsy, stenting, surgery, or conclusive prognostication. In these instances, the standard evidence requirement has been that management decisions follow as a result of test findings. Such a model has served the medical community well and set the standard for any emerging testing modality. The resulting action from these generally reimbursable common tests is often beneficial; however, they may also lead to unnecessary downstream diagnostic therapeutic cascades of interventions.15,16,27,28 For instance, a majority of carotid interventions in the United States are for asymptomatic carotid stenosis. A diagnostic test obviously preceded the detection of the stenosis, although the subsequent occlusion-preventive intervention is seldom a valid indication in asymptomatic patients.29 Because of the above cited considerations, payers are often cautious about utilization of certain tests and their consequent expensive treatments. Thus, circumspection on part of the payers, especially for tests based on novel technologies, would not be an unreasonable expectation. However, the emerging evidence suggests that the benefits of CES favor cost reduction by eliminating the need for subsequent diagnostic procedures and further detailed clinical evaluation.
The 3 technical issues most commonly debated with regard to exome sequencing include completeness of gene coverage, validation of results, and variant interpretation.21,22 As described above, coverage across the exome is determined on average because, typically due to structural differences, not all portions of the exome are sequenced equally and it is therefore possible that a mutation may exist in a region of reduced coverage. However, coverage and detection of variants can be improved by increasing the number of reads (the depth of exome sequencing)30 or using targeted next-generation strategies to improve capture of troubling regions31 so this concern is not inherent to the method itself and can be resolved through technical performance standards. As of this writing, technological improvements have already significantly reduced this issue to a level of minimal concern. Validation, usually by Sanger-based methodology, which is still considered the gold standard by some, has been shown to not be necessary for most identified variants of sufficient quality.10 Variant interpretation is the process by which the likelihood that a variant is pathogenic or not is determined. For variants that have never been reported in patients, the methods of assessing pathogenicity do not differ between Sanger and next-generation technologies, as they require human application of knowledge (e.g., bioinformatics). The process, however, becomes much more extensive for exome sequencing because of the larger dataset being evaluated and is complicated by technical issues of quality, data processing, genomic alignment, and variant analysis that can vary among laboratories.32 Because most laboratories performing exome sequencing utilize different methods for variant assessment, they may potentially interpret identical variants in different ways as a result (e.g., pathogenic, likely pathogenic, or benign vs uncertain significance, discussed further below). These differences in analytic method, when made transparent, can be assessed by the ordering clinician and utilized in the final clinical interpretation. Because of this aspect of uncertainty, unique to genomic sequencing methods, exome sequencing results can produce indeterminate yet still clinically relevant results (e.g., variants of uncertain significance). As the testing becomes more widespread, it is also likely that a degree of standardization of testing platforms and analytic methods will occur across laboratories.
Major considerations in exome sequencing: Variants of uncertain importance and incidental/secondary findings
The term variant of uncertain significance (VUS) refers to a genetic change in a gene of interest that is novel or rare in the population and has never been reported in connection with disease.33 Such variants are not unique to exome sequencing and can be observed with any sequencing method. Bioinformatic prediction methods currently are insufficient to solely guide clinical interpretation34–36 and the presence of a variant at a low frequency in a population (e.g., <1%) also does not equate to a lack of pathogenicity due to issues of incomplete penetrance, expressivity, improper phenotyping, modes of inheritance, or other considerations.8,33 These variants are not insignificant as they can (and in many cases do) represent actual pathogenic mutations that have yet to be verified. In some cases, but not all, this verification can be done through additional testing of other family members or through detection of associated biomarkers. Furthermore, as DNA sequencing does not change over time, exome sequencing results can be reinterpreted as new clinical data is reported, increasing the chances for an ultimately positive result even if initial testing is inconclusive.
Exome sequencing can also identify incidental or secondary findings (e.g., unexpected but medically significant results such as a predisposition to cancer). Such findings are not uncommon and have been seen in approximately 4%–5% of patients.24,26 The American College of Medical Genetics has established recommendations for specific genes that should be screened in all clinical exomes.37 Patients must be counseled regarding this possibility prior to testing and provided the opportunity to decide whether they would wish to know such results if any are found.38 It is important to note that although this technology is capable of finding unexpected genetic mutations, due to multiple issues such as the variability associated with gene expressivity and penetrance, as well as ethical considerations, there is currently no clinical indication to perform exome sequencing as an elective screening test for assessing the risk of future disease in asymptomatic healthy individuals in the absence of other concerns.
Indications, contraindications, and billing of CES
The current and generally accepted indications for exome sequencing testing and the relative contraindications of such testing are indicated in table 3. Accumulating evidence in this relatively new field may alter these indications and limitations in the future.
Table 3.
Indications, contraindications, and Current Procedural Terminology/Healthcare Common Procedure Coding System (CPT/HCPCS) codes for clinical exome sequencing

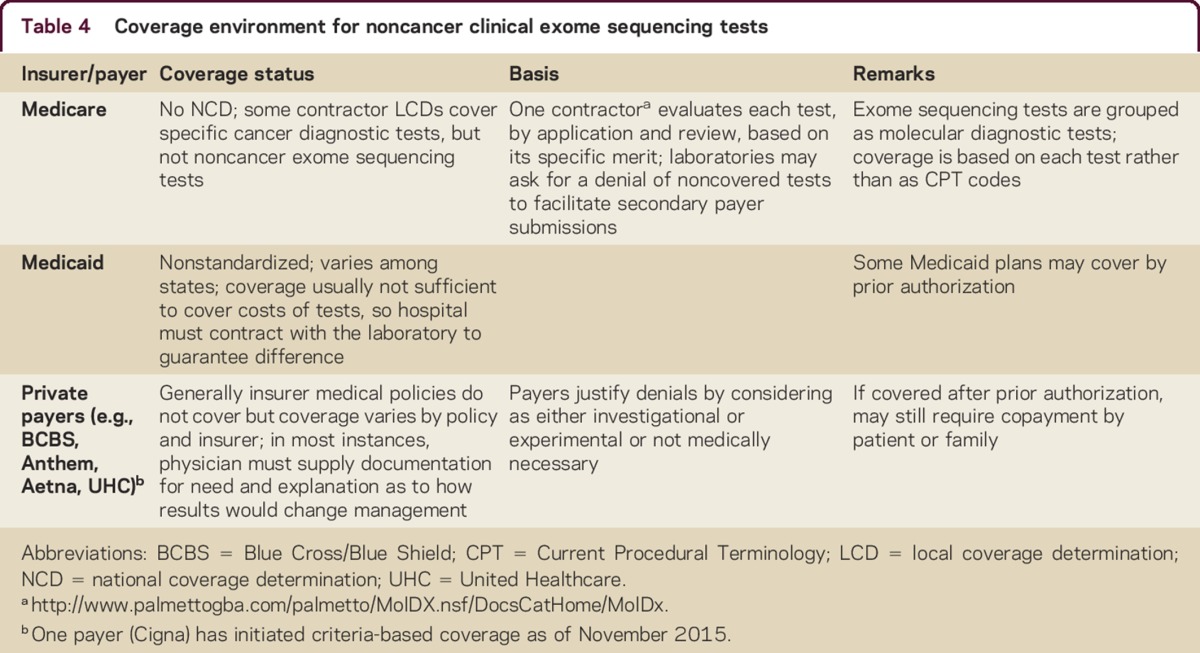
Not every payer accepts all submitted procedure codes for payment and not all providers submit the same set of codes. A new section in 2015 on genomic sequencing procedures describes DNA sequence analysis methods that simultaneously assay multiple genes or genetic regions and includes CES (table 3). These commonly used codes for exome sequencing should be used regardless of the technique employed, unless specifically noted in the code descriptor. Not all new molecular pathology diagnostic codes are recognized by common payers such as Medicare or Medicaid20 and coverage environment is variable (table 4),39,40 due in part to the lack of a comprehensive payer policy justifying the use of this testing clinically in neurologic disease.
Table 4.
Coverage environment for noncancer clinical exome sequencing tests
One Medicare contractor has established a Molecular Diagnostic Services Program (MolDx; Palmetto; http://www.palmettogba.com/palmetto/MolDX.nsf/DocsCatHome/MolDx). It acts as a repository and a registry primarily for coding help and it also details its expectations for coverage. It is advisable to consult with individual payers, diagnostic laboratories, and responsible billing agencies before selecting and submitting codes. The name of the specific test being performed may need to be provided either during submission of claim forms or subsequently. Similarly, prior clearance of the specific test by the payer contractor may also be necessary.
One payer has provided the helpful definition for a molecular diagnostic test (MDT) as “any test that involves the detection or identification of nucleic acid(s) (DNA/RNA), proteins, chromosomes, enzymes, cancer chemotherapy sensitivity and/or other metabolite(s). The test may or may not include multiple components. A MDT may consist of a single mutation analysis/identification, and/or may or may not rely upon an algorithm or other form of data evaluation/derivation” (MolDx; Palmetto; http://www.palmettogba.com/palmetto/MolDX.nsf/DocsCatHome/MolDx).
Regulatory status
No US Food and Drug Administration–approved genotyping tests are identifiable as of this writing. Therefore, genotyping by CES is offered as a laboratory-developed test. Clinical laboratories may develop and validate such tests in-house (e.g., home brew). These tests must comply with the regulatory standards of the Clinical Laboratory Improvement Act (CLIA) and the laboratory offering the service must be licensed by CLIA for high-complexity testing. At the time of this writing, only a select number of laboratories (<10) are currently performing CLIA-certified exome sequencing. Because of the sophisticated nature of this testing and analysis, diagnostic laboratories must invest major resources into this testing methodology (e.g., equipment, computers and software, and trained personnel). Consequently, the number of laboratories performing this testing is expected to remain limited and of high performance and quality.
Future directions
As illustrated above, CES is rapidly establishing itself as the standard for efficient and cost-effective diagnosis of complicated neurogenetic cases featuring broad differentials and clinically heterogeneous phenotypes. The field of human genetics and genomics is rapidly changing and this is likely a prelude to further advances including better methods of bioinformatic analysis, development of more extensive databases of normal human genetic variation, improvements in next-generation sequencing methodologies and equipment, and more widespread clinical use of genome sequencing.41 Establishing the appropriate clinical role for exome sequencing and integrating payer policies at this stage will ease the incorporation of these new advances as they become available and transition into the clinic to further improve diagnosis as well as patient management and care.
Take-home points
Next-generation sequencing methodology allows for the rapid and relatively inexpensive analysis of large amounts of DNA, up to entire genomes.
Exome sequencing utilizes next-generation technology to analyze all protein-coding sequences in the human genome, comprising approximately 21,000 genes.
Exome sequencing can broadly improve the diagnosis rate for suspected neurogenetic disorders with clinically heterogeneous phenotypes in both children and adults, leading to benefits in patient care.
Clinical exome sequencing should be utilized to complement, not supersede, a systematic comprehensive patient evaluation. Physicians should receive proper training on its appropriate use and interpretation of results.
We urge payers to utilize these recommendations in the development of comprehensive coverage policies for clinical exome sequencing.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Katie Shepard for administrative assistance with this manuscript. B.L.F. was supported by NIH grant R01NS082094.
Footnotes
Supplemental data at Neurology.org/cp
AUTHOR CONTRIBUTIONS
All authors contributed to the writing, editing, and critical review of this manuscript.
STUDY FUNDING
Supported by NIH (R01NS082094) to B.L.F.
DISCLOSURES
B. Fogel has received funding for travel or speaker honoraria from American Academy of Neurology, American Physician Institute for Advanced Professional Studies, and the National Ataxia Foundation and receives research support from NIH (NINDS, NIMH) and the National Ataxia Foundation. S. Satya-Murti has participated in telephone consultations or in-person medical advisory board meetings for United BioSource Corporation (UBC), Simon-Kucher consultants, AstraZeneca, Avalere LLC, Evidera consulting group, Covidien, Michael J. Fox Foundation, Foley Hoag LLP, Baxter, and Abiomed; has received funding for travel from UBC, AstraZeneca, Avalere LLC, Evidera consulting group, Covidien, Michael J. Fox Foundation, Foley Hoag LLP, and Baxter; serves on the editorial board of Neurology: Clinical Practice; served on the American Academy of Neurology Payment Policy Subcommittee; and served as panelist and later (2010–2011) Vice-Chair CMS-MEDCAC (Medicare Evidence Development and Coverage Advisory Committee). For the duration of the MEDCAC meeting, usually 1–2 days, S. Satya Murti was considered an SGE (Special Government Employee). B. Cohen serves on a DSMB for Stem Cell Transplantation for MNGIE and Chairman of the External Advisory Board of Clinical Protocols, Neurofibromatosis Consortium, Department of Defense; has received honoraria from AAN for speaking activities and serving on committees; serves on the editorial boards of Pediatric Neurology and Mitochondrion; serves as Director of The NeuroDevelopmental Science Center and Director of Neurology for Akron Children's Hospital; serves as a consultant for Motive Medical Intelligence (formally known as New Mentor), Stealth Biotherapeutics, and Mitobridge; serves on speakers' bureaus for Transgenomic Labs, Courtagen Labs, United Mitochondrial Disease Foundation, and Courtagen Labs; is a consultant to Health and Human Services for the Division of Vaccine Injury; receives research support from Edison Pharmaceuticals, Raptor Pharmaceuticals, Stealth Biotherapeutics, and Reata Pharma; serves on the Board of Trustees for the United Mitochondrial Disease Foundation; and has participated in medico-legal cases. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.

REFERENCES
- 1.Fogel BL, Lee H, Deignan JL, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol 2014;71:1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol 2014;76:473–483. [DOI] [PubMed] [Google Scholar]
- 3.Iglesias A, Anyane-Yeboa K, Wynn J, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med 2014;16:922–931. [DOI] [PubMed] [Google Scholar]
- 4.Fogel BL, Geschwind DH. Clinical neurogenetics. In: Daroff R, Jankovic J, Mazziotta J, Pomeroy S, eds. Neurology in Clinical Practice. 7th ed Philadelphia: Elsevier; 2015:648–675. [Google Scholar]
- 5.Neveling K, Feenstra I, Gilissen C, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 2013;34:1721–1726. [DOI] [PubMed] [Google Scholar]
- 6.Fogel BL, Vickrey BG, Walton-Wetzel J, Lieber E, Browner CH. Utilization of genetic testing prior to subspecialist referral for cerebellar ataxia. Genet Test Mol Biomarkers 2013;17:588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 2011;12:745–755. [DOI] [PubMed] [Google Scholar]
- 9.Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med 2014;370:2418–2425. [DOI] [PubMed] [Google Scholar]
- 10.Strom SP, Lee H, Das K, et al. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet Med 2014;16:510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelen M, Veltman JA. Genome and exome sequencing in the clinic: unbiased genomic approaches with a high diagnostic yield. Pharmacogenomics 2012;13:511–514. [DOI] [PubMed] [Google Scholar]
- 12.Miyatake S, Matsumoto N. Genetics: clinical exome sequencing in neurology practice. Nat Rev Neurol 2014;10:676–678. [DOI] [PubMed] [Google Scholar]
- 13.Gomez CM, Das S. Clinical exome sequencing: the new standard in genetic diagnosis. JAMA Neurol 2014;71:1215–1216. [DOI] [PubMed] [Google Scholar]
- 14.Riley RF, Don CW, Powell W, Maynard C, Dean LS. Trends in coronary revascularization in the United States from 2001 to 2009: recent declines in percutaneous coronary intervention volumes. Circ Cardiovasc Quality Outcomes 2011;4:193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lucas FL, Siewers AE, Malenka DJ, Wennberg DE. Diagnostic-therapeutic cascade revisited: coronary angiography, coronary artery bypass graft surgery, and percutaneous coronary intervention in the modern era. Circulation 2008;118:2797–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradley SM, Spertus JA, Kennedy KF, et al. Patient selection for diagnostic coronary angiography and hospital-level percutaneous coronary intervention appropriateness: insights from the National Cardiovascular Data Registry. JAMA Internal Medicine 2014;174:1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deverka PA, Kaufman D, McGuire AL. Overcoming the reimbursement barriers for clinical sequencing. JAMA 2014;312:1857–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deverka PA, Dreyfus JC. Clinical integration of next generation sequencing: coverage and reimbursement challenges. J Law Med Ethics 2014;42(suppl 1):22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014;6:265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satya-Murti S, Cohen B, Michelson D. Chromosomal microarray analysis for intellectual disabilities. Template Coverage Policy 2013. Available at: https://www.aan.com/uploadedFiles/Website_Library_Assets/Documents/3.Practice_Management/1.Reimbursement/1.Billing_and_Coding/5.Coverage_Policies/13%20ChromoMicroIntelDisabil.pdf Accessed November 10, 2015. [Google Scholar]
- 21.Ku CS, Cooper DN, Polychronakos C, Naidoo N, Wu M, Soong R. Exome sequencing: dual role as a discovery and diagnostic tool. Ann Neurol 2012;71:5–14. [DOI] [PubMed] [Google Scholar]
- 22.Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 2015;17:444–451. [DOI] [PubMed] [Google Scholar]
- 23.BCBS Tec Report. Special Report: Exome sequencing for clinical diagnosis of patients with suspected genetic disorders 2013. Available at: http://www.bcbs.com/blueresources/tec/vols/28/exome-sequencing.html Accessed March 30, 2015. [PubMed]
- 24.Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014;312:1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312:1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verrilli D, Welch HG. The impact of diagnostic testing on therapeutic interventions. JAMA 1996;275:1189–1191. [PubMed] [Google Scholar]
- 28.Hoffman JR, Kanzaria HK. Intolerance of error and culture of blame drive medical excess. Bmj 2014;349:g5702. [DOI] [PubMed] [Google Scholar]
- 29.Yang C, Bogiatzi C, Spence JD. Risk of Stroke at the time of carotid occlusion. JAMA Neurol 2015;72:1261–1267. [DOI] [PubMed] [Google Scholar]
- 30.Meynert AM, Ansari M, FitzPatrick DR, Taylor MS. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 2014;15:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sikkema-Raddatz B, Johansson LF, de Boer EN, et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat 2013;34:1035–1042. [DOI] [PubMed] [Google Scholar]
- 32.Bao R, Huang L, Andrade J, et al. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform 2014;13(suppl 2):67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fogel BL. Interpretation of genetic testing: variants of unknown significance. Continuum 2011;17:347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castellana S, Mazza T. Congruency in the prediction of pathogenic missense mutations: state-of-the-art web-based tools. Brief Bioinform 2013;14:448–459. [DOI] [PubMed] [Google Scholar]
- 35.Gray VE, Kukurba KR, Kumar S. Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations. Bioinformatics 2012;28:2093–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat 2011;32:358–368. [DOI] [PubMed] [Google Scholar]
- 37.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hallowell N, Hall A, Alberg C, Zimmern R. Revealing the results of whole-genome sequencing and whole-exome sequencing in research and clinical investigations: some ethical issues. J Med Ethics 2015;41:317–321. [DOI] [PubMed] [Google Scholar]
- 39.Prince AE. Prevention for those who can pay: insurance reimbursement of genetic-based preventive interventions in the liminal state between health and disease. J Law Biosciences 2015;2:365–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atwal PS, Brennan ML, Cox R, et al. Clinical whole-exome sequencing: are we there yet? Genet Med 2014;16:717–719. [DOI] [PubMed] [Google Scholar]
- 41.Fogel BL, Lee H, Strom SP, Deignan JL, Nelson SF. Clinical exome sequencing in neurogenetic and neuropsychiatric disorders. Ann NY Acad Sci (Epub ahead of print 2015). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.