

Abstract
Overexpression of EZH2 is frequently linked to the advanced and metastatic stage of cancers. The mechanisms of its oncogenic function can be context specific, and may vary depending on the protein complexes that EZH2 interacts with. To identify novel transcriptional collaborators of EZH2 in cancers, a computational approach was developed that integrates protein-DNA binding data, cell perturbation gene expression data, and compendiums of tumor expression profiles. This holistic approach identified E2F1, a known mediator of the retinoblastoma (Rb) tumor suppressor, as a transcriptional collaborator of EZH2 in castration resistant prostate cancer (CRPC). Subsequent analysis and experimental validation found EZH2 and E2F1 co-bind to a subset of chromatin sites lacking H3K27 trimethylation, and activate genes that are critical for prostate cancer progression. The collaboration of EZH2 and E2F1 in transcriptional regulation is also observed in diffuse large B-cell lymphoma (DLBCL) cell lines, where activation of the transcriptional network is concordant with the cellular response to the EZH2 inhibitor.
Implications
The direct collaboration between EZH2 and Rb/E2F1 pathway provides an innovative mechanism underlying the cascade of tumor progression, and lays the foundation for the development of new anticancer targets/strategies.
Keywords: EZH2, pRb/E2F pathway, transcriptional collaboration, castration resistant prostate cancer
INTRODUCTION
Deregulation of proteins involved in the epigenetic machineries is commonly observed in cancer, and the resultant aberrant gene expression leads to cancer initiation, progression and metastasis. One of the epigenetic regulators, the histone methyltransferase EZH2 (enhancer of zeste homolog 2), is frequently misregulated in a broad spectrum of tumors (1). As numerous studies have demonstrated that EZH2 plays a crucial role in multiple steps during tumor progression (2), targeting EZH2 represents a promising therapeutic option for advanced cancers. Several highly selective inhibitors of EZH2 methyltransferase activity have been generated (3-5). However, the biological processes affected by the EZH2 inhibitors are incompletely understood, and the molecular determinants of cancer cell responses to the drugs are still unclear.
EZH2 was originally identified as the catalytic subunit of the Polycomb repressive complex 2 (PRC2), which methylates K27 on histone H3 and leads to transcriptional silencing (6, 7). The oncogenic functions of EZH2 have been predominantly contributed to repression of tumor suppressor genes. However, increasing evidences, including our previous work, suggest that EZH2 may exert its oncogenic activities by transcriptional activation of genes critical for cancer progression (8-10). Under these scenarios, EZH2 physically interacts with signaling components other than PRC2 subunits, such as RelA/B, ER, and AR, to integrate the signaling circuitries for cancer-specific transcriptional programs. Therefore, it becomes intriguing and important to address the factors or pathways that cooperate with the oncogenic activity of EZH2.
To explore the transcriptional network in which EZH2 is involved to promote cancer progression, we developed a computational approach, named Gene Signature Association Analysis (GS2A), to infer the novel transcriptional collaborators of EZH2. The term “transcriptional collaborator” here refers to the factors that are co-localized with EZH2 at specific chromatin loci and cooperate to regulate the expression of a significant number of downstream target genes. The candidate collaborator predicted from GS2A was further validated using both experimental approaches and analysis of published datasets in other cancer types.
MATERIALS AND METHODS
Data collection
The candidate collaborator list of 2196 genes were curated by taking the union of (i) 1836 sequence-specific DNA binding TFs published by Vaquerizas et al (11), and (ii) 1469 TFs, 296 transcription cofactors, and 117 chromatin remodeling factors available at AnimalTFDB database (12). CistromeMap database (http://cistrome.org/pc/) was used for the query of publicly available ChIP-seq datasets (13). E2F1 and BRCA1 ChIP-seq datasets from ENCODE cell lines were downloaded from UCSC genome browser databases.
Identification of EZH2 solo peaks from ChIP-seq
EZH2 and H3K27me3 ChIP-seq data in abl cell line was processed using MACS v2.0 based on FDR<0.01 (14). To measure the EZH2 and H3K27me3 ChIP-seq intensity, we count the number of ChIP-seq reads within a 400 bp window centered by the summit of each EZH2 peak. If a read partially overlap with the window, the fraction of the read within the window is added to the count. To define the threshold for determining EZH2 solo peaks, we selected the top 1000 EZH2 peaks with highest EZH2 binding intensity, and plotted the cumulative distribution of H3K27me3 intensity (Supplemental Fig. 1). A plateau is observed in the curve, confirming the existence of bi-model. The threshold was then chosen at the point where the slope of the curve is minimal.
Determining signature genes
The EZH2 (E2F1)-activated signature genes were selected based on: (i) down-regulated upon EZH2 (E2F1) silencing with FDR<0.01 and fold-change>1.5; (ii) bound with EZH2 solo (E2F1) peaks at the promoter, i.e. +/− 2kb from Transcription Start Sites (TSS), in ChIP-seq data.
Preprocessing patient sample gene expression profile
For microarray datasets, raw measurements were normalized using RMA (Robust Multi-array Average). For RNA-seq data from TCGA, measurements in Reads Per Kilobases per Million reads (RPKM) were transformed into log-scale, where a pseudocount of 1 was added to the RPKM measure. The normalized data underwent Inverse Normal Transformation (INT) over the expression vector of each gene. INT transforms the distribution of variables into Gaussian, and represses the influence of the outliers on the correlation computation.
Association analysis between candidate gene and a set of signature genes
Suppose gc is a candidate gene, G is the set of all genes in a data matrix, and S = {g1, g2,..., gk} is a set of k signature genes such that Ss. We compute the Pearson correlation Cor(gc, x) between gc and any other gene x in G. Let mc and sdc be the mean and standard deviation of Cor(gc, x) for any x ∈ G – {gc}, the significance of coexpression association between gc and the signature gene set S is computed as:
This definition of association score is analogous to the modified GSEA score proposed by Irizarry et al, which takes the form of a z-score (15).
Robust Rank Aggregation
To integrate multiple rank lists from multiple datasets, we employed the Robust Rank Aggregation (RRA) algorithm proposed by Kolde et al (16). Suppose R = (r1, r2, = (rn) is a the vector of ranks of a gene in n lists, where the numbers of genes in the lists are represented as M = (m1, m2, = (mn). RRA first normalizes the ranks into percentiles U = (u1, u2, = Run), where ui = ri/mi (i = 1,2, irst. Under null hypotheses where the percentiles follow uniform distribution between 0 and 1, the kth smallest value among u1, u2, Uun is an order-statistic which follow Beta distribution B(k, n + 1 – k). RRA computes a p-value pk for the kth smallest value based on Beta distribution. The significance score of the gene, i.e. ρ-value, is ρ=min (p1, p2, ..., pn)
In GS2A, each candidate gene is assigned a “ρ-value” computed from order statistic, and a consensus rank list is generated in ascending order of the ρ-values. GS2A randomly permutes the order of candidates in each list to determine the distribution of ρ-value under the null hypothesis that the rank of a candidate is uniformly distributed. Empirical False Discovery Rates (FDRs) are then computed by comparing the observed ρ-values against the random distribution.
Motif enrichment analysis on ChIP-seq peaks
The sequences of 300-bp in length centered on summit were extracted for each ChIP-seq peak, and scanned with Position Weight Matrices. To determine the threshold for a motif “hit”, background sequences of same length were randomly extracted from promoter regions (+/− 2kb of TSS) of all RefSeq genes. Given a PWM, the threshold was chosen such that 10% of the background sequences have at least one hit.
Prostate cancer cell lines and culture conditions
The prostate cancer cell line LNCaP was obtained from the American Type Culture Collection, and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin. LNCaP-abl (abl) cell line was kindly provided by Zoran Culig (Innsbruck Medical University, Austria). It is maintained in phenol-red-free RPMI-1640 medium (Gibco) with the same supplements except for 10% charcoal-dextran-stripped FBS. Both cell lines have recently been authenticated at Bio-Synthesis Inc. and confirmed to be mycoplasma-free using MycoAlert Mycoplasma Detection Kit (Lonza).
Transfection and Western blot
Two different expression plasmids for HA-tagged E2F1 were purchased from Addgene. A total of 2 ug of each E2F1 construct were transfected into LNCaP cells using SuperFect (Qiagen) according to the manufacturer's instructions. 72 hrs after transfection, either total RNA or proteins were collected. For immunoblotting, cells were lysed in RIPA lysis buffer [25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS], and the following antibodies were used for Western blot: E2F1 (05-379; Millipore), HA-Tag (2367; Cell Signaling Technology), EZH2 (612666; BD Biosciences), and GAPDH (sc-32233, Santa Cruz Biotechnology).
RNA isolation and RT-PCR
RNA was extracted and purified using the TRIzol Reagent and RNeasy Mini Kit (Qiagen) according to manufacturer's protocols. Real-time quantitative RT-PCR was performed upon cDNA synthesis using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression was calculated relative to the level of GAPDH using the 2-ΔΔCt method as described previously (9). Sequences of primers were listed in Supplemental Table 5.
Standard ChIP and ChIP-seq assays
Chromatin immunoprecipitation (ChIP) experiments were performed as previously described (9). ChIP DNA was purified using PCR Purification Kit (Qiagen) and quantified by Quant-iT™ dsDNA HS Assay Kit (Invitrogen). 5-10 ng of ChIP enriched DNA were prepared for the ChIP-seq libraries using the ThruPLEX-FD Prep Kit (Rubicon Genomics) according to the manufacturer's protocol, and libraries were sequenced on the NextSeq 500. For targeted ChIP, extracted DNA was used for real-time quantitative PCR with the specific primers as listed in Supplemental Table 6. The antibodies used for ChIP and ChIP-seq assays include: normal rabbit IgG (sc-2027; Santa Cruz Biotechnology), E2F1 (554213, BD Pharmingen and sc-193, Santa Cruz Biotechnology) and EZH2 (39933, Active Motif).
Co-immunoprecipitation (co-IP) assays
The co-IP experiments of endogenous EZH2 and E2F1 proteins were performed in abl cells as describe previously (9). Antibodies used for the co-IP assays include 6 ug of E2F1 (sc-193; Santa Cruz Biotechnology), 8 uL of EZH2 (39933, Active Motif), and equal amount of normal rabbit IgG (sc-2027; Santa Cruz Biotechnology).
RNA-seq protocols and data analysis
RNA-seq library was prepared using Illumina True-seq RNA sample preparation kit and sequenced to 50bp using Illumina Hi-seq platform. The RNA-seq data was mapped to human genome build 37 (hg19) using TopHat version 2.0.6 (17). Gene expression level was calculated as gene-based FPKM (fragment count per kilobases per million reads) using Cufflinks version 2.0.2 (18), and the differential gene expression was measured as the fold change of the FPKM between knockdown and control conditions.
Genome re-sequencing of LNCaP and abl cell lines
Prostate cancer cell lines LNCaP and abl genomic DNA were extracted using QIAGEN QIAamp DNA Mini Kit. 100bp paired sequencing was performed using Illumina Hi-Seq platform. Paired reads of DNA sequences were aligned with BWA software (19), followed by realignment, re-calibration, quality control and genomic variant calling using GATK pipeline version 1.1 with default settings (20). Profile of relative copy number variations between LNCaP and abl were computed using VarScan version 2.2 (21).
Software availability
Source codes of GS2A modules are publicly accessible at https://github.com/xh1974/GS2A.
Data Access
RNA-seq and ChIP-seq datasets generated along with the work have been submitted to Gene Expression Omnibus (GEO) with accession numbers GSE56331 and GSE67809, respectively. The relative copy number changes between LNCaP and abl, as processed by VarScan on whole genome re-sequencing data, are available in Supplemental Table 7.
RESULTS
Bi-model of EZH2 chromatin binding in castration resistant prostate cancer cells
To explore the oncogenic function of EZH2 in cancer, we mapped EZH2 chromatin binding sites and H3K27 trimethylation (H3K27me3) signals in LNCaP-abl (abl) (9), a prostate cancer cell model that strongly resembles the clinical cases of metastatic, hormone-refractory prostate tumors (22). The ChIP-seq profiling revealed two types of EZH2 binding sites: “ensemble” peaks with both EZH2 and H3K27me3 enrichment (Fig. 1A), and “solo” peaks with only EZH2 binding signals (Fig. 1B). We analyzed the ChIP-seq data by quantitatively measuring the H3K27me3 signal across 26,645 EZH2 peaks in the genome, and confirmed the bi-model EZH2 binding in abl (Fig. 1C), with 22,042 (82.7%) ensemble peaks and 4,603 (17.3%) solo peaks (Supplemental Table 1).
Figure 1. Bi-model of EZH2 chromatin binding patterns in abl cells.
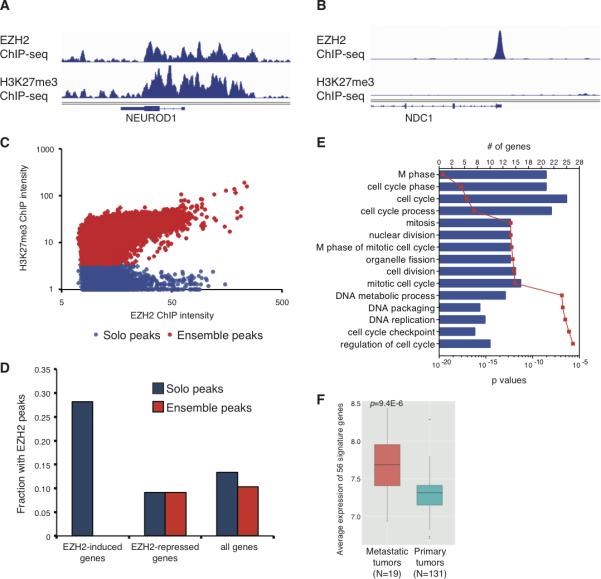
(A and B) ChIP-seq profiles demonstrating examples of EZH2 ensemble peaks (A) and solo peaks (B). (C) H3K27me3 signals at EZH2 binding sites. Each dot in the scatter plot represents one EZH2 binding peak. (D) Bar plots showing the association between EZH2 chromatin binding peaks and EZH2-dependent genes in abl cells. There are none EZH2-induced genes bound by the ensemble peaks. (E) Gene ontology analysis of the 56 EZH2-activated signature genes using DAVID functional analysis tool at https://david.ncifcrf.gov. Blue bars, the numbers of genes in the specific category; red dots, p values of each functional group. (F) Comparison of the expression levels of the 56 signature genes between metastatic and primary prostate tumors (26). N, numbers of clinical samples in the specific stage of tumors. P value was calculated based on Kolmogorov-Smirnov test.
To gain insights into the biological functions of these two distinct patterns of EZH2 chromatin binding, we examined the association of the peaks with EZH2-regulated genes (Fig. 1D). Using gene expression profiling in abl cells, we identified 197 downregulated and 28 upregulated genes upon EZH2 silencing. The over-representation of EZH2-induced genes suggests a gene activation function of EZH2 in castration resistant prostate cancer (CRPC) cells. Interestingly, 56 of EZH2-induced genes (28.4%) are bound by EZH2 solo peaks at their promoter regions, compared to only 15.5% of all RefSeq genes bound by the solo peaks (p=1.73E-6). Gene ontology analysis on the 56 genes showed significant enrichment of cell-cycle regulators (p=2.0E-6) and DNA packaging genes (p=1.7E-7) (Fig. 1E). It is interesting to note that no enrichment of ensemble peaks at EZH2-repressed genes was observed. Among these EZH2-activated genes, several have been indicated to play critical roles in cancer progression, such as ATAD2 (23), CDK1 (24) and FOXM1 (25). In prostate cancer patient samples (26), these signature genes are significantly upregulated in the metastatic prostate tumors comparing to the primary cancer samples (Fig. 1F). These lines of evidence imply a novel yet important function of EZH2 in transcriptional regulation, which employs different mechanisms from the canonical PRC2 complex.
Computational inference of transcriptional collaborator of EZH2 in prostate cancer
Because EZH2 binds to solo peaks independent of its Polycomb repressor activity, we hypothesized that a distinct set of transcriptional collaborators mediates the recruitment of EZH2 to solo peaks and its gene activation function in abl cells. Therefore, we performed computational analysis that integrates multiple data sources to systematically screen for the potential transcriptional collaborators of EZH2.
The workflow of our computational approach, named Gene Signature Association Analysis (GS2A), is illustrated in Fig. 2A. GS2A first selected the 56 EZH2-activated targets as “signature genes”, whose expressions are expected to reflect the activities of EZH2-involved transcriptional network in gene activation (Supplemental Table 2). Next, GS2A screened 2,196 candidates, including transcription factors, cofactors, and chromatin regulators, to identify those coexpressed with the signature genes in the compendiums of gene expression profiles from clinical prostate tumor samples. To measure the significance of coexpression association between a candidate regulator and a signature gene set, all the genes in a profiling dataset are ranked in order of their correlation with the candidate, followed by Gene Set Enrichment Analysis (GSEA) that measures the enrichment of signature genes in the top of the ranked list (15). A proof-of-concept hit is EZH2 itself, as most signature genes are at the top of the list sorted in descending order of the correlation with EZH2 (Fig. 2B). In contrast, the signature genes are almost uniformly distributed with a housekeeping gene ACTB (Fig. 2C).
Figure 2. Integrative analysis to infer transcriptional collaborators of EZH2 in prostate cancer cells.

(A) A schematic view of the GS2A computational analysis. (B and C) Ranks of the EZH2-activated signature genes in the RefSeq gene list in descending order of correlation with EZH2 (B) and ACTB (C). Red areas represent Pearson correlation coefficients. Black vertical bars represent the occurrence of signature genes in the list. P values were computed based on permutation test. (D) Motif enrichment at EZH2 solo peaks for 7 top-ranked candidates with known motif matrices.
To improve the accuracy and reproducibility of the association analysis, we included two independent gene expression profiling datasets: a compendium of 131 primary prostate tumor samples on Affymetrix exon arrays (26), and The Cancer Genome Atlas (TCGA) RNA-seq data that includes 140 primary prostate tumor samples (27). Since these expression cohorts have different profiling platforms, batch effects, and sample collection bias, GS2A generates one rank-ordered list for each cohort based on GSEA score, and employs a Robust Rank Aggregation (RRA) approach to compute a consensus rank list that integrates evidence from both cohorts. To this end, GS2A identified 35 genes that are reproducibly coexpressed with the signature genes (Supplemental Table 3). As expected, EZH2 itself is top-ranked.
Among the 35 candidate genes, 6 have established sequence-specific motif matrixes. BRCA1 does not directly bind to DNA, but a de novo BRCA1 motif can be derived from published BRCA1 ChIP-seq data. We scanned the motifs of these 7 factors in the pool of EZH2 solo peaks, and found that E2F1 binding sequence to be the most significantly enriched (Fig. 2D). Indeed, the coexpression association result showed a positive correlation of E2F1 with EZH2-activated signature genes (Supplemental Fig. 2A). Interestingly, the expression levels of EZH2 and E2F1 are only marginally correlated (Supplemental Fig. 2B), implying a direct correlation of E2F1 with EZH2-activated signature genes. Since E2F1 directly binds to DNA, investigation on its role in EZH2-associated pathway may clarify the molecular mechanisms underlying the recruitment of EZH2 to solo peaks. Therefore, we focused on E2F1 for subsequent analysis.
Co-localization of EZH2 and E2F1 at chromatin regulatory elements lacking of H3K27me3 signals
To validate the genomic co-localization of EZH2 and E2F1, we first performed direct ChIP against either EZH2 or E2F1 at a number of selected locations. While EZH2 is enriched at both types of sites, E2F1 can only be detected at solo peaks (Fig. 3A). We then generated the E2F1 ChIP-seq profile in abl cell, and the global chromatin binding profiling confirmed the enrichment of E2F1 binding at the proximal regions of EZH2 solo peaks, but not at the ensemble peaks (Fig. 3B). We also retrieved two public available E2F1 ChIP-seq datasets, one in the cervical cancer cell line Hela and the other in the breast cancer cell line MCF-7 (28). We found that more than 60% of EZH2 solo peaks overlap with E2F1 binding sites in at least one of the cell lines mentioned above, whereas less than 10% of the ensemble peaks overlap with E2F1 localization (Fig. 3C). This suggests that the EZH2 solo peaks are mostly conserved E2F1 binding sites in human cancers. We next performed the coimmunoprecipitation assays, and detected a clear interaction between endogenous EZH2 and E2F1 proteins in abl cells (Fig. 3D), suggesting that these two factors bind to each other physically. To this end, we conclude that EZH2 and E2F1 co-localize within a transcriptional complex at a specific subset of chromatin regulatory elements that lack the signals of repressive histone mark H3K27me3.
Figure 3. Co-localization of EZH2 and E2F1 at solo peaks.

(A) ChIP qPCR validation of EZH2 and E2F1 binding at solo or ensemble peaks in abl cells. Error bars represent the standard deviation from three biological replicates. (B) Heat maps showing ChIP-seq intensities of EZH2, H3K27me3, and E2F1 in a window size of 2 kb centered at the summits of EZH2 solo or ensemble peaks in abl. (C) Bar charts showing the percentages of EZH2 solo or ensemble peaks overlapping with E2F1 binding sites in Hela and MCF-7 cells. (D) Western blots of co-immunoprecipitation using indicated antibodies in abl cells. NE, nuclear extracts.
Mutual activation of downstream targets by the transcriptional complex consisting of EZH2 and E2F1
To validate the cooperation between EZH2 and E2F1 on transcriptional control, we compared the transcriptional programs induced by either protein. We found significant overlap between EZH2- and E2F1-activated genes (Fig. 4A), but observed little overlap between the repressed genes (Supplemental Fig. 3), suggesting that both proteins are involved in the same transcriptional network leading to similar gene activation patterns. Specifically, most EZH2-stimulated signature genes are downregulated upon E2F1 knockdown (Fig. 4B). When we overexpressed E2F1 in prostate cancer cells, the transcriptional levels of EZH2-induced genes are significantly increased (Supplemental Fig. 4). Similarly, the expression levels of genes directly activated by E2F1 (Supplemental Table 4) are also decreased when silencing EZH2 (Fig. 4C). The EZH2-mediated regulation of E2F1 targets is not likely through the modulation of E2F1 level, since the E2F1 protein expression did not show dramatic changes upon EZH2 silencing (Supplemental Fig. 5). These results further imply the direct crosstalk between EZH2 and E2F1 in transcriptional control. The collaboration between EZH2 and E2F1 can also be demonstrated in some other biological systems. Data in mouse fibroblast showed similar upregulation pattern of EZH2-activated signature genes upon E2F1 overexpression (29) (Supplementary Fig. 6A). Likewise, E2F1-activated signature genes were significantly downregulated when EZH2 was depleted in the primary erythroid progenitor cells (30) (Supplemental Fig. 6B).
Figure 4. Collaboration between EZH2 and E2F1 in transcriptional activation of downstream targets.

(A) Venn diagram showing overlap of differential genes downregulated by silencing of EZH2 or E2F1 in abl cells. Numbers, number of genes that are activated by either EZH2 or E2F1 or both. P value was computed based on hyper-geometric distribution. (B and C) Box plots showing downregulation of EZH2-activated signature genes upon E2F1 knockdown (B) or E2F1-activated signature genes upon EZH2 knockdown (C) in abl cells. P values were computed using Kolmogorov-Smirnov test. (D) Bar charts showing the enrichment of EZH2 solo peaks and/or E2F1 chromatin binding within the promoter regions of genes activated by either/both proteins. The random expectations were computed by assuming independence between binding events and gene activation in 3*4 contingency table. P values were computed based on hyper-geometric distribution. N.s., not significant.
To determine the functional significance of the collaboration between EZH2 and E2F1, we correlated the chromatin binding profiles of EZH2 and E2F1 with the gene patterns activated by either protein in abl cells (Fig. 4D). Since 98.8% of the genes containing EZH2 solo peaks are also bound by E2F1 (Supplemental Fig. 7), we included unique E2F1 binding sites and background sequences as controls. It is noteworthy that the enrichment of binding sites near activated genes became the most prominent when considering the co-localization of EZH2 solo and E2F1 peaks at target genes coactivated by both. These data further confirmed the significance of EZH2 and E2F1 collaboration on direct activation of downstream genes.
Association of EZH2-E2F1 pathway with genomic aberrations in prostate cancer
Somatic mutations of EZH2 have been identified in various lymphoid and myeloid neoplasms (31). However, EZH2 is frequently overexpressed but rarely mutated in prostate tumors (32). It is thereby compelling to explore the genetic alterations that activate EZH2-associated signaling. The Rb/E2F pathway has been well established as the upstream regulator of EZH2 expression (33). MYC coordinately stimulates EZH2 expression (34) and induces the expression as well as the transcriptional activity of E2F1 (35). Therefore, MYC amplification and RB1 deletion, the genomic abnormalities that are frequently observed in a wide range of cancer types, may drive an EZH2/E2F1-dependent transcriptional program for cancer progression.
We firstly compared the sequence of castration resistant abl cells against the parental androgen-dependent LNCaP using whole genome re-sequencing. MYC amplification and RB1 deletion were both observed in abl compared with LNCaP (Fig. 5A), while no copy number aberrations were detected at EZH2 and E2F1 loci. We next asked if the activation of EZH2-E2F1 pathway is associated with MYC amplification and RB1 deletion in prostate tumors. We categorized the TCGA prostate cancer samples into four groups based on the copy number statuses of MYC and RB1 (MYC+/RB1−, MYC+/RB1wt, MYCwt/RB1−, MYCwt/RB1wt) (27). The signature genes are marginally overexpressed in MYC+/RB1wt and MYCwt/RB1− categories when comparing with the MYCwt/RB1wt category, and are most significantly upregulated when MYC is amplified and RB1 is deleted (Fig. 5B). These observations suggest that the combination of MYC amplification and RB1 deletion may contribute to the activation of EZH2 and E2F1 signaling during prostate cancer progression.
Figure 5. Activation of EZH2-E2F1 transcriptional complex is associated with MYC and RB1 genetic aberrations.

(A) Genome-scale view of relative copy number alterations in abl cells compared with LNCaP cells. Arrows, the genetic loci of indicated genes. (B) The average expression levels of EZH2-activated signature genes in prostate tumor samples with the indicated copy number alterations of MYC and RB1. P values were computed from t-test. +, gene amplification; −, gene deletion; wt, wild type. N, number of clinical samples available in each corresponding category. (C) The graphic view of the proposed model for oncogenic signaling pathway in prostate cancer.
In summary, we propose a schematic model of EZH2-E2F1 oncogenic pathway in prostate cancer, as shown in Fig. 5C. The MYC amplification and RB1 deletion drive the activation of the transcriptional network consisting of EZH2 and E2F1. Co-binding of EZH2 and E2F1 at a specific set of chromatin regulatory sites further activates downstream genes that play critical roles in cancer cell proliferation.
Conservation of EZH2-E2F1 pathway in DLBCLs and its clinical implication
To investigate the conservation of EZH2-E2F1 pathway in other cancer types and extend our discoveries to potential clinical applications, we collected the expression profiles in diffuse large B-cell lymphomas (DLBCLs) (3), and examined the expression patterns of EZH2-activated signature genes derived from prostate cancer cells under this scenario (Fig. 6A). This dataset includes the transcriptional profiles in 10 DLBCL cell lines treated with the small-molecule EZH2 inhibitor GSK126, as well as the profiles upon EZH2 knockdown in two of the GSK126-sensitive DLBCL cell lines, Pfeiffer and KARPAS-422.
Figure 6. Collaboration between EZH2 and E2F1 in transcriptional control is conserved in DLBCL.

(A) Heat maps showing the relative gene expression levels upon EZH2 knockdown (shEZH2) or EZH2 inhibitor (GSK126) treatment in DLBCL cell lines. For EZH2 silencing, cells were infected with either control shRNA or specific shRNA against EZH2 (shEZH2) for 10 days; for inhibitor treatment, cells were incubated with either 0.1% DMSO or 500 nM GSK126 for 3 days. Upper panel, EZH2-activated signature genes; bottom panel, EZH2-repressed genes identified in abl cells. Genes were sorted from top to bottom according to the expression change levels upon EZH2 knockdown in Pfeiffer cells. DLBCL cell lines with red fonts bear EZH2 mutations, and those with black fonts are EZH2 wild type. Cells were sorted from left to right according to the sensitivity to GSK126. PCa, prostate cancer. (B) Changes of average log2-ratio expression levels of EZH2-activated signature genes (blue bars) or EZH2-repressed genes (red bars) upon GSK126 treatment in DLBCL cell lines. (C and D) Box plots showing the correlation of EZH2-activated signature genes with EZH2 (C) and E2F1 (D) in DLBCL tumor samples. P values were computed based on Kolmogorov-Smirnov test.
We found that majority of the signature genes derived from abl cells are also downregulated upon EZH2 silencing or inhibitor treatment in DLBCL lines. However, the EZH2-repressed genes do not show a trend of derepression upon EZH2 perturbation. The magnitudes of the average expression changes for the activated signature genes are maximal in GSK126-sensitive cell lines, and are attenuated in GSK126-insensitive cell lines. In contrast, the expression changes of the canonical EZH2-repressed genes do not agree with the cell responsiveness to GSK126 (Fig. 6B). We specifically examined the levels of three previously reported EZH2-repressed genes, CDKN1A, IRF4 and PRDM1 (36, 37). These genes do not show significant or consistent upregulation in both DLBCL and prostate cancer cells upon either GSK126 treatment or EZH2 knockdown (Supplemental Fig. 8). We found similar correlation between the efficacy of GSK126 and gene expression changes in prostate cancer cells. GSK126 displayed relatively minor effects in LNCaP cells, but dramatically retarded the androgen-independent growth of abl (Supplemental Fig. 9). Interestingly, the genes that are co-activated by EZH2 and E2F1 were significantly downregulated by GSK126 in abl cells, and their levels were barely changed in LNCaP (Supplemental Fig. 10). Taken together, these results suggest that the activity of EZH2-E2F1 complex may determine the cancer cell response to the pharmacological inhibition of EZH2.
To further confirm the conclusion in clinical scenarios, we computed the gene expression correlation in the dataset from DLBCL patient samples (38). Both EZH2 and E2F1 are significantly correlated with the signature genes in clinical samples (Fig. 6C and D). The close correlation of EZH2 and E2F1 with their coactivated genes in clinical cases as well as with the cell responsiveness to EZH2 inhibitors support the ideas that the regulatory network between EZH2 and E2F1 plays an important role in determining the cancer-specific transcriptional profiles, and that activation of this signaling may contribute to the cellular behavior in response to EZH2-targeting drugs for cancer treatment.
DISCUSSION
In this study, we demonstrated that EZH2 and E2F1 form a transcriptional complex co-localizing at specific chromatin sites to activate the downstream target genes. By integrative analysis using the GS2A pipeline, we found E2F1 as the top candidate that significantly correlates with the expression levels of EZH2-activated signature genes, and E2F1 co-localizes with EZH2 at specific chromatin sites in cancer cells. Most importantly, our discoveries from the cell models fit well with the sceneries in clinical tumors, and the findings provide clinical implications for EZH2-targeting inhibitors as anticancer therapy.
As a strong regulator of cell growth, the Rb/E2F pathway is commonly deregulated in many types of human cancers, such as Rb loss and E2F1 amplification/overexpression. Although being originally recognized to induce apoptosis after DNA damage, E2F1 is frequently upregulated in high-grade tumors and the increased level is closely associated with poor patient outcomes (39). The downstream target genes of E2F1, in particular those involved in cell cycle progression, are abnormally overexpressed in cancers. Although regulation of E2F1 activity by EZH2 has been indicated in human cancers (40, 41), our findings provided a novel insight into the mechanisms of how perturbations of the Rb/E2F1 signaling lead to lethal tumor phenotypes through interaction with EZH2. The direct crosstalk between EZH2 and Rb/E2F1 pathway reinforce the gene expression patterns critical for cancer cell growth and thus constitutes a deadly alliance that underpins tumor progression.
Our previous work demonstrated the interplay between EZH2 and AR signaling critical for prostate tumor progression (9). Although the androgen response element (ARE) is not listed among the top transcription factor binding motifs enriched in EZH2 ChIP-seq dataset, we confirmed its significant enrichment in the proximal region of EZH2 solo peaks. It is interesting to note that the Rb/E2F1 pathway has been shown as a critical regulator of AR expression and transcriptional activity (42-44). Although the activator E2Fs are generally believed to have a great deal of functional redundancy, only E2F1 and E2F3 were shown to enhance AR activity, while E2F2 was insufficient for this function (42). Interestingly, besides E2F1, our GS2A analysis implied the potential involvement of E2F3 in the transcriptional network with EZH2 as well, but not E2F2. This result not only confirms that GS2A is a powerful and practical approach to infer the biologically important signaling, but also indicates a decisive role of EZH2-E2F-AR axis in controlling downstream genes important for cancer progression.
Several questions remain unsolved for the mechanisms underpinning this critical transcriptional network for cancer progression. First, it is unclear about the precise molecular events that link the interplay between EZH2 and E2F1 to the transcriptional activation. Non-histone substrates for EZH2, such as the transcription factors GATA4 and STAT3, have been identified (45, 46), and the modifications influence the transcriptional activities by modulating either the protein stability or other post-translational modifications critical for transactivation. Since the enzymatic activity of EZH2 is required for the gene expression programs co-regulated by E2F1, it is possible that E2F1 is another non-histone substrate of EZH2. Second, it is intriguing that de-repression of EZH2-repressed genes were not always observed in response to either EZH2 silencing or inhibitor treatment. In contrast, the genes that are directly activated by EZH2 were consistent downstream targets of the EZH2-E2F1 complex. The activities of EZH2 as either a transcriptional repressor or a transcriptional inducer have been both indicated, and the potential mechanisms of its conflicting functions may be the involvement of EZH2 in specific transcriptional complexes or pathways (47). It is therefore worth exploring the signaling/factors that determine the diverse and context-specific functions of EZH2. Third, although we showed strong correlation between MYC amplification and EZH2/E2F1 co-regulated genes, the connection of the genetic alterations with the activities of EZH2-E2F1 network is yet incomplete. It is plausible that such genetic aberration induces the transactivation of EZH2 and E2F1 through modulation of critical cis-acting elements, and therefore amplifies the cascades of the transcriptional network (48).
In summary, our work uncovered the collaboration between EZH2 and E2F1 in controlling downstream genes that are highly expressed in aggressive tumors. The mechanisms underlying EZH2-E2F1 complex for cancer progression and their therapeutic potential await future research to be better understood and utilized.
Supplementary Material
ACKNOWLEDGMENTS
The authors sincerely thank Graham McVicker, Cliff Meyer and Xiaoxing Wang for their helpful discussions.
Grant support: This work was funded by National Institutes of Health grants (GM056211 and HG4069 to X.S.L., 1K99CA178199-01A1 to K.X., K99CA172948 to H.H.H., and K99CA175290 to Y.C.), Prostate Cancer Foundation Young Investigator award (to K.X.), Dana-Farber Cancer Institute supports (to the Center of Functional Cancer Epigenetics), and National Science Foundation of China (NSFC 31329003 to X.S.L.).
Footnotes
Disclosure of potential conflicts of interest: The authors disclose no potential conflicts of interest.
REFERENCES
- 1.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nature reviews. Cancer. 2006 Nov;6:846. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 2.Chang CJ, Hung MC. The role of EZH2 in tumour progression. British journal of cancer. 2012 Jan 17;106:243. doi: 10.1038/bjc.2011.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCabe MT, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012 Dec 6;492:108. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 4.Qi W, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012 Dec 26;109:21360. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knutson SK, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nature chemical biology. 2012 Nov;8:890. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 6.Cao R, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002 Nov 1;298:1039. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 7.Muller J, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002 Oct 18;111:197. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 8.Lee ST, et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011 Sep 2;43:798. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 9.Xu K, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012 Dec 14;338:1465. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi B, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007 Jul;27:5105. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nature reviews. Genetics. 2009 Apr;10:252. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 12.Zhang HM, et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 2012 Jan;40:D144. doi: 10.1093/nar/gkr965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin B, et al. CistromeMap: a knowledgebase and web server for ChIP-Seq and DNase-Seq studies in mouse and human. Bioinformatics. 2012 May 15;28:1411. doi: 10.1093/bioinformatics/bts157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS). Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005 Oct 25;102:15545. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012 Feb 15;28:573. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009 May 1;25:1105. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012 Mar;7:562. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009 Jul 15;25:1754. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010 Sep;20:1297. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koboldt DC, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009 Sep 1;25:2283. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Culig Z, et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. British journal of cancer. 1999 Sep;81:242. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caron C, et al. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene. 2010 Sep 16;29:5171. doi: 10.1038/onc.2010.259. [DOI] [PubMed] [Google Scholar]
- 24.Willder JM, et al. Androgen receptor phosphorylation at serine 515 by Cdk1 predicts biochemical relapse in prostate cancer patients. British journal of cancer. 2013 Jan 15;108:139. doi: 10.1038/bjc.2012.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aytes A, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014 May 12;25:638. doi: 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010 Jul 13;18:11. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.N. Cancer Genome Atlas Research Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 Oct 23;455:1061. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012 Sep 6;489:57. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Y, Croxton R, Moorer RL, Jr., Cress WD. Identification of novel E2F1-regulated genes by microarray. Archives of biochemistry and biophysics. 2002 Mar 15;399:212. doi: 10.1006/abbi.2002.2761. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, et al. Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Mol Cell. 2015 Jan 22;57:304. doi: 10.1016/j.molcel.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hock H. A complex Polycomb issue: the two faces of EZH2 in cancer. Genes Dev. 2012 Apr 15;26:751. doi: 10.1101/gad.191163.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bachmann N, et al. Mutation screen and association study of EZH2 as a susceptibility gene for aggressive prostate cancer. The Prostate. 2005 Nov 1;65:252. doi: 10.1002/pros.20296. [DOI] [PubMed] [Google Scholar]
- 33.Bracken AP, et al. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. The EMBO journal. 2003 Oct 15;22:5323. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sander S, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008 Nov 15;112:4202. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 35.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005 Jun 9;435:839. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 36.Beguelin W, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013 May 13;23:677. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caganova M, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. The Journal of clinical investigation. 2013 Dec;123:5009. doi: 10.1172/JCI70626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monti S, et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell. 2012 Sep 11;22:359. doi: 10.1016/j.ccr.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engelmann D, Putzer BM. The dark side of E2F1: in transit beyond apoptosis. Cancer Res. 2012 Feb 1;72:571. doi: 10.1158/0008-5472.CAN-11-2575. [DOI] [PubMed] [Google Scholar]
- 40.Gao SB, et al. EZH2 represses target genes through H3K27-dependent and H3K27-independent mechanisms in hepatocellular carcinoma. Molecular cancer research : MCR. 2014 Oct;12:1388. doi: 10.1158/1541-7786.MCR-14-0034. [DOI] [PubMed] [Google Scholar]
- 41.Wu ZL, et al. Polycomb protein EZH2 regulates E2F1-dependent apoptosis through epigenetically modulating Bim expression. Cell death and differentiation. 2010 May;17:801. doi: 10.1038/cdd.2009.162. [DOI] [PubMed] [Google Scholar]
- 42.Sharma A, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. The Journal of clinical investigation. 2010 Dec;120:4478. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis JN, et al. Elevated E2F1 inhibits transcription of the androgen receptor in metastatic hormone-resistant prostate cancer. Cancer Res. 2006 Dec 15;66:11897. doi: 10.1158/0008-5472.CAN-06-2497. [DOI] [PubMed] [Google Scholar]
- 44.Mallik I, Davila M, Tapia T, Schanen B, Chakrabarti R. Androgen regulates Cdc6 transcription through interactions between androgen receptor and E2F transcription factor in prostate cancer cells. Biochimica et biophysica acta. 2008 Oct;1783:1737. doi: 10.1016/j.bbamcr.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 45.Kim E, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013 Jun 10;23:839. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He A, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012 Jan 1;26:37. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deb G, Singh AK, Gupta S. EZH2: not EZHY (easy) to deal. Molecular cancer research : MCR. 2014 May;12:639. doi: 10.1158/1541-7786.MCR-13-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013 Apr 11;153:307. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.