

Abstract
Background
Sequencing of both healthy and disease singletons yields many novel and low frequency variants of uncertain significance (VUS). Complete gene and genome sequencing by next generation sequencing (NGS) significantly increases the number of VUS detected. While prior studies have emphasized protein coding variants, non-coding sequence variants have also been proven to significantly contribute to high penetrance disorders, such as hereditary breast and ovarian cancer (HBOC). We present a strategy for analyzing different functional classes of non-coding variants based on information theory (IT) and prioritizing patients with large intragenic deletions.
Methods
We captured and enriched for coding and non-coding variants in genes known to harbor mutations that increase HBOC risk. Custom oligonucleotide baits spanning the complete coding, non-coding, and intergenic regions 10 kb up- and downstream of ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, and TP53 were synthesized for solution hybridization enrichment. Unique and divergent repetitive sequences were sequenced in 102 high-risk, anonymized patients without identified mutations in BRCA1/2. Aside from protein coding and copy number changes, IT-based sequence analysis was used to identify and prioritize pathogenic non-coding variants that occurred within sequence elements predicted to be recognized by proteins or protein complexes involved in mRNA splicing, transcription, and untranslated region (UTR) binding and structure. This approach was supplemented by in silico and laboratory analysis of UTR structure.
Results
15,311 unique variants were identified, of which 245 occurred in coding regions. With the unified IT-framework, 132 variants were identified and 87 functionally significant VUS were further prioritized. An intragenic 32.1 kb interval in BRCA2 that was likely hemizygous was detected in one patient. We also identified 4 stop-gain variants and 3 reading-frame altering exonic insertions/deletions (indels).
Conclusions
We have presented a strategy for complete gene sequence analysis followed by a unified framework for interpreting non-coding variants that may affect gene expression. This approach distills large numbers of variants detected by NGS to a limited set of variants prioritized as potential deleterious changes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12920-016-0178-5) contains supplementary material, which is available to authorized users.
Keywords: Information theory, Hereditary breast and ovarian cancer, Transcription factor binding, RNA-binding protein, Prioritization, Variants of uncertain significance, Splicing, Non-coding, Next-generation sequencing
Background
Advances in NGS have enabled panels of genes, whole exomes, and even whole genomes to be sequenced for multiple individuals in parallel. These platforms have become so cost-effective and accurate that they are beginning to be adopted in clinical settings, as evidenced by recent FDA approvals [1, 2]. However, the overwhelming number of gene variants revealed in each individual has challenged interpretation of clinically significant genetic variation [3–5].
After common variants, which are rarely pathogenic, are eliminated, the number of VUS in the residual set remains substantial. Assessment of pathogenicity is not trivial, considering that nearly half of the unique variants are novel, and cannot be resolved using published literature and variant databases [6]. Furthermore, loss-of-function variants (those resulting in protein truncation are most likely to be deleterious) represent a very small proportion of identified variants. The remaining variants are missense and synonymous variants in the exon, single nucleotide changes, or in frame insertions or deletions in intervening and intergenic regions. Functional analysis of large numbers of these variants often cannot be performed, due to lack of relevant tissues, and the cost, time, and labor required for each variant. Another problem is that in silico protein coding prediction tools exhibit inconsistent accuracy and are thus problematic for clinical risk evaluation [7–9]. Consequently, many HBOC patients undergoing genetic susceptibility testing will receive either an inconclusive (no BRCA variant identified) or an uncertain (BRCA VUS) result. The former has been reported in up to 80 % of cases and depends on the number of genes tested [10]. The occurrence of uncertain BRCA mutations varies greatly (as high as 46 % in African American populations and as low as 2.1 %) among tested individuals depending on the laboratory and the patient’s ethnicity [11–13]. The inconsistency in diagnostic yield is significant, considering that HBOC accounts for 5–10 % of all breast/ovarian cancer [14, 15].
One strategy to improve variant interpretation in patients is to reduce the full set of variants to a manageable list of potentially pathogenic variants. Evidence for pathogenicity of VUS in genetic disease is often limited to amino acid coding changes [16, 17], and mutations affecting splicing, transcriptional activation, and mRNA stability tend to be underreported [18–24]. Splicing errors are estimated to represent 15 % of disease-causing mutations [25], but may be much higher [26, 27]. The impact of a single nucleotide change in a recognition sequence can range from insignificant to complete abolition of a protein binding site. Aberrant splicing events causing frameshifts often disrupt protein function; in-frame changes are dependent on gene context. The complexity of interpretation of non-coding sequence variants benefits from computational approaches [28] and direct functional analyses [29–33] that may each support evidence of pathogenicity.
Ex vivo transfection assays developed to determine the pathogenicity of VUS predicted to lead to splicing aberrations (using in silico tools) have been successful in identifying pathogenic sequence variants [34, 35]. IT-based analysis of splicing variants has proven to be robust and accurate (as determined by functional assays for mRNA expression or binding assays) at analyzing splice site (SS) variants, including splicing regulatory factor binding sites (SRFBSs), and in distinguishing them from polymorphisms in both rare and common diseases [36–39]. However, IT can be applied to any sequence recognized and bound by another factor [40], such as with transcription factor binding sites (TFBSs) and RNA-binding protein binding sites (RBBSs). IT is used as a measure of sequence conservation and is more accurate than consensus sequences [41]. The individual information (Ri) of a base is related to thermodynamic entropy, and therefore free energy of binding, and is measured on a logarithmic scale (in bits). By comparing the change in information (ΔRi) for a nucleotide variation of a bound sequence, the resulting change in binding affinity is ≥ 2ΔRi, such that a 1 bit change in information will result in at least a 2-fold change in binding affinity [42].
IT measures nucleotide sequence conservation and does not provide information on effects of variants on mRNA secondary (2°) structure, nor can it accurately predict effects of amino acid sequence changes. Associations of structural changes in untranslated regions (UTR) of mRNA with disease justifies including predicted effects of these changes on 2° structure in the comprehensive analysis of sequence variants [43]. Other in silico methods have attempted to address these deficiencies. For example, Halvorsen et al. (2010) introduced an algorithm called SNPfold, which computes the potential effect of a single nucleotide variant (SNV) on mRNA 2° structure [20]. Predictions made by SNPfold can be tested by the SHAPE assay (Selective 2’-Hydroxyl Acylation analyzed by Primer Extension) [44], which provides evidence for sequence variants that lead to structural changes in mRNA by detection of covalent adducts in mRNA.
The implications of improved VUS interpretation are particularly relevant for HBOC due to its incidence and the adoption of panel testing for these individuals [45, 46]. It has been suggested that patients with a high risk profile receiving uninformative results would imply that deleterious variants lie in untested regions of BRCA1/2, untested genes, or are unrecognized [47, 48]. This is also supported by studies where families with linkage to BRCA1/2 had no detectable pathogenic mutation (however it is noteworthy that detection rates of BRCA mutations in families with documented linkage to these loci appears to vary by ascertainment, inclusion criteria, and technology used to identify the mutations) [49, 50]. The concept of non-BRCA gene association has been demonstrated by the identification of low-to-moderate risk HBOC genes, and variants within coding and non-coding regions affecting splicing and regulatory factor binding [51, 52]. Consequently, VUS, which include rare missense changes, other coding and non-coding changes in all of these genes, greatly outnumber the catalog of known deleterious mutations [53].
Here, we develop and evaluate IT-based models to predict potential non-coding sequence mutations in SSs, TFBSs, and RBBSs in 7 genes sequenced in their entirety. These models were used to analyze 102 anonymous HBOC patients who did not exhibit known BRCA1/2 coding mutations at the time of initial testing, despite meeting the criteria for BRCA genetic testing. The genes are: ATM, BRCA1, BRCA2, CDH1, CHEK2, PALB2, and TP53, and have been reported to harbor mutations that increase HBOC risk [54–76]. We apply these IT-based methods to analyze variants in the complete sequences of coding, non-coding, and up- and downstream regions of the 7 genes. In this study, we established and applied a unified IT-based framework, first filtering out common variants, then to “flag” potentially deleterious ones. Then, using context-specific criteria and information from the published literature, we prioritized likely candidates.
Methods
Design of tiled capture array for HBOC gene panel
Nucleic acid hybridization capture reagents designed from genomic sequences generally avoid repetitive sequence content to avoid cross hybridization [77]. Complete gene sequences harbor numerous repetitive sequences, and an excess of denatured C0t-1 DNA is usually added to hybridization to prevent inclusion of these sequences [78]. RepeatMasker software completely masks all repetitive and low-complexity sequences [79]. We increased sequence coverage in complete genes with capture probes by enriching for both single-copy and divergent repeat (>30 % divergence) regions, such that, under the correct hybridization and wash conditions, all probes hybridize only to their correct genomic locations [77]. This step was incorporated into a modified version of Gnirke and colleagues’ (2009) in-solution hybridization enrichment protocol, in which the majority of library preparation, pull-down, and wash steps were automated using a BioMek® FXP Automation Workstation (Beckman Coulter, Mississauga, Canada) [80].
Genes ATM (RefSeq: NM_000051.3, NP_000042.3), BRCA1 (RefSeq: NM_007294.3, NP_009225.1), BRCA2 (RefSeq: NM_000059.3, NP_000050.2), CDH1 (RefSeq: NM_004360.3, NP_004351.1), CHEK2 (RefSeq: NM_145862.2, NP_665861.1), PALB2 (RefSeq: NM_024675.3, NP_078951.2), and TP53 (RefSeq: NM_000546.5, NP_000537.3) were selected for capture probe design by targeting single copy or highly divergent repeat regions (spanning 10 kb up- and downstream of each gene relative to the most upstream first exon and most downstream final exon in RefSeq) using an ab initio approach [77]. If a region was excluded by ab initio but lacked a conserved repeat element (i.e. divergence > 30 %) [79], the region was added back into the probe-design sequence file. Probe sequences were selected using PICKY 2.2 software [81]. These probes were used in solution hybridization to capture our target sequences, followed by NGS on an Illumina Genome Analyzer IIx (Additional file 1: Methods).
Genomic sequences from both strands were captured using overlapping oligonucleotide sequence designs covering 342,075 nt among the 7 genes (Fig. 1). In total, 11,841 oligonucleotides were synthesized from the transcribed strand consisting of the complete, single copy coding, and flanking regions of ATM (3513), BRCA1 (1587), BRCA2 (2386), CDH1 (1867), CHEK2 (889), PALB2 (811), and TP53 (788). Additionally, 11,828 antisense strand oligos were synthesized (3497 ATM, 1591 BRCA1, 2395 BRCA2, 1860 CDH1, 883 CHEK2, 826 PALB2, and 776 TP53). Any intronic or intergenic regions without probe coverage are most likely due to the presence of conserved repetitive elements or other paralogous sequences.
Fig. 1.
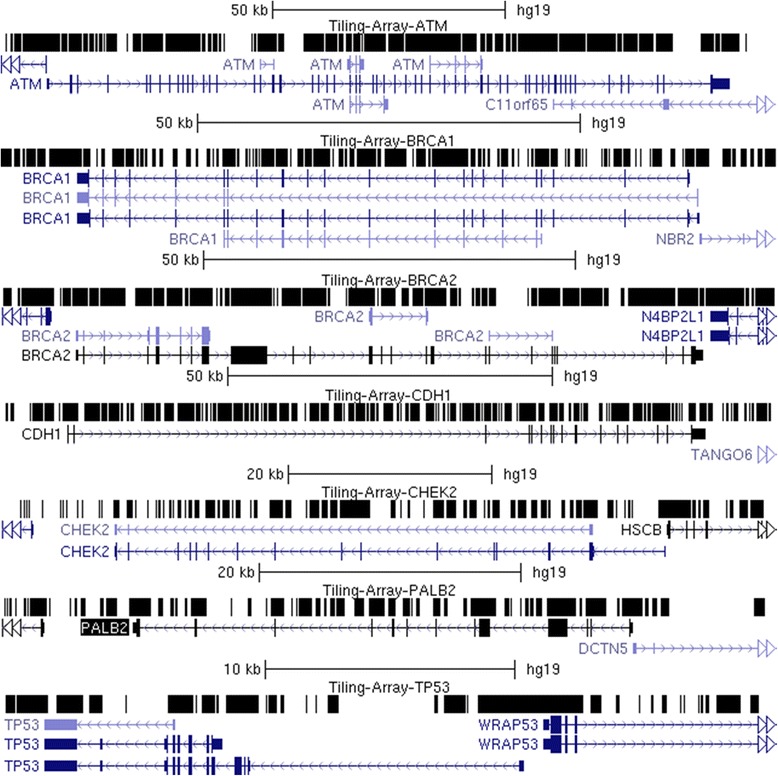
Capture Probe Coverage over Sequenced Genes. The genomic structure of the 7 genes chosen are displayed with the UCSC Genome Browser. Top row for each gene is a custom track with the “dense” visualization modality selected with black regions indicating the intervals covered by the oligonucleotide capture reagent. Regions without probe coverage contain conserved repetitive sequences or correspond to paralogous sequences that are unsuitable for probe design
For regions lacking probe coverage (of ≥ 10 nt, N = 141; 8 in ATM, 26 in BRCA1, 10 in BRCA2, 29 in CDH1, 36 in CHEK2, 15 in PALB2, and 17 in TP53), probes were selected based on predicted Tms similar to other probes, limited alignment to other sequences in the transcriptome (<10 times), and avoidance of stable, base-paired 2° structures (with unaFOLD) [82, 83]. The average coverage of these sequenced regions was 14.1–24.9 % lower than other probe sets, indicating that capture was less efficient, though still successful.
HBOC samples for oligo capture and high-throughput sequencing
Genomic DNA from 102 patients previously tested for inherited breast/ovarian cancer without evidence of a predisposing genetic mutation, was obtained from the Molecular Genetics Laboratory (MGL) at the London Health Sciences Centre in London, Ontario, Canada. Patients qualified for genetic susceptibility testing as determined by the Ontario Ministry of Health and Long-Term Care BRCA1 and BRCA2 genetic testing criteria [84] (see Additional file 2). The University of Western Ontario research ethics board (REB) approved this anonymized study of these individuals to evaluate the analytical methods presented here. BRCA1 and BRCA2 were previously analyzed by Protein Truncation Test (PTT) and Multiplex Ligation-dependent Probe Amplification (MLPA). The exons of several patients (N = 14) had also been Sanger sequenced. No pathogenic sequence change was found in any of these individuals. In addition, one patient with a known pathogenic BRCA variant was re-sequenced by NGS as a positive control.
Sequence alignment and variant calling
Variant analysis involved the steps of detection, filtering, IT-based and coding sequence analysis, and prioritization (Fig. 2). Sequencing data were demultiplexed and aligned to the specific chromosomes of our sequenced genes (hg19) using both CASAVA (Consensus Assessment of Sequencing and Variation; v1.8.2) [85] and CRAC (Complex Reads Analysis and Classification; v1.3.0) [86] software. Alignments were prepared for variant calling using Picard [87] and variant calling was performed on both versions of the aligned sequences using the UnifiedGenotyper tool in the Genome Analysis Toolkit (GATK) [88]. We used the recommended minimum phred base quality score of 30, and results were exported in variant call format (VCF; v4.1). A software program was developed to exclude variants called outside of targeted capture regions and those with quality scores < 50. Variants flagged by bioinformatic analysis (described below) were also assessed by manually inspecting the reads in the region using the Integrative Genomics Viewer (IGV; version 2.3) [89, 90] to note and eliminate obvious false positives (i.e. variant called due to polyhomonucleotide run dephasing, or PCR duplicates that were not eliminated by Picard). Finally, common variants (≥1 % allele frequency based on dbSNP 142 or > 5 individuals in our study cohort) were not prioritized.
Fig. 2.
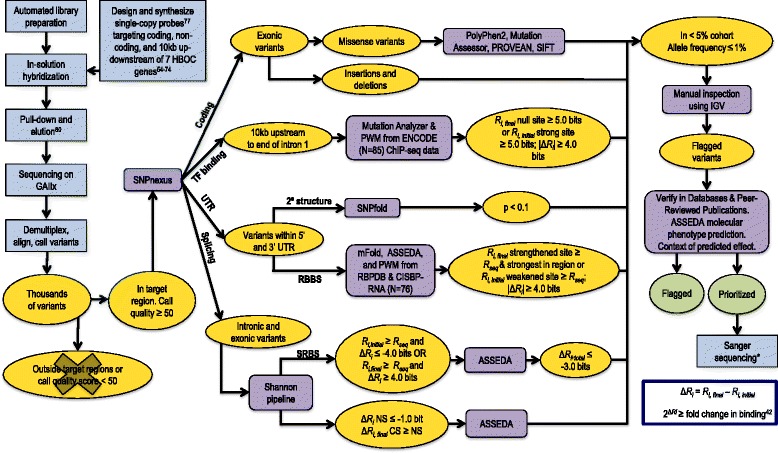
Framework for the Identification of Potentially Pathogenic Variants. Integrated laboratory processing and bioinformatic analysis procedures for comprehensive complete gene variant determination and analysis. Intermediate datasets resulting from filtering are represented in yellow and final datasets in green. Non-bioinformatic steps, such as sample preparation are represented in blue and prediction programs in purple. Sequencing analysis yields base calls for all samples. CASAVA [85] and CRAC [86] were used to align these sequencing results to hg19. GATK [88] was used to call variants from this data against GRCh37 release of the reference human genome. Variants with a quality score < 50 and/or call confidence score < 30 were eliminated along with variants falling outside of our target regions. SNPnexus [112–114] was used to identify the genomic location of the variants. Nonsense and indels were noted and prediction tools were used to assess the potential pathogenicity of missense variants. The Shannon Pipeline [91] evaluated the effect of a variant on natural and cryptic SSs, as well as SRFBSs. ASSEDA [38] was used to predict the potential isoforms as a result of these variants. PWMs for 83 TFs were built using an information weight matrix generator based on Bipad [106]. Mutation Analyzer evaluated the effect of variants found 10 kb upstream up to the first intron on protein binding. Bit thresholds (R i values) for filtering variants on software program outputs are indicated. Variants falling within the UTR sequences were assessed using SNPfold [20], and the most probable variants that alter mRNA structure (p < 0.1) were then processed using mFold to predict the effect on stability [83]. All UTR variants were scanned with a modified version of the Shannon Pipeline, which uses PWMs computed from nucleotide frequencies for 28 RBPs in RBPDB [109] and 76 RBPs in CISBP-RNA [110]. All variants meeting these filtering criteria were verified with IGV [89, 90]. *Sanger sequencing was only performed for protein truncating, splicing, and selected missense variants
IT-based variant analysis
All variants were analyzed using the Shannon Human Splicing Mutation Pipeline, a genome-scale variant analysis program that predicts the effects of variants on mRNA splicing [91, 92]. Variants were flagged based on criteria reported in Shirley et al. (2013): weakened natural site ≥ 1.0 bits, or strengthened cryptic site (within 300 nt of the nearest exon) where cryptic site strength is equivalent or greater than the nearest natural site of the same phase [91]. The effects of flagged variants were further analyzed in detail using the Automated Splice Site and Exon Definition Analysis (ASSEDA) server [38].
Exonic variants and those found within 500 nt of an exon were assessed for their effects, if any, on SRFBSs [38]. Sequence logos for splicing regulatory factors (SRFs) (SRSF1, SRSF2, SRSF5, SRSF6, hnRNPH, hnRNPA1, ELAVL1, TIA1, and PTB) and their Rsequence values (the mean information content [93]) are provided in Caminsky et al. (2015) [36]. Because these motifs occur frequently in unspliced transcripts, only variants with large information changes were flagged, notably those with a) ≥ 4.0 bit decrease, i.e. at least a 16-fold reduction in binding site affinity, with Ri,initial ≥ Rsequence for the particular factor analyzed, or b) ≥ 4.0 bit increase in a site where Ri,final ≥ 0 bits. ASSEDA was used to calculate Ri,total, with the option selected to include the given SRF in the calculation. Variants decreasing Ri,total by < 3.0 bits (i.e. 8-fold) were predicted to potentially have benign effects on expression, and were not considered further.
Activation of pseudoexons through creating/strengthening of an intronic cryptic SS was also assessed [94]. Changes in intronic cryptic sites, where ΔRi > 1 bit and Ri,final ≥ (Rsequence – 1 standard deviation [S.D.] of Rsequence), were identified. A pseudoexon was predicted if a pre-existing cryptic site of opposite polarity (with Ri > [Rsequence - 1 S.D.]) and in the proper orientation for formation of exons between 10–250 nt in length was present. In addition, the minimum intronic distance between the pseudoexon and either adjacent natural exon was 100 nt. The acceptor site of the pseudoexon was also required to have a strong hnRNPA1 site located within 10 nt (Ri ≥ Rsequence) [38] to ensure accurate proofreading of the exon [37].
Next, variants affecting the strength of SRFs were analyzed by a contextual exon definition analysis of ΔRi,total. The context refers to the documented splicing activity of an SRF. For example, TIA1 has been shown to be an intronic enhancer of exon definition, so only intronic sites were considered. Similarly, hnRNPA1 proofreads the 3’ SS (acceptor) and inhibits exon recognition elsewhere [95]. Variants that lead to redundant SRFBS changes (i.e. one site is abolished and another proximate site [≤2 nt] of equivalent strength is activated) were assumed to have a neutral effect on splicing. If the strength of a site bound by PTB (polypyrimidine tract binding protein) was affected, its impact on binding by other factors was analyzed, as PTB impedes binding of other factors with overlapping recognition sites, but does not directly enhance or inhibit splicing itself [96].
To determine effects of variants on transcription factor (TF) binding, we first established which TFs bound to the sequenced regions of the gene promoters (and first exons) in this study by using ChIP-seq data from 125 cell types (Additional file 1: Methods) [97]. We identified 141 TFs with evidence for binding to the promoters of the genes we sequenced, including c-Myc, C/EBPβ, and Sp1, shown to transcriptionally regulate BRCA1, TP53, and ATM, respectively [98–100]. Furthermore, polymorphisms in TCF7L2, known to bind enhancer regions of a wide variety of genes in a tissue-specific manner [101], have been shown to increase risk of sporadic [102] and hereditary breast [103], as well as other types of cancer [104, 105].
IT-based models of the 141 TFs of interest were derived by entropy minimization of the DNase accessible ChIP-seq subsets [106]. Details are provided in Lu R, Mucaki E, and Rogan PK (BioRxiv; http://dx.doi.org/10.1101/042853). While some data sets would only yield noise or co-factor motifs (i.e. co-factors that bind via tethering, or histone modifying proteins [107]), techniques such as motif masking and increasing the number of Monte Carlo cycles yielded models for 83 TFs resembling each factor’s published motif. Additional file 3: Table S1 contains the final list of TFs and the models we built (described below) [108].
These TFBS models (N = 83) were used to scan all variants called in the promoter regions (10 kb upstream of transcriptional start site to the end of IVS1) of HBOC genes for changes in Ri. Binding site changes that weaken interactions with the corresponding TF (to Ri ≤ Rsequence) are likely to affect regulation of the adjacent target gene. Stringent criteria were used to prioritize the most likely variants and thus only changes to strong TFBSs (Ri,initial ≥ Rsequence), where reduction in strength was significant (ΔRi ≥ 4.0 bits), were considered. Alternatively, novel or strengthened TFBSs were also considered sources of dysregulated transcription. These sites were defined as having Ri,final ≥ Rsequence and as being the strongest predicted site in the corresponding genomic interval (i.e. exceeding the Ri values of adjacent sites unaltered by the variant). Variants were not prioritized if the TF was known to a) enhance transcription and IT analysis predicted stronger binding, or b) repress transcription and IT analysis predicted weaker binding.
Two complementary strategies were used to assess the possible impact of variants within UTRs. First, SNPfold software was used to assess the effect of a variant on 2° structure of the UTR (Additional file 1: Methods) [20]. Variants flagged by SNPfold with the highest probability of altering stable 2° structures in mRNA (where p-value < 0.1) were prioritized. To evaluate these predictions, oligonucleotides containing complete wild-type and variant UTR sequences (Additional file 4: Table S2) were transcribed in vitro and followed by SHAPE analysis, a method that can confirm structural changes in mRNA [44].
Second, the effects of variants on the strength of RBBSs were predicted. Frequency-based, position weight matrices (PWMs) for 156 RNA-binding proteins (RBPs) were obtained from the RNA-Binding Protein DataBase (RBPDB) [109] and the Catalog of Inferred Sequence Binding Preferences of RNA binding proteins (CISBP-RNA) [110, 111]. These were used to compute information weight matrices (based on the method described by Schneider et al. 1984; N = 147) (see Additional file 1: Methods) [40]. All UTR variants were assessed using a modified version of the Shannon Pipeline [91] containing the RBPDB and CISBP-RNA models. Results were filtered to include a) variants with |ΔRi| ≥ 4.0 bits, b) variants creating or strengthening sites (Ri,final ≥ Rsequence and the Ri,initial < Rsequence), and c) RBBSs not overlapping or occurring within 10 nt of a stronger, pre-existing site of another RBP.
Exonic protein-altering variant analysis
The predicted effects of all coding variants were assessed with SNPnexus [112–114], an annotation tool that can be applied to known and novel variants using up-to-date dbSNP and UCSC human genome annotations. Variants predicted to cause premature protein truncation were given higher priority than those resulting in missense (or synonymous) coding changes. Missense variants were first cross referenced with dbSNP 142 [115]. Population frequencies from the Exome Variant Server [116] and 1000Genomes [117] are also provided. The predicted effects on protein conservation and function of the remaining variants were evaluated by in silico tools: PolyPhen-2 [118], Mutation Assessor (release 2) [119, 120], and PROVEAN (v1.1.3) [121, 122]. Default settings were applied and in the case of PROVEAN, the “PROVEAN Human Genome Variants Tool” was used, which includes SIFT predictions as a part of its output. Variants predicted by all four programs to be benign were less likely to have a deleterious impact on protein activity; however this did not exclude them from mRNA splicing analysis (described above in IT-Based Variant Analysis). All rare and novel variants were cross-referenced with general mutation databases (ClinVar [123, 124], Human Gene Mutation Database [HGMD] [125, 126], Leiden Open Variant Database [LOVD] [127–134], Domain Mapping of Disease Mutations [DM2] [135], Expert Protein Analysis System [ExPASy] [136] and UniProt [137, 138]), and gene-specific databases (BRCA1/2: the Breast Cancer Information Core database [BIC] [139] and Evidence-based Network for the Interpretation of Germline Mutant Alleles [ENIGMA] [140]; TP53: International Agency for Research on Cancer [IARC] [141]), as well as published reports to prioritize them for further workup.
Variant classification
Flagged variants were prioritized if they were likely to encode a dysfunctional protein (indels, nonsense codon > 50 amino acids from the C-terminus, or abolition of a natural SS resulting in out-of-frame exon skipping) or if they exceeded established thresholds for fold changes in binding affinity based on IT (see Methods above). In several instances, our classification was superseded by previous functional or pedigree analyses (reported in published literature or databases) that categorized these variants as pathogenic or benign.
Positive control
We identified the BRCA1 exon 17 nonsense variant c.5136G > A (chr17:41215907C > T; rs80357418; 2-5A) [142] in the sample that was provided as a positive control. This was the same mutation identified by the MGL as pathogenic for this patient. We also prioritized another variant in this patient (Table 1) [143].
Table 1.
Prioritized variants in the positive control
| Gene | mRNA Protein | rsID (dbSNP 142) | Category | Consequence | Ref |
|---|---|---|---|---|---|
| BRCA1 | c.5136G > A | rs80357418 | Nonsense | 151 AA short | [142] |
| p.Trp1712Ter | |||||
| BRCA2 | c.3218A > G | rs80358566 | SRFBS | Repressor action of hnRNPA1 at this site abolished (5.2 to 0.4 bits). Blocking action of PTB removed as site is abolished (5.5 to -7.5 bits) and may uncover binding sites of other SRFs. | |
| p.Gln1073Arg | |||||
| Missense | Listed in ClinVar as conflicting interpretations (likely benign, unknown) and in BIC as unknown clinical importance. 2 in silico programs called deleterious. The variant occurs between repeat motifs BRC1 and BRC2 of BRCA2, a region in which pathogenic missense mutations have not yet been identified. | [143] | |||
| SRFBS | Repressor action of hnRNPA1 at this site abolished (5.2 to 0.4 bits). Blocking action of PTB removed as site is abolished (5.5 to -7.5 bits) and may uncover binding sites of other SRFs. |
Variant validation
Protein-truncating, prioritized splicing, and selected prioritized missense variants were verified by Sanger sequencing. Primers of PCR amplicons are indicated in Additional file 5: Table S3.
Deletion analysis
Junctional read detection
Potential large rearrangements were detected with BreakDancer software [144], which identifies novel genomic rearrangements based on the respective orientation and distance between ends of the same read (and exceeding the lengths of NGS library inserts). This approach can, in theory, approximately localize deletions, duplications, or other types of breakpoints within exons, introns, and regulatory regions (eg. promoters) that could affect gene expression and function. We required at least 4 reads per suspected rearrangement in a sample separated by >700 nt, with each end mapping to proximate genomic reference coordinates to infer a potential deletion. Synthetic and cost limitations in the maximum genomic real estate covered by the capture reagent led to a tradeoff between extending the span of captured genomic intervals and higher tiling densities over shorter sequences, ie. exons, to achieve the level of coverage to reliably detect deletions based on read counts alone.
Prioritization based on potential hemizygosity
Our complete gene enrichment strategy with independent capture of both genomic strands enabled and facilitated development of a new algorithm to identify potential hemizygous genomic intervals in these individuals. In each subject, we first searched for contiguous long stretches (usually > > 1 kb) of non-polymorphic segments with diminished repetitive element content (<10 %), which is consistent with the possibility of these regions harboring a deletion. Then, we determined the likelihood of homo- or hemizygosity by comparing the degree of heterozygosity of variants in each of these intervals in for an individual with all of the other individuals sequenced with this protocol in this population. Regions containing haplotype blocks in strong linkage disequilibrium (LD; from HapMap [145]) were then excluded as candidate deletion intervals. Some individuals without a deletion are expected to be non-polymorphic, because detection of heterozygosity depends on genomic length of the region, marker informativeness, and the level of LD for those markers. We required that > 80 % of the control individuals be heterozyogous for at least two well-distributed loci within these intervals. Highly informative SNPs with a random genomic distribution in the controls (and other public databases) and which were non-polymorphic in the individual with the suspected deletion were weighted more heavily in inferring potential hemizygosity. This analysis was implemented using a Perl script that identified the most likely intervals of hemizygosity, which were then crossreferenced with the corresponding genomic intervals in HapMap.
Results
Capture, sequencing, and alignment
The average coverage of capture region per individual was 90.8x (range of 53.8 to 118.2x between 32 samples) with 98.8 % of the probe-covered nucleotides having ≥ 10 reads. Samples with fewer than 10 reads per nucleotide were re-sequenced and the results of both runs were combined. The combined coverage of these samples was, on average, 48.2x (±36.2).
The consistency of both library preparation and capture protocols was improved from initial runs, which significantly impacted sequence coverage (Additional file 1: Methods). Of the 102 patients tested, 14 had been previously Sanger sequenced for BRCA1 and BRCA2 exons. Confirmation of previously discovered SNVs served to assess the methodological improvements introduced during NGS and ultimately, to increase confidence in variant calling. Initially, only 15 of 49 SNVs in 3 samples were detected. The detection rate of SNVs was improved to 100 % as the protocol progressed. All known SNVs (N = 157) were called in subsequent sequencing runs where purification steps were replaced with solid phase reversible immobilization beads and where RNA bait was transcribed the same day as capture. To minimize false positive variant calls, sequence read data were aligned with CASAVA and CRAC, variants were called for each alignment with GATK, and discrepancies were then resolved by manual review.
GATK called 14,164 unique SNVs and 1147 indels. Only 3777 (15.3 %) SNVs were present in both CASAVA and CRAC-alignments for at least one patient, and even fewer indel calls were concordant between both methods (N = 110; 6.2 %). For all other SNVs and indels, CASAVA called 6871 and 1566, respectively, whereas CRAC called 13,958 and 110, respectively. Some variants were counted more than once if they were called by different alignment programs in two or more patients. Intronic and intergenic variants proximate to low complexity sequences tend to generate false positive variants due to ambiguous alignment, a well known technical issue in short read sequence analysis [146, 147], contributing to this discrepancy. For example, CRAC correctly called a 19 nt deletion of BRCA1 (rs80359876; also confirmed by Sanger sequencing) but CASAVA flagged the deleted segment as a series of false-positives (Additional file 6: Figure S1). For these reasons, all variants were manually reviewed.
IT-based variant identification and prioritization
Natural SS variants
The Shannon Pipeline reported 99 unique variants in natural donor or acceptor SSs. After technical and frequency filtering criteria were applied, 12 variants remained (Additional file 7: Table S4). IT analysis allowed for the prioritization of 3 variants, summarized in Table 2.
Table 2.
Variants prioritized by IT analysis
| Patient ID | Gene | mRNA | rsID (dbSNP 142) | Information Change | Consequencef or Binding Factor Affected | ||
|---|---|---|---|---|---|---|---|
| R i,initial | R i,final | ΔR i or R i e | |||||
| Allele Frequency (%)d | (bits) | (bits) | (bits) | ||||
| Abolished Natural SS | |||||||
| 7-4 F | ATM | c.3747-1G > Aa | Novel | 11.0 | 0.1 | −10.9 | Exon skipping and use of alternative splice forms |
| 4-1 F | ATM | c.6347 + 1G > Tb | Novel | 10.4 | −8.3 | −18.6 | Exon skipping |
| Leaky Natural SS | |||||||
| 4-2B | CHEK2 | c.320-5 T > Aa | rs121908700 | 6.8 | 4.1 | −2.7 | Leaky splicing with intron inclusion |
| 0.08 | |||||||
| Activated Cryptic SS | |||||||
| 7-3E | BRCA1 | c.548-293G > A | rs117281398 | −12.1 | 2.6 | 14.7 | Cryptic site not expected to be used. Total information for natural exon is stronger than cryptic exon. |
| 0.74 | |||||||
| 7-4A | BRCA2 | c.7618-269_7618-260del10 | Novel | 3.9 | 9.4 | 5.5 | Cryptic site not expected to be used. Total information for natural exon is stronger than cryptic exon. |
| Pseudoexon formation due to activated acceptor SS | |||||||
| 7-3 F | BRCA2 | c.8332-805G > A | Novel | −9.3 | 5.4 | 5.6e | 6065/211/592f |
| 7-3D | CDH1 | c.164-2023A > G | rs184740925 | −6.6 | 4.3 | 6.5e | 61,236/224/1798f |
| 0.3 | |||||||
| 5-3H | CDH1 | c.2296-174 T > A | rs565488866 | 7.3 | 8.5 | 5.0e | 1175/50/124f |
| 0.02 | |||||||
| Pseudoexon formation due to activated donor SS | |||||||
| 3-6A | BRCA1 | c.212 + 253G > A | rs189352191 | 4.1 | 6.7 | 5.2e | 186/63/1250f |
| 0.08 | |||||||
| 5-2G | BRCA2 | c.7007 + 2691G > A | rs367890577 | 4.7 | 7.2 | 7.7e | 2589/103/5272f |
| 0.02 | |||||||
| Affected TFBSs | |||||||
| 7-4B | BRCA1 | c.-8895G > A | Novel | 10.9 | −0.2 | −11.1 | GATA-3 (GATA3) |
| 5-3E | CDH1 | c.-54G > C | rs5030874 | 1.7 | 12.0 | 10.4 | E2F-4 (E2F4) |
| 7-4E | 0.16 | ||||||
| 5-2B | PALB2 | c.-291C > G | rs552824227 | 12.1 | −1.3 | −13.4 | GABPα (GABPA) |
| 0.1 | |||||||
| 7-2 F | TP53 | c.-28-3132 T > C | rs17882863 | −6.3 | 10.9 | 17.2 | RUNX3 (RUNX3) |
| 0.3 | |||||||
| 4-1A | TP53 | c.-28-1102 T > C | rs113451673 | 5.1 | 12.3 | 7.2 | E2F-4 (E2F4) |
| 0.4 | 8.0 | 12.9 | 4.8 | Sp1 (SP1) | |||
| Affected RBBSs | |||||||
| 7-4G | ATM | c.-244 T > A | rs539948218 | 9.8 | −19.9 | −29.7 | RBFOX |
| c.-744 T > A | 0.04 | ||||||
| c.-1929 T > A | |||||||
| c.-3515 T > A | |||||||
| 5-3C | CDH1 | c.*424 T > A | Novel | −20.3 | 9.6 | 29.9 | SF3B4 |
| 8.2 | 1.8 | −6.4 | CELF4 | ||||
| 7-2E | CHEK2 | c.-588G > A | rs141568342 | 10.9 | 3.7 | −7.2 | BX511012.1 |
| 4-3C.5-4G | CHEK2 | c.-345C > Tc | rs137853007 | 3.3 | 11.4 | 8.2 | SF3B4 |
| 3-1A | TP53 | c.-107 T > C | rs113530090 | 10.5 | 4.5 | −6.0 | ELAVL1 |
| 4-1H | c.-188 T > C | 0.72 | |||||
| 4-2H | TP53 | c.*1175A > C | rs78378222 | 10.7 | 4.1 | −6.6 | KHDRBS1 |
| 7-2 F | c.*1376A > C | 0.26 | |||||
| c.*1464A > C | |||||||
aConfirmed by Sanger sequencing
bAmbiguous Sanger sequencing results
cPrioritized under missense change and was therefore verified with Sanger sequencing. Variant was confirmed
dIf available
e R i of site of opposite polarity in the pseudoexon
fConsequences for pseudoexon formation describe how the intron is divided: “new intron A length/pseudoexon length/new exon B length
None of the variants have been previously reported by other groups with the exception of CHEK2 c.320-5T>A [148]
First, the novel ATM variant c.3747-1G > A (chr11:108,154,953G > A; sample number 7-4 F) abolishes the natural acceptor of exon 26 (11.0 to 0.1 bits). ASSEDA reports the presence of a 5.3 bit cryptic acceptor site 13 nt downstream of the natural site, but the effect of the variant on a pre-existing cryptic site is negligible (~0.1 bits). The cryptic exon would lead to exon deletion and frameshift (Fig. 3a). ASSEDA also predicts skipping of the 246 nt exon, as the Ri,final of the natural acceptor is now below Ri,minimum (1.6 bits), altering the reading frame. Second, the novel ATM c.6347 + 1G > T (chr11:108188249G > T; 4-1 F) abolishes the 10.4 bit natural donor site of exon 44 (ΔRi = -18.6 bits), and is predicted to cause exon skipping. Finally, the previously reported CHEK2 variant, c.320-5A > T (chr22:29,121,360 T > A; rs121908700; 4-2B) [148] weakens the natural acceptor of exon 3 (6.8 to 4.1 bits), and may activate a cryptic acceptor (7.4 bits) 92 nt upstream of the natural acceptor site which would shift the reading frame (Fig. 4). A constitutive, frameshifted alternative isoform of CHEK2 lacking exons 3 and 4 has been reported, but skipping of exon 3 alone is not normally observed.
Fig. 3.
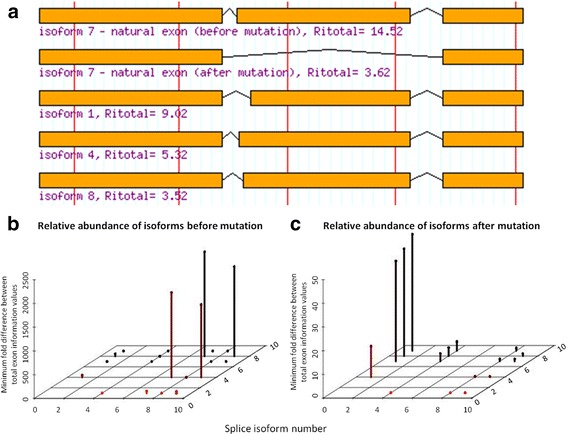
Predicted Isoforms and Relative Abundances as a Consequence of ATM splice variant c.3747-1G > A. Intronic ATM variant c.3747-1G > A abolishes (11.0 to 0.1 bits) the natural acceptor of exon 26 (total of 63 exons). a ASSEDA predicts skipping of the natural exon (R i,total from 14.5 to 3.6 bits [a 1910 fold decrease in exon strength]; isoform 7) and/or activation of a pre-existing cryptic acceptor site 13 nt downstream (R i,total for cryptic exon = 9.0 bits; isoform 1) of the natural site leading to exon truncation. The reading frame is altered in both mutant isoforms. The other isoforms use weak, alternate acceptor/donor sites leading to cryptic exons with much lower total information. b Before the mutation, isoform 7 is expected to be the most abundant splice form. c After the mutation, isoform 1 is predicted to become the most abundant splice form and the wild-type isoform is not expected to be expressed
Fig. 4.
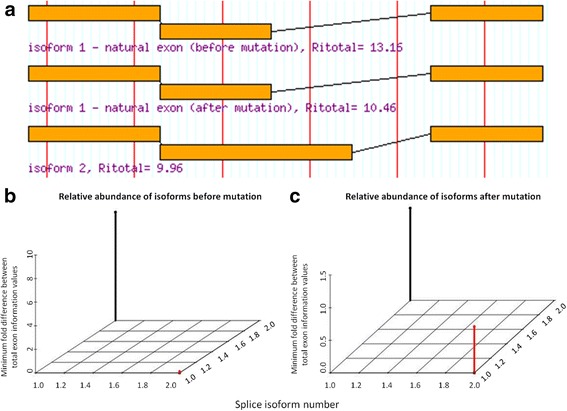
Predicted Isoforms and Relative Abundances as a Consequence of CHEK2 splice variant c.320-5 T > A. Intronic CHEK2 variant c.320-5 T > A weakens (6.8 to 4.1 bits) the natural acceptor of exon 3 (total of 15 exons). a ASSEDA reports the weakening of the natural exon strength (R i,total reduced from 13.2 to 10.5 bits), which would result in reduced splicing of the exon otherwise known as leaky splicing. A pre-existing cryptic acceptor exists 92 nt upstream of the natural site, leading to a cryptic exon with similar strength to the mutated exon (R i,total = 10.0 bits). This cryptic exon would contain 92 nt of the intron. b Before the mutation, isoform 1 is expected to be the only isoform expressed. c After the mutation, isoform 1 (wild-type) is predicted to become relatively less abundant and isoform 2 is expected to be expressed, although less abundant in relation to isoform 1
Variants either strengthening (N = 4) or slightly weakening (ΔRi < 1.0 bits; N = 4) a natural site were not prioritized. In addition, we rejected the ATM variant (c.1066-6 T > G; chr11:108,119,654 T > G; 4-1E and 7-2B), which slightly weakens the natural acceptor of exon 9 (11.0 to 8.1 bits). Although other studies have shown leaky expression as a result of this variant [149], a more recent meta-analysis concluded that this variant is not associated with increased breast cancer risk [150].
Cryptic SS activation
Two variants produced information changes that could potentially impact cryptic splicing, but were not prioritized for the following reasons (Table 2). The first variant, novel BRCA2 deletion c.7618-269_7618-260del10 (chr13:32931610_32931619del10; 7-4A) strengthens a cryptic acceptor site 245 nt upstream from the natural acceptor of exon 16 (Ri,final = 9.4 bits, ΔRi = 5.5 bits). Being 5.7-fold stronger than the natural site (6.9 bits), two potential cryptic isoforms were predicted, however the exon strengths of both are weaker than the unaffected natural exon (Ri,total = 6.6 bits) and thus neither were prioritized. The larger gap surprisal penalties explain the differences in exon strength. The natural donor SS may still be used in conjunction with the abovementioned cryptic SS, resulting in an exon with Ri,total = 3.5 bits. Alternatively, the cryptic site and a weak donor site 180 nt upstream of the natural donor (Ri = 0.7 vs 1.4, cryptic and natural donors, respectively) result in an exon with Ri,total = 6.5 bits. The second variant, BRCA1 c.548-293G > A (chr17:41249599C > T; 7-3E), creates a weak cryptic acceptor (Ri,final = 2.6 bits, ΔRi = 6.2 bits) 291 nt upstream of the natural acceptor for exon 8 (Ri = 0.5). Although the cryptic exon is strengthened (final Ri,total = 6.9 bits, ΔRi = 14.7 bits), ASSEDA predicts the level of expression of this exon to be negligible, as it is weaker than the natural exon (Ri,total = 8.4 bits) due to the increased length of the predicted exon (+291 nt) [38].
Pseudoexon formation
The Shannon Pipeline initially reported 1583 unique variants creating or strengthening intronic cryptic sites. We prioritized 5 variants, 1 of which is novel (BRCA2 c.8332-805G > A; 7-3 F), that were within 250 nt of a pre-existing complementary cryptic site and have an hnRNPA1 site within 5 nt of the acceptor (Table 2). If used, 3 of these pseudoexons would lead to a frameshifted transcript.
SRF binding
Variants within 500 nt of an exon junction and all exonic variants (N = 4015) were investigated for their potential effects on affinity of sites to corresponding SRFs [38]. IT analysis flagged 54 variants significantly altering the strength of at least one binding site (Additional file 8: Table S5). A careful review of the variants, the factor affected, and the position of the binding site relative to the natural SS, prioritized 36 variants (21 novel), of which 4 are in exons and 32 are in introns. As an example, a novel CHEK2 exon 2 variant c.69C > A (p.Gly23=) is predicted to increase the strength of an hnRNP A1 site (0.7 to 5.3 bits) and decrease total exon strength (ΔRi,total = -5.7 bits). A similar type of exonic variant in FANCM, which was predicted to create an exonic hnRNP A1 site by IT, has been shown to bind this exonic repressor and induce exon skipping [37].
TF binding
We assessed SNVs with models of 83 TFs experimentally shown to bind (Additional file 3: Table S1) upstream or within the first exon and intron of our sequenced genes (N = 2177). Thirteen variants expected to significantly affect TF binding were flagged (Additional file 9: Table S6). The final filtering step considered the known function of the TF in transcription, resulting in 5 variants (Table 2) in 6 patients (one variant was identified in two patients). Four of these variants have been previously reported (rs5030874, rs552824227, rs17882863, rs113451673) and one is novel (c.-8895G > A; 7-4B).
UTR structure and protein binding
There were 364 unique UTR variants found by sequencing. These variants were evaluated for their effects on mRNA 2° structure (including that of splice forms with alternate UTRs in the cases of BRCA1 and TP53) through SNPfold, resulting in 5 flagged variants (Table 3), all of which have been previously reported.
Table 3.
Variants predicted by SNPfold to affect UTR structure
| Classa | Patient ID | Gene | mRNA | UTR position | rsID (dbSNP 142) | Ranke | p-value |
|---|---|---|---|---|---|---|---|
| Allele Frequency (%)d | |||||||
| F | In 26 patients | BRCA2 b | c.-52A > G | 5’ UTR | rs206118 | 2/900 | 0.002 |
| 14.86 | |||||||
| F | In 40 patients | BRCA2 b | c.*532A > G | 3’ UTR | rs11571836 | 239/2700 | 0.089 |
| 19.75 | |||||||
| P | 7-4C | CDH1 c | c.-71C > G | 5’ UTR | rs34033771 | 69/600 | 0.115 |
| 0.56 | |||||||
| F | 4-2E | TP53 b | c.*485G > A | 3’ UTR | rs4968187 | 169/4500 | 0.038 |
| 5-4A | |||||||
| 5.11 | |||||||
| F | 2-1A, 7-1B, 5-2A.7-1D, 7-2B, 7-2F | TP53 b | c.*826G > A | 3’ UTR | rs17884306 | 371/4500 | 0.082 |
| 7-4C | |||||||
| 5.71 |
aF:Flagged; P:Prioritized
bLong Range UTR SNPfold Analysis
cLocal Range SNPfold Analysis
dIf available
eRank of the SNP, in terms of how much it changes the mRNA structure compared to all other possible mutations
Analysis of three variants using mFOLD [83] revealed likely changes to the UTR structure (Fig. 5). Two variants with possible 2° structure effects were common (BRCA2 c.-52A > G [N = 26 samples] and c.*532A > G [N = 40]) and not prioritized. The 5’ UTR CDH1 variant c.-71C > G (chr16:68771248C > G; rs34033771; 7-4C) disrupts a double-stranded hairpin region to create a larger loop structure, thus increasing binding accessibility (Fig. 5a and b). Analysis using RBPDB and CISBP-RNA-derived IT models suggests this variant affects binding by NCL (Nucleolin, a transcription coactivator) by decreasing binding affinity 14-fold (Ri,initial = 6.6 bits, ΔRi = -3.8 bits) (Additional file 10: Table S7). This RBP has been shown to bind to the 5’ and 3’ UTR of p53 mRNA and plays a role in repressing its translation [151].
Fig. 5.
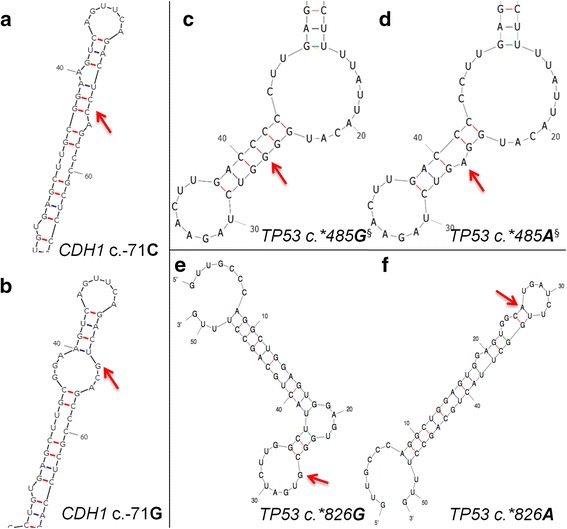
Predicted Alteration in UTR Structure Using mFOLD for Variants Flagged by SNPfold. Wild-type and variant structures are displayed, with the variant indicated by a red arrow. a Predicted wild-type structure of CDH1 5’UTR surrounding c.-71. b Predicted CDH1 5’UTR structure due to c.-71C > G variant. c Predicted wild-type TP53 3’UTR structure surrounding c.*485. d Predicted TP53 5’UTR structure due to c.*485G > A variant. e Predicted wild-type TP53 3’UTR structure surrounding c.*826. f Predicted TP53 5’UTR structure due to c.*826G > A variant. §SHAPE analysis revealed differences in reactivity between mutant and variant mRNAs, confirming alterations to 2° structure
In addition, the TP53 variant c.*485G > A (NM_000546.5: chr17:7572442C > T; rs4968187) is found at the 3’ UTR and was identified in two patients (4-2E and 5-4A). In silico mRNA folding analysis demonstrated this variant disrupts a G/C bond of a loop in the highest ranked potential mRNA structure (Fig. 5c and d). Also, SHAPE analysis showed a difference in 2° structure between the wild-type and mutant (data not shown). IT analysis with RBBS models indicated that this variant significantly increases the binding affinity of SF3B4 by > 48-fold (Ri,final = 11.0 bits, ΔRi = 5.6 bits) (Additional file 10: Table S7). This RBP is one of four subunits comprising the splice factor 3B, which binds upstream of the branch-point sequence in pre-mRNA [152].
The third flagged variant also occurs in the 3’ UTR of TP53 (c.*826G > A; chr17:7572,101C > T; rs17884306), and was identified in 6 patients (2-1A, 7-1B, 5-2A.7-1D, 7-2B, 7-2 F, and 7-4C). It disrupts a potential loop structure, stabilizing a double-stranded hairpin, and possibly making it less accessible (Fig. 5e and f). Analysis using RBPDB-derived models suggests this variant could affect the binding of both RBFOX2 and SF3B4 (Additional file 10: Table S7). A binding site for RBFOX2, which acts as a promoter of alternative splicing by favoring the inclusion of alternative exons [153], is created (Ri,final = 9.8 bits; ΔRi = -6.5 bits). This variant is also expected to simultaneously abolish a SF3B4 binding site (Ri,final = -20.3 bits; ΔRi = -29.9 bits).
RBPDB- and CISBP-RNA-derived information model analysis of all UTR variants resulted in the prioritization of 1 novel, and 5 previously-reported variants (Table 2). No patient within the cohort exhibited more than one prioritized RBBS variant.
To evaluate the background rate of prioritizing variants flagged by this method, all 5’ and 3’ UTR SNVs in dbSNP144 for the 7 genes sequenced (excluding those already flagged in Table 3) were evaluated by SNPfold and our RBP information models. Of 1207 SNVs, only 10 were prioritized with both methods, which results in a background rate of 0.83 %.
Exonic variants altering protein sequence
Exonic variants called by GATK (N = 245) included insertions, deletions, nonsense, missense, and synonymous changes.
Protein-truncating variants
We identified 3 patients with different indels (Table 4). One was a PALB2 insertion c.1617_1618insTT (chr16:23646249_23646250insAA; 5-3A) in exon 4, previously reported in ClinVar as pathogenic. This mutation results in a frameshift and premature translation termination by 626 residues, abolishing domain interactions with RAD51, BRCA2, and POLH [137]. We also identified two known frameshift mutations in BRCA1: c.4964_4982del19 in exon 15 (chr17:41222949_41222967del19; rs80359876; 5-1B) and c.5266_5267insC in exon 19 (chr17:41209079_41209080insG; rs397507247; 5-3C) [148, 154]. Both are indicated as pathogenic and common in the BIC Database due to the loss of one or both C-terminal BRCT repeat domains [137]. Truncation of these domains produces instability and impairs nuclear transcript localization [155], and this bipartite domain is responsible for binding phosphoproteins that are phosphorylated in response to DNA damage [156, 157].
Table 4.
Variants resulting in premature protein truncation
| Patient ID | Gene | Exon | mRNA Protein | rsID (dbSNP 142) | ClinVard,e,f | Details | Ref |
|---|---|---|---|---|---|---|---|
| Allele Frequency (%)c | |||||||
| Insertions/Deletions | |||||||
| 5-1B | BRCA1 | 15 of 23 | c.4964_4982del19a | rs80359876 | 6d; Pathogenic/likely pathogenice; Familial breast and breast-ovarian cancer, Hereditary cancer-predisposing syndromef. | STOP at p.1670 | - |
| p.Ser1655Tyrfs | 193 AA short | ||||||
| 5-3C | BRCA1 | 19 of 23 | c.5266_5267insCa | rs397507247 | 13d; Pathogenic, risk factore; Familial breast, breast-ovarian, and pancreatic cancer, Hereditary cancer-predisposing syndromef. | STOP at p.1788 | [148, 154] |
| p.Gln1756Profs | 75 AA short | ||||||
| 5-3A | PALB2 | 4 of 13 | c.1617_1618insTTa | - | 1d; Pathogenice; Hereditary cancer-predisposing syndromef. | STOP at p.561 | - |
| p.Asn540Leufs | 626 AA short | ||||||
| Stop Codons | |||||||
| 7-1G | BRCA2 | 15 of 27 | c.7558C > Tb | rs80358981 | 5d; Pathogenice; Familial breast, and breast-ovarian cancer, Hereditary cancer-predisposing syndromef. | 899 AA short | [158] |
| p.Arg2520Ter | |||||||
| 4-4A | BRCA2 | 25 of 27 | c.9294C > Ga | rs80359200 | 3d; Pathogenice; Familial breast and breast-ovarian cancerf. | 321 AA short | [159] |
| p.Tyr3098Ter | |||||||
| 7-3A | PALB2 | 4 of 13 | c.1240C > Ta | rs180177100 | 3d; Pathogenice; Familial breast cancer, Hereditary cancer-predisposing syndromef. | 773 AA short | [58] |
| p.Arg414Ter | |||||||
| 4-4D | PALB2 | 4 of 13 | c.1042C > Ta | Novel | - | 839 AA short | - |
| p.Gln348Ter | |||||||
aConfirmed by Sanger sequencing
bNot confirmed by Sanger sequencing
cIf available
dNumber of submissions
eClinical significance
fCondition(s)
We also identified 4 nonsense mutations, one of which was novel in exon 4 of PALB2 (c.1042C > T; chr16:23646825G > A; 4-4D). Another in PALB2 has been previously reported (c.1240C > T; chr16:23646627G > A; rs180177100; 7-3A) [58]. As a consequence, functional domains of PALB2 that interact with BRCA1, RAD51, BRCA2, and POLH are lost [137]. Two known nonsense mutations were found in BRCA2, c.7558C > T in exon 15 [158] and c.9294C > G in exon 25 [159]. The first (chr13:32930687C > T; rs80358981; 7-1G) causes the loss of the BRCA2 region that binds FANCD2, responsible for loading BRCA2 onto damaged chromatin [160]. The second (chr13:32968863C > G, rs80359200; 4-4A) does not occur within a known functional domain, however the transcript is likely to be degraded by nonsense mediated decay [161].
Missense
GATK called 61 missense variants, of which 18 were identified in 6 patients or more and 19 had allele frequencies > 1.0 % (Additional file 11: Table S8). The 40 remaining variants (15 ATM, 8 BRCA1, 9 BRCA2, 2 CDH1, 2 CHEK2, 3 PALB2, and 1 TP53) were assessed using a combination of gene specific databases, published classifications, and 4 in silico tools (Additional file 12: Table S9). We prioritized 27 variants, 2 of which were novel. None of the non-prioritized variants were predicted to be damaging by more than 2 of 4 conservation-based software programs.
Variant classification
Initially, 15,311 unique variants were identified by complete gene sequencing of 7 HBOC genes. Of these, 132 were flagged after filtering, and further reduced by IT-based variant analysis and consultation of the published literature to 87 prioritized variants. Figure 6 illustrates the decrease in the number of unique variants per patient at each step of our identification and prioritization process. The distribution of prioritized variants by gene is 34 in ATM, 13 in BRCA1, 11 in BRCA2, 8 in CDH1, 6 in CHEK2, 10 in PALB2, and 5 in TP53 (Additional file 13: Table S10), which are categorized by type in Table 5.
Fig. 6.
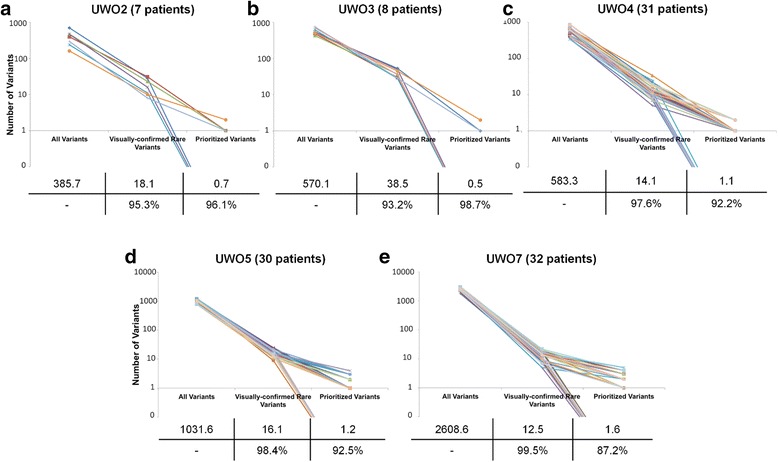
Ladder Plot Representing Variant Identification and Prioritization. Each line is representative of a different sample in each sequencing run (a-e), illustrating the number of unique variants at important steps throughout the variant prioritization process. The left-most point indicates the total number of unique variants. The second point represents the number of unique variants remaining after common (>5 patients within cohort and/or ≥ 1.0 % allele frequency) and false-positive variants were removed. The right-most point represents the final number of unique. No variants were prioritized in the following patients: 2-1A, 2-5A, 2-6A, 3-2A, 3-3A, 3-4A, 3-5A, 3-8A, 4-1B, 4-2C, 4-2 F, 4-3B, 4-3D, 4-4B, 4-4E, 5-1G, 5-1H, 5-3D, 5-4C, 5-4D, 5-4 F, 5-4G, 5-4H, 7-1B, 7-1C, 7-1D, 7-1H, 7-2B, 7-2C, 7-2H, 7-3H, 7-4A, 7-4D, 7-4H. The average number of variants per patient at each step is indicated in a table below each plot, along with the percent reduction in variants from one step to another
Table 5.
Summary of prioritized variants by gene
| Indel | Nonsense | Missense | Natural Splicing | Cryptic Splicing | Pseudoexon | SR Factor | TF | UTR Structure | UTR Binding | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATM | 0 | 0 | 14 | 2 | 0 | 0 | 18 | 0 | 0 | 1 | 34 a |
| BRCA1 | 2 | 0 | 2 | 0 | 0 | 1 | 7 | 1 | 0 | 0 | 13 |
| BRCA2 | 0 | 2 | 3 | 0 | 0 | 2 | 4 | 0 | 0 | 0 | 11 |
| CDH1 | 0 | 0 | 2 | 0 | 0 | 2 | 1 | 1 | 1 | 1 | 8 |
| CHEK2 | 0 | 0 | 2 | 1 | 0 | 0 | 3 | 0 | 0 | 2 | 6 a |
| PALB2 | 1 | 2 | 3 | 0 | 0 | 0 | 3 | 1 | 0 | 0 | 10 |
| TP53 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 2 | 5 |
| Total | 3 | 4 | 27 | 3 | 0 | 5 | 36 | 5 | 1 | 6 |
Three variants were prioritized under multiple categories: ATM chr11:108121730A > G (missense and SRFBS), CHEK2 chr22:29121242G > A (missense, UTR binding), and CHEK2 chr22:29130520C > T (missense, UTR binding)
a Counts represent the number of unique variants identified (i.e. a variant is not counted twice if it appeared in multiple individuals)
Three prioritized variants have multiple predicted roles: ATM c.1538A > G in missense and SRFBS, CHEK2 c.190G > A in missense and UTR binding, and CHEK2 c.433C > T in missense and UTR binding. Of the 102 patients that were sequenced, 72 (70.6 %) exhibited at least one prioritized variant, and some patients harbored more than one prioritized variant (N = 33; 32 %). Additional file 14: Table S11 presents a summary of all flagged and prioritized variants for patients with at least one prioritized variant.
Prioritization of potential deletions
Using BreakDancer, none of the individuals analyzed exhibited large rearrangements that met the level of stringency required, but a small intragenic rearrangement in BRCA1 was identified and confirmed by Sanger sequencing. Attempts to detect deletions with BreakDancer only flagged single, non-contiguous paired-end reads, rather than a series of reads clustered within the same region within the same individual, which would be necessary to indicate the presence of a true deletion or structural rearrangement.
After prioritizing individuals for potential hemizygosity in the sequenced regions, potential deletions were detected in BRCA2 and CDH1. Patient UWO5-4D exhibited a non-polymorphic 32.1 kb interval in BRCA2, spanning introns 1 to 13, that was absent from all of the other individuals (chr13:32890227-32922331). Haploview (hapmap.org) showed very low levels of LD in this region. The potential deletion may extend further downstream, however the presence of a haploblock, covering the entire sequenced interval beyond exon 11, with significant LD precludes delineation of the telomeric breakpoint. We also flagged a non-polymorphic 2.6 kb interval near the 3’ end of CDH1 in 6 individuals (UWO3-5, UWO4-2C, UWO4-4E, UWO4-4 F, UWO4-2G, UWO5-2H). This is a low LD region spanning chr16:68861286-68863887 that includes exons 14 and 15, and is polymorphic in all of the other individuals sequenced. CDH1 mutations are characteristically present in families with predisposition to gastric cancer, however breast cancer frequently co-occurs [69]. A study of CDH1 deletions in inherited gastric cancer identified two families with deletions that overlap the intervals prioritized in the present study [162].
Comparison to combined annotation dependent depletion
The analysis and prioritization of non-coding variants can also be accomplished using Combined Annotation Dependent Depletion (CADD; [163]), which uses known and simulated variants to compute a C-score, an ad hoc measure of how deleterious is likely to be. The suggested C-score cutoff is between 10 and 20, though it is stated that any selected cutoff value would be arbitrary (http://cadd.gs.washington.edu/info). This contrasts with information-based methods, which are based on thermodynamically-defined thresholds. To directly compare methods, CADD scores were obtained for all prioritized or flagged SNVs. Half of prioritized variants met this cutoff (C > 10), while only 28.6 % of flagged variants did the same. All prioritized nonsense variants (4/4) and 26/27 missense variants had strong C-scores. Prioritized non-coding variant categories that correlated well with CADD include natural splicing variants (4/4), UTR structure variants (1/1), and RBPs (4/6). Weakly correlated variants included those affecting SRFBPs (5/36), TFBS (2/5), and pseudoexon activating variants (0/5). Missense mutations comprised 75 % of the flagged variants with C > 10. The aforementioned flagged splicing variant ATM c.1066-6 T > G also exceeded the threshold C value (C = 11.9). Meanwhile, the flagged TP53 variant, shown by SHAPE analysis to alter UTR structure, did not (C = 5.3). Despite consistency between some variant categories, the underlying assumptions of each approach probably explain why these results differ for non-coding variants. The limited numbers validated, deleterious non-coding variants also contributes to the accuracy of these predictions [163].
Variant verification
We verified prioritized protein-truncating (N = 7) and splicing (N = 4) variants by Sanger sequencing (Tables 2 and 4, respectively). In addition, two missense variants (BRCA2 c.7958 T > C and CHEK2 c.433C > T) were re-sequenced, since they are indicated as likely pathogenic/pathogenic in ClinVar (Additional file 12: Table S9). All protein-truncating variants were confirmed, with one exception (BRCA2 c.7558C > T, no evidence for the variant was present for either strand). Two of the mRNA splicing mutations were confirmed on both strands, while the other two were confirmed on a single strand (ATM c.6347 + 1G > T and ATM c.1066-6 T > G). Both documented pathogenic missense variants were also confirmed.
Discussion
NGS technology offers advantages in throughput and variant detection [126], but the task of interpreting the sheer volume of variants in complete gene or genome data can be daunting. The whole genome of a Yoruban male contained approximately 4.2 million SNVs and 0.4 million structural variants [164]. The variant density in the present study (average 948 variants per patient) was 5.3-fold lower than the same regions in HapMap sample NA12878 in Illumina Platinum Genomes Project (5029 variants) [165]. The difference can be attributed primarily to the exclusion of polymorphisms in highly repetitive regions in our study.
Conventional coding sequence analysis, combined with an IT-based approach for regulatory and splicing-related variants, reduced the set to a manageable number of prioritized variants. Unification of non-coding analysis of diverse protein-nucleic acid interactions using the IT framework accomplishes this by applying thermodynamic-based thresholds to binding affinity changes and by selecting the most significant binding site information changes, regardless of whether the motifs of different factors overlap.
Previously, rule-based systems have been proposed for variant severity classification [166, 167]. Functional validation and risk analyses of these variants are a prerequisite for classification, but this would not be practical to accomplish without first limiting the subset of variants analyzed. With the exception of some (but not all [37]) protein truncating variants, classification is generally not achievable by sequence analysis alone. Only a minority of variants with extreme likelihoods of pathogenic or benign phenotypes are clearly delineated because only these types of variants are considered actionable [166, 167]. The proposed classification systems preferably require functional, co-segregation, and risk analyses to stratify patients. Nevertheless, the majority of variants are VUS, especially in the case of variants occurring beyond exon boundaries. Of the 5713 variants in the BIC database, the clinical significance of 4102 BRCA1 and BRCA2 variants are either unknown (1904) or pending (2198), and only 1535 have been classified as pathogenic (Class 5) [168]. Our results cannot be considered equivalent to validation, which usually include expression assays [36] or the use of RNA-seq data [169] (splicing), qRT-PCR [170] (transcription), SHAPE analysis (mRNA 2° structure) [44], or binding assays to determine functional effects of variants. Classification of VUS in BRCA1 and BRCA2 by the ENIGMA Consortium addresses mRNA splicing and missense variants. Criteria define risk based on whether the variant occurs within a protein structural domain, the impact on protein function, and the segregation pattern of variant with disease in pedigrees [171]. These guidelines cannot be fully implemented here for several reasons: a) patients were anonymized in this study, precluding segregation analysis, b) the splicing mutation guideline does not take into account predicted leaky or cryptic SS mutations, nor other non-canonical changes that have been demonstrated to alter the expression of these and numerous other genes, c) conserved domains have not been identified in regions of the proteins encoded by these genes, especially BRCA2, where many missense mutations reside, and d) the guidelines are currently silent as to the potential impact of regulatory variants affecting splicing, RNA stability, and transcriptional regulation.
While the miRNA variant prediction program mrSNP [172] was used to evaluate all of the 3’ UTR variants, 41.4 % of the variants were predicted to alter the stability of the miRNA-target mRNA duplex for at least one miRNA expressed in breast tissue. However, only 2 of these interactions could be confirmed using TarBase [173], and these variants could not be prioritized for disruption of miRNA regulation. Other post-transcriptional processes, including miRNA regulation, that were not addressed in this study, may also be amenable to such IT-based modeling. With the proposed approach, functional prediction of such variants could precede or at least inform the classification of VUS.
It is unrealistic to expect all variants to be functionally analyzed, just as it may not be feasible to assess family members for a suspected pathogenic variant detected in a proband. The prioritization procedure reduces the chance that significant variants have been overlooked. Capturing coding and non-coding regions of HBOC-related genes, combined with the framework for assessing variants, balances the need to comprehensively detect all variation in a gene panel with the goal of identifying variants likely to be phenotypically relevant.
The location of variants in relation to known protein domains was documented in this study, but was not directly incorporated into our prioritization method. The locations and impact of splicing mutations in BRCA1 and BRCA2 were mapped to the known functional domains of the encoded proteins [174]. A high concentration of variants predicted to result in splicing changes occurred in the BRCT, RING finger, and NLS domains of BRCA1. However, BRCA2 variants were generally concentrated outside of known functional domains (aside from the C-terminal domain). Because of these inconsistencies, domain-mapping was not integrated with IT based prioritization. However, where adequate information on structure-function relationships is available (eg. TP53), we suggest that such analysis be carried out subsequent to IT-based variant prioritization.
Non-coding variants
Although coding variants are typically the sole focus of a molecular diagnostic laboratory (with the exception of the canonical dinucleotide positions within SS), non-coding mutations have long been known to be disease causing [19, 36, 175–183]. In this study, variant density in non-coding regions significantly exceeded exonic variants by > 60-fold, which, in absolute terms, constituted 1.6 % of the 15,311 variants. This is comparable to whole genome sequencing studies, which typically result in 3-4 million variants per individual, with < 2 % occurring in protein coding regions [184]. IT analysis prioritized 3 natural SS, 36 SRFBS, 5 TFBS, and 6 RBBS variants and 5 predicted to create pseudoexons. Two SS variants in ATM (c.3747-1G > A and c.6347 + 1G > T) were predicted to completely abolish the natural site and cause exon skipping. A CHEK2 variant (c.320-5A > T) was predicted to result in leaky splicing.
The IT-based framework evaluates all variants on a common scale, based on bit values, the universal unit that predicts changes in binding affinity [185]. A variant can alter the strength of one or a “set” of binding sites; the magnitude and direction of these changes is used to rank their significance. The models used to derive information weight matrices take into account the frequency of all observed bases at a given position of a binding motif, making them more accurate than consensus sequence and conservation-based approaches [36].
IT has been widely used to analyze natural and cryptic SSs [36], but its use in SRFBS analysis was only introduced recently [38]. For this reason, we assigned conservative, minimum thresholds for reporting information changes. Although there are examples of disease-causing variants resulting in small changes in Ri [174, 186–192], the majority of deleterious splicing mutations that have been verified functionally, produce large information changes. Among 698 experimentally verified deleterious variants in 117 studies, only 1.96 % resulted in < 1.0 bit change [36]. For SRFBS variants, the absolute information changes for deleterious variants ranged from 0.2 to 17.1 bits (mean 4.7 ± 3.8). This first application of IT in TFBS and RBBS analysis, however, lacks a large reference set of validated mutations for the distribution of information changes associated with deleterious variants. The release of new ChIP-seq datasets will enable IT models to be derived for TFs currently unmodeled and will improve existing models [193].
Pseudoexon activation results in disease-causing mutations [194], however such consequences are not customarily screened for in mRNA splicing analysis. IT analysis was used to detect variants that predict pseudoexon formation and 5 variants were prioritized. Previously, we have predicted experimentally proven pseudoexons with IT (Ref 42: Table 2, No #2; and Ref 195: Table 2, No #7) [42, 195]. Although it was not possible to confirm prioritized variants in the current study predicted to activate pseudoexons because of their low allele frequencies, common intronic variants that were predicted to form pseudoexons were analyzed. We then searched for evidence of pseudoexon activation in mapped human EST and mRNA tracks [196] and RNA-seq data of breast normal and tumour tissue from the Cancer Genome Atlas project [15]. One of these variants (rs6005843) appeared to splice the human EST HY160109 [197] at the predicted cryptic SS and is expressed within the pseudoexon boundaries.
Variants that were common within our population sample (i.e. occurring in > 5 individuals) and/or common in the general population (>1.0 % allele frequency) reduced the list of flagged variants substantially. This is now a commonly accepted approach for reducing candidate disease variants [166], based on the principle that the disease-causing variants occur at lower population frequencies. Variants occurring in > 5 patients all either had allele frequencies above 1.0 % or, as shown previously, resulted in very small ΔRi values [198].
The genomic context of sequence changes can influence the interpretation of a particular variant [36]. For example, variants causing significant information changes may be interpreted as inconsequential if they are functionally redundant or enhancing existing binding site function (see IT-Based Variant Analysis for details). Our understanding of the roles and context of these cognate protein factors is incomplete, which affects confidence in interpretation of variants that alter binding. Also, certain factors with important roles in the regulation of these genes, but that do not bind DNA directly or in a sequence-specific manner (eg. CtBP2 [199]), could not be included. Therefore, some variants may have been incorrectly excluded.
Prioritization of potential deletions
Although individuals can be prioritized based on potential hemizygosity, this does not definitively identify deletions. Nevertheless, it should be possible to prioritize those individuals worthy of further detailed diagnostic workup. It has not escaped our attention that the weighted probabilities obtained from this analysis could be represented and formalized using the same units of Shannon information (in bits) as the other sequence changes we have described, analogous to single or multinucleotide gene variants predicted to affect nucleic acid binding sites. Full development and validation of this method is in progress.
Coding sequence changes
We also identified 4 nonsense and 3 indels in this cohort. In one individual, a 19 nt BRCA1 deletion in exon 15 causes a frameshift leading to a stop codon within 14 codons downstream. This variant, rs80359876, is considered clinically relevant. Interestingly, this deletion overlaps two other published deletions in this exon (rs397509209 and rs80359884). This raises the question as to whether this region of the BRCA1 gene is a hotspot for replication errors. DNA folding analysis indicates a possible 15 nt long stem-loop spanning this interval as the most stable predicted structure (data not shown). This 15 nt structure occurs entirely within the rs80359876 and rs397509209 deletions and partially overlaps rs80359884 (13 of 15 nt of the stem loop). It is plausible that the 2° structure of this sequence predisposes to a replication error that leads to the observed deletion.
Missense coding variants were also assessed using multiple in silico tools and evaluated based on allele frequency, literature references, and gene-specific databases. Of the 27 prioritized missense variants, the previously reported CHEK2 variant c.433G > A (chr22:29121242G > A; rs137853007) stood out, as it was identified in one patient (4-3C.5-4G) and is predicted by all 4 in silico tools to have a damaging effect on protein function. Accordingly, Wu et al. (2001) demonstrated reduced in vitro kinase activity and phosphorylation by ATM kinase compared to the wild-type CHEK2 protein [200], presumably due to the variant’s occurrence within the forkhead homology-associated domain, involved in protein-phosphoprotein interactions [201]. Implicated in Li-Fraumeni syndrome, known to increase the risk of developing several types of cancer including breast [202, 203], the CHEK2: c.433G > A variant is expected to result in a misfolded protein that would be targeted for degradation via the ubiquitin-proteosome pathway [204]. Another important missense variant is c.7958 T > C (chr13:32,936,812 T > C; rs80359022; 4-4C) in exon 17 of BRCA2. Although classified as being of unknown clinical importance in both BIC and ClinVar, it has been classified as pathogenic based on posterior probability calculations [205].
It is unlikely that all prioritized variants are pathogenic in patients carrying more than one prioritized variant. Nevertheless, a polygenic model for breast cancer susceptibility, whereby multiple moderate and low-risk alleles contribute to increased risk of HBOC may also account for multiple prioritized variants [206, 207]. There was a significant fraction of patients (29.4 %) in whom no variants were prioritized. This could be due to a) the inability of the analysis to predict a variant affecting the binding sites analyzed, b) a pathogenic variant affecting a function that was not analyzed or in a gene that was not sequenced, c) a large rearrangement/deletion where both breakpoints occur beyond the captured genomic intervals (which is unlikely, as this would have been observed as an extended non-polymorphic sequence), or d) the significant family history was not due to heritable, but instead to shared environmental influences.
BRCA coding variants were found in individuals who were previously screened for lesions in these genes, suggesting this NGS protocol is a more sensitive approach for detecting coding changes. However, previous testing of a number of these patients had been predominantly based on PTT and MLPA, which have lower sensitivity for detecting mutations than sequence analysis. Nevertheless, we identified 2 BRCA1 and 2 BRCA2 variants predicted to encode prematurely truncated proteins. Fewer non-coding BRCA variants were prioritized (15.7 %) than expected by linkage analysis [49], however this presumes at least 4 affected breast cancer diagnoses per pedigree, and, in the present study, the number of affected individuals per family was not known.
Prioritization of a variant does not equate with pathogenicity. Some prioritized variants may not increase risk, but may simply modify a primary unrecognized pathogenic mutation. A patient with a known BRCA1 nonsense variant, used as a positive control, was also found to possess an additional prioritized variant in BRCA2 (missense variant chr13:32911710A > G), which was flagged by PROVEAN and SIFT as damaging, as well as flagged for changing an SRFBS for abolishing a PTB site (while simultaneously abolished an exonic hnRNPA1 site). This variant has been identified in cases of early onset prostate cancer and is considered a VUS in ClinVar [143]. Similarly, variants prioritized in multiple patients may act as risk modifiers rather than pathogenic mutations. A larger cohort of patients with known pathogenic mutations would be necessary to calculate a background/basal rate of falsely flagged variants.
Other groups have attempted to develop comprehensive approaches for variant analysis, analogous to the one proposed here [208–210]. While most employ high-throughput sequencing and classify variants, either the sequences analyzed or the types of variants assessed tend to be limited. In particular, non-coding sequences have not been sequenced or studied to the same extent, and none of these analytical approaches have adopted a common framework for mutation analysis.
Our published oligonucleotide design method [77] produced an average sequence coverage of 98.8 %. The capture reagent did not overlap conserved highly repetitive regions, but included divergent repetitive sequences. Nevertheless, neighboring probes generated reads with partial overlap of repetitive intervals. As previously reported [147], we noted that false positive variant calls within intronic and intergenic regions were the most common consequence of dephasing in low complexity, pyrimidine-enriched intervals. This was not alleviated by processing data with software programs based on different alignment or calling algorithms. Manual review of all intronic or intergenic variants became imperative. As these sequences can still affect functional binding elements detectable by IT analysis (i.e. 3’ SSs and SRFBSs), it may prove essential to adopt or develop alignment software that explicitly and correctly identifies variants in these regions [147]. Most variants were confirmed with Sanger sequencing (10/13), and those that could not be confirmed are not necessarily false positives. A recent study demonstrated that NGS can identify variants that Sanger sequencing cannot, and reproducing sequencing results by NGS may be worthwhile before eliminating such variants [211].
Conclusions
Through a comprehensive protocol based on high-throughput, IT-based and complementary coding sequence analyses, the numbers of VUS can be reduced to a manageable quantity of variants, prioritized by predicted function. While exonic variants corresponded to a small fraction of prioritized variants, there is considerably more evidence for their pathogenicity because clinical sequencing has concentrated in these regions. Our sequencing approach illustrates the importance of sequencing non-coding regions of genes to establish pathogenic mutations not already evident from changes in the amino acid based genetic code. We suggest our approach for variant flagging and prioritization bridges the phase between high-throughput sequencing, variant detection with the time-consuming process of variant classification, including pedigree analysis and functional validation. Subsequent to completion of the present study, ethics approval was obtained for a similar analysis of consented patients with clinical information. This work has since been described elsewhere [212].
Availability of supporting data
Variants will be deposited with the ENIGMA Consortium (www.enigmaconsortium.org), which is a designated organization for curation of HBOC mutations and which is charged with protection of genetic privacy of participants. Additionally, all likely pathogenic variants were submitted to ClinVar (submission ID: SUB1332053) while other novel variants were submitted to dbSNP (ss1966658584-1966658622).
Acknowledgements
We thank Dr. Peter Ainsworth and Alan Stuart of the MGL, Molecular Diagnostics Division, Dept of Pathology and Laboratory Medicine at the London Health Sciences Centre for access to patient DNA samples and the use of the BioMek® FXP Automation Workstation. Information models for protein binding were computed with PoWeMaGen (Nathan Bryans) and variants were scanned with Mutation Analyzer, a modification of the Shannon Human Splicing Mutation Pipeline (Coby Viner). Edwin Dovigi contributed to design and synthesis of the custom capture reagent. PKR is supported by the Canadian Breast Cancer Foundation, Canadian Foundation for Innovation, Canada Research Chairs Secretariat and the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant 371758-2009). NGC is funded by the CIHR Strategic Training Program in Cancer Research and Technology Transfer (CaRTT) and the Pamela Greenaway-Kohlmeier Translational Breast Cancer Research Unit (TBCRU) awards. Our work was made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET) and Compute/Calcul Canada.
Abbreviations
- ASSEDA
Automated Splice Site and Exon Definition Analysis
- BIC
Breast Cancer Information Core Database
- CASAVA
Consensus Assessment of Sequencing and Variation
- CIS-BP-RNA
Catalog of Inferred Sequence Binding Preferences of RNA binding proteins
- CRAC
Complex Reads Analysis and Classification
- DM2
Domain Mapping of Disease Mutations
- ENIGMA
Evidence-based Network for the Interpretation of Germline Mutant Alleles
- ExPASy
Expert Protein Analysis System
- GATK
Genome Analysis Toolkit
- HBOC
Hereditary Breast and Ovarian Cancer
- HGMD
Human Gene Mutation Database
- IARC
International Agency for Research on Cancer
- IGV
Integrative Genomics Viewer
- Indel
Insertion/deletion
- IT
Information theory
- LD
Linkage Disequilibrium
- LOVD
Leiden Open Variant Database
- MGL
Molecular Genetics Laboratory
- MLPA
Multiplex Ligation Probe Amplification
- NGS
Next-Generation Sequencing
- PTB
Polypyrimidine tract binding protein
- PTT
Protein Truncation Test
- PWM
Position Weight Matrix
- RBBS
RNA-Binding protein Binding Site
- RBP
RNA-Binding Protein
- RBPDB
RNA-Binding Protein DataBase
- REB
Research Ethics Board
- Ri
Individual information
- Rsequence
Mean information content
- SHAPE
Selective 2’-Hydroxyl Acylation analyzed by Primer Extension
- SNV
Single Nucleotide Variant
- SRF
Splicing Regulatory Factor
- SRFBS
Splicing Regulatory Factor Binding Site
- SS
Splice Site
- TF
Transcription Factor
- TFBS
Transcription Factor Binding Site
- UTR
Untranslated Region
- VCF
Variant Call File
- VUS
Variants of Uncertain Significance
- ΔRi
Change in individual information; Patient Sample IDs are assigned in following manner: number-number + letter (i.e. 1-1A). If a sample was repeated, the IDs are separated by a “.” (i.e. 1-1A.2-1A)
Additional files
Supplementary Methods. (DOCX 243 kb)
Provincial Eligibility Criteria. Risk Categories for Individuals Eligible for Screening for a Genetic Susceptibility to Breast or Ovarian Cancer as determined by the Ontario Ministry of Health and Long Tern-Care Referral Criteria for Genetic Counseling (PDF 10 kb)
TFs For Which Information Weight Matrices Were Built And Factor’s Role in Transcription (XLSX 17 kb)
UTR Sequences Used for SHAPE Analysis on SNPfold-flagged Variants (XLSX 9 kb)
Primer Sequences for Sanger Sequencing of Likely Pathogenic Variants (XLSX 10 kb)
BRCA1 Deletion Inaccurately Aligned by CASAVA (PDF 22 kb)
Variants identified within natural donor or acceptor splice sites (XLSX 14 kb)
Variants Predicted by IT to Affect SRFBSs (XLSX 30 kb)
Variants Predicted by IT to Affect TFBSs (XLSX 14 kb)
Top Changes in RBBSs Predicted by IT for Variants Predicted to Significantly Alter RNA Structure (XLSX 10 kb)
Missense Variants Identified In 6 Patients Or More (XLSX 11 kb)
Missense Variants and Their Classification (XLSX 27 kb)
Prioritized Variants by Gene (XLSX 16 kb)
All Flagged and Prioritized Variants by Patient (XLSX 26 kb)
Footnotes
EJM and NGC should be considered to be joint first authors.
Competing interests
PKR is the inventor of US Patent 5,867,402 and other patents pending, which underlie the prediction and validation of mutations. He and JHMK founded Cytognomix Inc., which is developing software based on this technology for complete genome or exome mutation analysis.
Authors’ contribution
PKR designed, coordinated, and supervised the study, which was motivated by discussions with JHMK regarding prioritization of VUS. EJM performed probe design and synthesis. EJM and NGC performed sample preparation and sequencing. EJM wrote software and performed bioinformatic analysis. EJM, NGC, and AMP conducted variant analysis and prioritization. RL generated the TFBS information models and EJM generated the RBBS, SRF, and splicing information models. AMP confirmed prioritized variants by Sanger sequencing. MH and AL conducted the SHAPE analysis. EJM, NGC, AMP, JHMK, and PKR wrote the manuscript, which has been approved by all authors.
References
- 1.Collins FS, Hamburg MA. First FDA authorization for next-generation sequencer. N Engl J Med. 2013;369:2369–2371. doi: 10.1056/NEJMp1314561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green ED, Guyer MS, National Human Genome Research Institute Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470:204–213. doi: 10.1038/nature09764. [DOI] [PubMed] [Google Scholar]
- 3.Cassa CA, Savage SK, Taylor PL, Green RC, McGuire AL, Mandl KD. Disclosing pathogenic genetic variants to research participants: Quantifying an emerging ethical responsibility. Genome Res. 2012;22:421–428. doi: 10.1101/gr.127845.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Domchek SM, Bradbury A, Garber JE, Offit K, Robson ME. Multiplex genetic testing for cancer susceptibility: out on the high wire without a net? J Clin Oncol. 2013;31:1267–1270. doi: 10.1200/JCO.2012.46.9403. [DOI] [PubMed] [Google Scholar]
- 5.Yorczyk A, Robinson LS, Ross TS. Use of panel tests in place of single gene tests in the cancer genetics clinic. Clin Genet. 2015;88:278–282. doi: 10.1111/cge.12488. [DOI] [PubMed] [Google Scholar]
- 6.Foley SB, Rios JJ, Mgbemena VE, Robinson LS, Hampel HL, Toland AE, Durham L, Ross TS. Use of whole genome sequencing for diagnosis and discovery in the cancer genetics clinic. EBioMed. 2015;2:74–81. doi: 10.1016/j.ebiom.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwartz GF, Hughes KS, Lynch HT, Fabian CJ, Fentiman IS, Robson ME, Domchek SM, Hartmann LC, Holland R, Winchester DJ, Consensus Conference Committee The International Consensus Conference Committee Proceedings of the international consensus conference on breast cancer risk, genetics, & risk management, April, 2007. Cancer. 2008;113:2627–2637. doi: 10.1002/cncr.23903. [DOI] [PubMed] [Google Scholar]
- 8.Kavanagh D, Anderson HE. Interpretation of genetic variants of uncertain significance in atypical hemolytic uremic syndrome. Kidney Int. 2012;81:11–13. doi: 10.1038/ki.2011.330. [DOI] [PubMed] [Google Scholar]
- 9.Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB, Group IUGVW In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29:1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat Genet. 2008;40:17–22. doi: 10.1038/ng.2007.53. [DOI] [PubMed] [Google Scholar]
- 11.Ready K, Gutierrez-Barrera AM, Amos C, Meric-Bernstam F, Lu K, Hortobagyi G, Arun B. Cancer risk management decisions of women with BRCA1 or BRCA2 variants of uncertain significance. Breast J. 2011;17:210–212. doi: 10.1111/j.1524-4741.2010.01055.x. [DOI] [PubMed] [Google Scholar]
- 12.Eggington JM, Bowles KR, Moyes K, Manley S, Esterling L, Sizemore S, Rosenthal E, Theisen A, Saam J, Arnell C, Pruss D, Bennett J, Burbidge LA, Roa B, Wenstrup RJ. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. 2014;86:229–237. doi: 10.1111/cge.12315. [DOI] [PubMed] [Google Scholar]
- 13.Nanda R, Schumm LP, Cummings S, Fackenthal JD, Sveen L, Ademuyiwa F, Cobleigh M, Esserman L, Lindor NM, Neuhausen SL, Olopade OI. Genetic testing in an ethnically diverse cohort of high-risk women: a comparative analysis of BRCA1 and BRCA2 mutations in American families of European and African ancestry. JAMA. 2005;294:1925–1933. doi: 10.1001/jama.294.15.1925. [DOI] [PubMed] [Google Scholar]
- 14.U.S. Cancer Statistics Working Group . United States Cancer Statistics: 1999–2012 Incidence and Mortality Web-based Report. Atlanta: Department of Health and Human Services, Centers for Disease Control and Prevention, and National Cancer Institute; 2015. [Google Scholar]
- 15.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domchek S, Weber BL. Genetic variants of uncertain significance: flies in the ointment. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:16–17. doi: 10.1200/JCO.2007.14.4154. [DOI] [PubMed] [Google Scholar]
- 17.Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, Deluca AP, Fishman GA, Lam BL, Weleber RG, Cideciyan AV, Jacobson SG, Sheffield VC, Tucker BA, Stone EM. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. 2013;22:5136–5145. doi: 10.1093/hmg/ddt367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castello A, Fischer B, Hentze MW, Preiss T. RNA-binding proteins in Mendelian disease. Trends Genet TIG. 2013;29:318–327. doi: 10.1016/j.tig.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Chatterjee S, Berwal SK, Pal JK. Pathological Mutations in 5' Untranslated Regions of Human Genes. 2001; In: eLS. John Wiley & Sons Ltd; Chichester.
- 20.Halvorsen M, Martin JS, Broadaway S, Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet. 2010;6:e1001074. doi: 10.1371/journal.pgen.1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misquitta CM, Iyer VR, Werstiuk ES, Grover AK. The role of 3’-untranslated region (3’-UTR) mediated mRNA stability in cardiovascular pathophysiology. Mol Cell Biochem. 2001;224:53–67. doi: 10.1023/A:1011982932645. [DOI] [PubMed] [Google Scholar]
- 22.Latchman DS. Transcription-factor mutations and disease. N Engl J Med. 1996;334:28–33. doi: 10.1056/NEJM199601043340108. [DOI] [PubMed] [Google Scholar]
- 23.Ward AJ, Cooper TA. The pathobiology of splicing. J Pathol. 2010;220:152–163. doi: 10.1002/path.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Araujo PR, Yoon K, Ko D, Smith AD, Qiao M, Suresh U, Burns SC, Penalva LOF. Before it gets started: regulating translation at the 5’ UTR. Comp Funct Genomics. 2012;2012:475731. doi: 10.1155/2012/475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cáceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet TIG. 2002;18:186–193. doi: 10.1016/S0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 26.Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengüt S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA, Concannon P. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64:1617–1631. doi: 10.1086/302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ars E, Serra E, García J, Kruyer H, Gaona A, Lázaro C, Estivill X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9:237–247. doi: 10.1093/hmg/9.2.237. [DOI] [PubMed] [Google Scholar]
- 28.Paul DS, Soranzo N, Beck S. Functional interpretation of non-coding sequence variation: Concepts and challenges. Bioessays. 2014;36:191–199. doi: 10.1002/bies.201300126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Y, Jamison DC. The distribution of SNPs in human gene regulatory regions. BMC Genomics. 2005;6:140. doi: 10.1186/1471-2164-6-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horvath A, Pakala SB, Mudvari P, Reddy SDN, Ohshiro K, Casimiro S, Pires R, Fuqua SAW, Toi M, Costa L, Nair SS, Sukumar S, Kumar R. Novel insights into breast cancer genetic variance through RNA sequencing. Sci Rep. 2013;3:2256. doi: 10.1038/srep02256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pavithra L, Rampalli S, Sinha S, Sreenath K, Pestell RG, Chattopadhyay S. Stabilization of SMAR1 mRNA by PGA2 involves a stem loop structure in the 5’ UTR. Nucleic Acids Res. 2007;35:6004–6016. doi: 10.1093/nar/gkm649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pérez-Cabornero L, Infante M, Velasco E, Lastra E, Miner C, Durán M. Evaluating the effect of unclassified variants identified in MMR genes using phenotypic features, bioinformatics prediction, and RNA assays. J Mol Diagn JMD. 2013;15:380–390. doi: 10.1016/j.jmoldx.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Zeng T, Dong Z-F, Liu S-J, Wan R-P, Tang L-J, Liu T, Zhao Q-H, Shi Y-W, Yi Y-H, Liao W-P, Long Y-S. A novel variant in the 3’ UTR of human SCN1A gene from a patient with Dravet syndrome decreases mRNA stability mediated by GAPDH’s binding. Hum Genet. 2014;133:801–811. doi: 10.1007/s00439-014-1422-8. [DOI] [PubMed] [Google Scholar]
- 34.Gaildrat P, Krieger S, Théry J-C, Killian A, Rousselin A, Berthet P, Frébourg T, Hardouin A, Martins A, Tosi M. The BRCA1 c.5434C- > G (p.Pro1812Ala) variant induces a deleterious exon 23 skipping by affecting exonic splicing regulatory elements. J Med Genet. 2010;47:398–403. doi: 10.1136/jmg.2009.074047. [DOI] [PubMed] [Google Scholar]
- 35.Tournier I, Vezain M, Martins A, Charbonnier F, Baert-Desurmont S, Olschwang S, Wang Q, Buisine MP, Soret J, Tazi J, Frébourg T, Tosi M. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mutat. 2008;29:1412–1424. doi: 10.1002/humu.20796. [DOI] [PubMed] [Google Scholar]
- 36.Caminsky NG, Mucaki EJ, Rogan PK. Interpretation of mRNA splicing mutations in genetic disease: review of the literature and guidelines for information-theoretical analysis. F1000Res. 2015;3:282. doi: 10.12688/f1000research.5654.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterlongo P, Catucci I, Colombo M, Caleca L, Mucaki E, Bogliolo M, Marin M, Damiola F, Bernard L, Pensotti V, Volorio S, Dall’Olio V, Meindl A, Bartram C, Sutter C, Surowy H, Sornin V, Dondon M-G, Eon-Marchais S, Stoppa-Lyonnet D, Andrieu N, Sinilnikova OM, Genesis. Mitchell G, James PA, Thompson E, kConFab. Swe-Brca. Marchetti M, Verzeroli C, et al. FANCM c.5791C > T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum Mol Genet. 2015;24:5345–5355. doi: 10.1093/hmg/ddv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mucaki EJ, Shirley BC, Rogan PK. Prediction of Mutant mRNA Splice Isoforms by Information Theory-Based Exon Definition. Hum Mutat. 2013;34:557–565. doi: 10.1002/humu.22277. [DOI] [PubMed] [Google Scholar]
- 39.Olsen RKJ, Brøner S, Sabaratnam R, Doktor TK, Andersen HS, Bruun GH, Gahrn B, Stenbroen V, Olpin SE, Dobbie A, Gregersen N, Andresen BS. The ETFDH c.158A > G variation disrupts the balanced interplay of ESE- and ESS-binding proteins thereby causing missplicing and multiple Acyl-CoA dehydrogenation deficiency. Hum Mutat. 2014;35:86–95. doi: 10.1002/humu.22455. [DOI] [PubMed] [Google Scholar]
- 40.Schneider TD, Stormo GD, Yarus MA, Gold L. Delila system tools. Nucleic Acids Res. 1984;12(1 Pt 1):129–140. doi: 10.1093/nar/12.1Part1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mutat. 1998;12:153–171. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 43.Chen J-M, Férec C, Cooper DN. A systematic analysis of disease-associated variants in the 3’ regulatory regions of human protein-coding genes II: the importance of mRNA secondary structure in assessing the functionality of 3’ UTR variants. Hum Genet. 2006;120:301–333. doi: 10.1007/s00439-006-0218-x. [DOI] [PubMed] [Google Scholar]
- 44.Steen K-A, Siegfried NA, Weeks KM. Selective 2’-hydroxyl acylation analyzed by protection from exoribonuclease (RNase-detected SHAPE) for direct analysis of covalent adducts and of nucleotide flexibility in RNA. Nat Protoc. 2011;6:1683–1694. doi: 10.1038/nprot.2011.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 46.Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, Vaccari EM, Bissonnette J, Booker JK, Cremona ML, Gibellini F, Murphy PD, Pineda-Alvarez DE, Pollevick GD, Xu Z, Richard G, Bale S, Klein RT, Hruska KS, Chung WK: Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med 2015. doi: 10.1038/gim.2015.166. [DOI] [PMC free article] [PubMed]
- 47.Levy-Lahad E, Plon SE. Cancer. A risky business--assessing breast cancer risk. Science. 2003;302:574–575. doi: 10.1126/science.1091465. [DOI] [PubMed] [Google Scholar]
- 48.Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FBL, Hoogerbrugge N, Spurdle AB, Tavtigian SV, IARC Unclassified Genetic Variants Working Group Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, Arason A, Scherneck S, Peto J, Rebbeck TR, Tonin P, Neuhausen S, Barkardottir R, Eyfjord J, Lynch H, Ponder BA, Gayther SA, Zelada-Hedman M. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah PD, Garber JE, Stopfer JE, Powers J, Nathanson KL, Domchek S. Sensitivity of clinical BRCA1 testing compared with linkage analysis. J Clin Oncol. 2012 ASCO Annual Meeting Abstacts. Vol 30, No 15_suppl (May 20 Supplement), 2012; 1506.
- 51.Bakker JL, Thirthagiri E, van Mil SE, Adank MA, Ikeda H, Verheul HMW, Meijers-Heijboer H, de Winter JP, Sharan SK, Waisfisz Q. A novel splice site mutation in the noncoding region of BRCA2: implications for Fanconi anemia and familial breast cancer diagnostics. Hum Mutat. 2014;35:442–446. doi: 10.1002/humu.22505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menéndez M, Castellsagué J, Mirete M, Pros E, Feliubadaló L, Osorio A, Calaf M, Tornero E, Valle J del, Fernández-Rodríguez J, Quiles F, Salinas M, Velasco A, Teulé A, Brunet J, Blanco I, Capellá G, Lázaro C. Assessing the RNA effect of 26 DNA variants in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat. 2011;132:979–92. [DOI] [PubMed]
- 53.Borg A, Haile RW, Malone KE, Capanu M, Diep A, Torngren T, Teraoka S, Begg CB, Thomas DC, Concannon P, Mellemkjaer L, Bernstein L, Tellhed L, Xue S, Olson ER, Liang X, Dolle J, Borresen-Dale AL, Bernstein JL. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: the WECARE study. Hum Mutat. 2010;31:E1200–40. doi: 10.1002/humu.21202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adank MA, Jonker MA, Kluijt I, Mil SE van, Oldenburg RA, Mooi WJ, Hogervorst FBL, Ouweland AMW van den, Gille JJP, Schmidt MK, Vaart AW van der, Meijers-Heijboer H, Waisfisz Q. CHEK2*1100delC homozygosity is associated with a high breast cancer risk in women. J Med Genet. 2011;48:860–3. [DOI] [PubMed]
- 55.Baloch AH, Daud S, Raheem N, Luqman M, Ahmad A, Rehman A, Shuja J, Rasheed S, Ali A, Kakar N, Naseeb HK, Mengal MA, Awan MA, Wasim M, Baloch DM, Ahmad J. Missense mutations (p.H371Y, p.D438Y) in gene CHEK2 are associated with breast cancer risk in women of Balochistan origin. Mol Biol Rep. 2014;41:1103–1107. doi: 10.1007/s11033-013-2956-x. [DOI] [PubMed] [Google Scholar]
- 56.Benusiglio PR, Malka D, Rouleau E, De Pauw A, Buecher B, Noguès C, Fourme E, Colas C, Coulet F, Warcoin M, Grandjouan S, Sezeur A, Laurent-Puig P, Molière D, Tlemsani C, Di Maria M, Byrde V, Delaloge S, Blayau M, Caron O. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J Med Genet. 2013;50:486–489. doi: 10.1136/jmedgenet-2012-101472. [DOI] [PubMed] [Google Scholar]
- 57.Brooks-Wilson AR, Kaurah P, Suriano G, Leach S, Senz J, Grehan N, Butterfield YSN, Jeyes J, Schinas J, Bacani J, Kelsey M, Ferreira P, MacGillivray B, MacLeod P, Micek M, Ford J, Foulkes W, Australie K, Greenberg C, LaPointe M, Gilpin C, Nikkel S, Gilchrist D, Hughes R, Jackson CE, Monaghan KG, Oliveira MJ, Seruca R, Gallinger S, Caldas C, et al. Germline E-cadherin mutations in hereditary diffuse gastric cancer: assessment of 42 new families and review of genetic screening criteria. J Med Genet. 2004;41:508–517. doi: 10.1136/jmg.2004.018275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, Stamatoyannopoulos JA, King M-C. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–2229. doi: 10.1158/0008-5472.CAN-10-3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.CHEK2 Breast Cancer Case-Control Consortium CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004;74:1175–1182. doi: 10.1086/421251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23:276–292. doi: 10.1200/JCO.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 61.Kangelaris KN, Gruber SB. Clinical implications of founder and recurrent CDH1 mutations in hereditary diffuse gastric cancer. JAMA. 2007;297:2410–2411. doi: 10.1001/jama.297.21.2410. [DOI] [PubMed] [Google Scholar]
- 62.Kaurah P, MacMillan A, Boyd N, Senz J, De Luca A, Chun N, Suriano G, Zaor S, Van Manen L, Gilpin C, Nikkel S, Connolly-Wilson M, Weissman S, Rubinstein WS, Sebold C, Greenstein R, Stroop J, Yim D, Panzini B, McKinnon W, Greenblatt M, Wirtzfeld D, Fontaine D, Coit D, Yoon S, Chung D, Lauwers G, Pizzuti A, Vaccaro C, Redal MA, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297:2360–2372. doi: 10.1001/jama.297.21.2360. [DOI] [PubMed] [Google Scholar]
- 63.Kluijt I, Sijmons RH, Hoogerbrugge N, Plukker JT, de Jong D, van Krieken JH, van Hillegersberg R, Ligtenberg M, Bleiker E, Cats A, Dutch Working Group on Hereditary Gastric Cancer Familial gastric cancer: guidelines for diagnosis, treatment and periodic surveillance. Fam Cancer. 2012;11:363–369. doi: 10.1007/s10689-012-9521-y. [DOI] [PubMed] [Google Scholar]
- 64.Martin A-M, Kanetsky PA, Amirimani B, Colligon TA, Athanasiadis G, Shih HA, Gerrero MR, 1089 Calzone K, Rebbeck TR, Weber BL. Germline TP53 mutations in breast cancer families with multiple primary cancers: is TP53 a modifier of BRCA1? J Med Genet. 2003;40:e34–4. [DOI] [PMC free article] [PubMed]
- 65.Masciari S, Larsson N, Senz J, Boyd N, Kaurah P, Kandel MJ, Harris LN, Pinheiro HC, Troussard A, Miron P, Tung N, Oliveira C, Collins L, Schnitt S, Garber JE, Huntsman D. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet. 2007;44:726–731. doi: 10.1136/jmg.2007.051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, Rathbun K, Stopfer JE, Zhu J, Bradbury AR, Simon MS, DeMichele A, Domchek SM, Nathanson KL. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med Off J Am Coll Med Genet. 2015;17:630–638. doi: 10.1038/gim.2014.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Minion LE, Dolinsky JS, Chase DM, Dunlop CL, Chao EC, Monk BJ. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol Oncol. 2015;137:86–92. doi: 10.1016/j.ygyno.2015.01.537. [DOI] [PubMed] [Google Scholar]
- 68.Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- 69.Pharoah PD, Guilford P, Caldas C, International Gastric Cancer Linkage Consortium Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001;121:1348–1353. doi: 10.1053/gast.2001.29611. [DOI] [PubMed] [Google Scholar]
- 70.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Breast Cancer Susceptibility Collaboration (UK) Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Breast Cancer Susceptibility Collaboration (UK), Easton DF, Stratton MR, Rahman N. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. [DOI] [PubMed]
- 72.Sidransky D, Tokino T, Helzlsouer K, Zehnbauer B, Rausch G, Shelton B, Prestigiacomo L, Vogelstein B, Davidson N. Inherited p53 gene mutations in breast cancer. Cancer Res. 1992;52:2984–2986. [PubMed] [Google Scholar]
- 73.Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, Neoptolemos JP, Greenhalf W, Bartsch DK. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78:490–494. doi: 10.1111/j.1399-0004.2010.01425.x. [DOI] [PubMed] [Google Scholar]
- 74.Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 75.Tischkowitz M, Capanu M, Sabbaghian N, Li L, Liang X, Vallée MP, Tavtigian SV, Concannon P, Foulkes WD, Bernstein L, WECARE Study Collaborative Group. Bernstein JL, Begg CB. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat. 2012;33:674–680. doi: 10.1002/humu.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, Foretova L, Soucek P, King M-C. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006;295:1379–1388. doi: 10.1001/jama.295.12.1379. [DOI] [PubMed] [Google Scholar]
- 77.Dorman SN, Shirley BC, Knoll JHM, Rogan PK. Expanding probe repertoire and improving reproducibility in human genomic hybridization. Nucleic Acids Res. 2013;41:e81. doi: 10.1093/nar/gkt048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pinkel D, Landegent J, Collins C, Fuscoe J, Segraves R, Lucas J, Gray J. Fluorescence in situ hybridization with human chromosome-specific libraries: detection of trisomy 21 and translocations of chromosome 4. Proc Natl Acad Sci U S A. 1988;85:9138–9142. doi: 10.1073/pnas.85.23.9138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0. 2013-2015 <http://www.repeatmasker.org>.
- 80.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chou H-H, Hsia A-P, Mooney DL, Schnable PS. Picky: oligo microarray design for large genomes. Bioinforma Oxf Engl. 2004;20:2893–2902. doi: 10.1093/bioinformatics/bth347. [DOI] [PubMed] [Google Scholar]
- 82.Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol Clifton NJ. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 83.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Predictive Cancer Genetics Steering Committee. Ontario physicians’ guide to referral of patients with family history of cancer to a familial cancer genetics clinic or genetics clinic. Ont Med Rev 2001, 68:24–30.
- 85.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Philippe N, Salson M, Commes T, Rivals E. CRAC: an integrated approach to the analysis of RNA-seq reads. Genome Biol. 2013;14:R30. doi: 10.1186/gb-2013-14-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Picard [http://broadinstitute.github.io/picard/]. Accessed 1 June 2015.
- 88.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shirley BC, Mucaki EJ, Whitehead T, Costea PI, Akan P, Rogan PK. Interpretation, stratification and evidence for sequence variants affecting mRNA splicing in complete human genome sequences. Genomics Proteomics Bioinformatics. 2013;11:77–85. doi: 10.1016/j.gpb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mutation Forecaster [https://www.mutationforecaster.com/index.php]. Accessed 1 June 2015.
- 93.Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 94.Dhir A, Buratti E. Alternative splicing: role of pseudoexons in human disease and potential therapeutic strategies. FEBS J. 2010;277:841–855. doi: 10.1111/j.1742-4658.2009.07520.x. [DOI] [PubMed] [Google Scholar]
- 95.Tavanez JP, Madl T, Kooshapur H, Sattler M, Valcárcel J. hnRNP A1 proofreads 3’ splice site recognition by U2AF. Mol Cell. 2012;45:314–329. doi: 10.1016/j.molcel.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 96.Paradis C, Cloutier P, Shkreta L, Toutant J, Klarskov K, Chabot B. hnRNP I/PTB can antagonize the splicing repressor activity of SRp30c. RNA N Y N. 2007;13:1287–1300. doi: 10.1261/rna.403607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boggs K, Reisman D. Increased p53 transcription prior to DNA synthesis is regulated through a novel regulatory element within the p53 promoter. Oncogene. 2005;25:555–565. doi: 10.1038/sj.onc.1209076. [DOI] [PubMed] [Google Scholar]
- 99.Chen Y, Xu J, Borowicz S, Collins C, Huo D, Olopade OI. c-Myc activates BRCA1 gene expression through distal promoter elements in breast cancer cells. BMC Cancer. 2011;11:246. doi: 10.1186/1471-2407-11-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gueven N, Keating K, Fukao T, Loeffler H, Kondo N, Rodemann HP, Lavin MF. Site-directed mutagenesis of the ATM promoter: Consequences for response to proliferation and ionizing radiation. Genes Chromosomes Cancer. 2003;38:157–167. doi: 10.1002/gcc.10261. [DOI] [PubMed] [Google Scholar]
- 101.Frietze S, Wang R, Yao L, Tak YG, Ye Z, Gaddis M, Witt H, Farnham PJ, Jin VX. Cell type-specific binding patterns reveal that TCF7L2 can be tethered to the genome by association with GATA3. Genome Biol. 2012;13:R52. doi: 10.1186/gb-2012-13-9-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Connor AE, Baumgartner RN, Baumgartner KB, Kerber RA, Pinkston C, John EM, Torres-Mejia G, Hines L, Giuliano A, Wolff RK, Slattery ML. Associations between TCF7L2 polymorphisms and risk of breast cancer among Hispanic and non-Hispanic white women: the Breast Cancer Health Disparities Study. Breast Cancer Res Treat. 2012;136:593–602. doi: 10.1007/s10549-012-2299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Burwinkel B, Shanmugam KS, Hemminki K, Meindl A, Schmutzler RK, Sutter C, Wappenschmidt B, Kiechle M, Bartram CR, Frank B. Transcription factor 7-like 2 (TCF7L2) variant is associated with familial breast cancer risk: a case-control study. BMC Cancer. 2006;6:268. doi: 10.1186/1471-2407-6-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen J, Yuan T, Liu M, Chen P. Association between TCF7L2 Gene Polymorphism and Cancer Risk: A Meta-Analysis. PLoS One. 2013;8:e71730. doi: 10.1371/journal.pone.0071730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Purrington KS, Slager S, Eccles D, Yannoukakos D, Fasching PA, Miron P, Carpenter J, Chang-Claude J, Martin NG, Montgomery GW, Kristensen V, Anton-Culver H, Goodfellow P, Tapper WJ, Rafiq S, Gerty SM, Durcan L, Konstantopoulou I, Fostira F, Vratimos A, Apostolou P, Konstanta I, Kotoula V, Lakis S, Dimopoulos MA, Skarlos D, Pectasides D, Fountzilas G, Beckmann MW, Hein A, et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis. 2014;35:1012–1019. doi: 10.1093/carcin/bgt404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bi C, Rogan PK. Bipartite pattern discovery by entropy minimization-based multiple local alignment. Nucleic Acids Res. 2004;32:4979–4991. doi: 10.1093/nar/gkh825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, Rando OJ, Birney E, Myers RM, Noble WS, Snyder M, Weng Z. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet TIG. 1997;13:163. doi: 10.1016/S0168-9525(97)01103-7. [DOI] [PubMed] [Google Scholar]
- 109.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39(Database issue):D301–8. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, Na H, Irimia M, Matzat LH, Dale RK, Smith SA, Yarosh CA, Kelly SM, Nabet B, Mecenas D, Li W, Laishram RS, Qiao M, Lipshitz HD, Piano F, Corbett AH, Carstens RP, Frey BJ, Anderson RA, Lynch KW, Penalva LOF, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, Zheng H, Goity A, van Bakel H, Lozano J-C, Galli M, Lewsey MG, Huang E, Mukherjee T, Chen X, Reece-Hoyes JS, Govindarajan S, Shaulsky G, Walhout AJM, Bouget F-Y, Ratsch G, Larrondo LF, Ecker JR, Hughes TR. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dayem Ullah AZ, Lemoine NR, Chelala C. SNPnexus: a web server for functional annotation of novel and publicly known genetic variants (2012 update) Nucleic Acids Res. 2012;40:W65–W70. doi: 10.1093/nar/gks364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dayem Ullah AZ, Lemoine NR, Chelala C. A practical guide for the functional annotation of genetic variations using SNPnexus. Brief Bioinform. 2013;14:437–447. doi: 10.1093/bib/bbt004. [DOI] [PubMed] [Google Scholar]
- 114.Chelala C, Khan A, Lemoine NR. SNPnexus: a web database for functional annotation of newly discovered and public domain single nucleotide polymorphisms. Bioinformatics. 2009;25:655–661. doi: 10.1093/bioinformatics/btn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.dbSNP [http://www.ncbi.nlm.nih.gov/SNP/]. Accessed 1 June 2015.
- 116.Exome Variant Server [http://evs.gs.washington.edu/EVS/]. Accessed 1 June 2015.
- 117.1000Genomes [http://www.1000genomes.org/]. Accessed 1 June 2015.
- 118.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Choi Y. Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine. New York: ACM; 2012. A Fast Computation of Pairwise Sequence Alignment Scores Between a Protein and a Set of Single-locus Variants of Another Protein; pp. 414–417. [Google Scholar]
- 122.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.ClinVar [http://www.ncbi.nlm.nih.gov/clinvar/]. Accessed 1 June 2015.
- 124.Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42: D980-985. [DOI] [PMC free article] [PubMed]
- 125.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, Abeysinghe S, Krawczak M, Cooper DN. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 126.Human Gene Mutation Database (HGMD) [http://hgmd/cf/ac/uk/ac/index.php]. Accessed 1 June 2015.
- 127.Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. LOVD v. 2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- 128.Leiden Open Variation Database (LOVD) - Ataxia Telangiectasia Mutated (ATM) [http://chromium.lovd.nl/LOVD2/variants.php?action=search_unique&select_db=ATM]. Accessed 1 June 2015.
- 129.LOVD - IARC Breast Cancer Type 1 susceptibility protein (BRCA1) [http://brca.iarc.fr/LOVD/variants.php?action=view_unique&select_db=BRCA1]. Accessed 1 June 2015.
- 130.LOVD - IARC Breast Cancer Type 2 susceptibility protein (BRCA2) [http://brca.iarc.fr/LOVD/variants.php?action=view_unique&select_db=BRCA2]. Accessed 1 June 2015.
- 131.LOVD - Leiden Open Variation Database Partner and localizer of BRCA2 (FANCN) (PALB2) [https://grenada.lumc.nl/LOVD2/shared1/variants.php?action=search_unique&select_db=PALB2]. Accessed 1 June 2015.
- 132.LOVD - Leiden Open Variation Database tumour protein p53 (TP53) [http://proteomics.bio21.unimelb.edu.au/lovd/variants/TP53]. Accessed 1 June 2015.
- 133.Zhejiang University Center for Genetic and Genomic Medicine (ZJU-CGGM) cadherin 1, type 1, E-cadherin (epithelial) (CDH1) [http://www.genomed.org/lovd2/variants.php?action=search_unique&select_db=CDH1]. Accessed 1 June 2015.
- 134.Zhejiang University Center for Genetic and Genomic Medicine (ZJU-CGGM) checkpoint kinase 2 (CHEK2) [http://www.genomed.org/lovd2/variants.php?action=search_unique&select_db=CHEK2]. Accessed 1 June 2015.
- 135.Domain Mapping of Disease Mutations (DM2) [http://bioinf.umbc.edu/dmdm]. Accessed 1 June 2015.
- 136.Expert Protein Analysis System (ExPASy) [http://www.expasy.org/]. Accessed 1 June 2015.
- 137.The UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.UniProt [http://uniprot.org/]. Accessed 1 June 2015.
- 139.Breast Cancer Information Core (BIC) Database [https://research.nhgri.nih.gov/projects/bic/Member/index/shtml]. Accessed 1 June 2015.
- 140.Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) [http://enigmaconsortium.org/]. Accessed 1 June 2015.
- 141.International Agency for Research on Cancer (IARC) TP53 Database [http://p53.iarc.fr/tp53genevariations.aspx]. Accessed 1 June 2015.
- 142.Ozcelik H, Knight JA, Glendon G, Yazici H, Carson N, Ainsworth PJ, Taylor S a M, Feilotter H, Carter RF, Boyd NF, Andrulis IL, Ontario Cancer Genetics Network Individual and family characteristics associated with protein truncating BRCA1 and BRCA2 mutations in an Ontario population based series from the Cooperative Family Registry for Breast Cancer Studies. J Med Genet. 2003;40:e91. doi: 10.1136/jmg.40.8.e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Maier C, Herkommer K, Luedeke M, Rinckleb A, Schrader M, Vogel W. Subgroups of familial and aggressive prostate cancer with considerable frequencies of BRCA2 mutations. Prostate. 2014;74:1444–1451. doi: 10.1002/pros.22860. [DOI] [PubMed] [Google Scholar]
- 144.Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, Shi X, Fulton RS, Ley TJ, Wilson RK, Ding L, Mardis ER. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gibbs RA, Belmont JW, Hardenbol P, Willis TD, Yu F, Yang H, Ch’ang L-Y, Huang W, Liu B, Shen Y, Tam PK-H, Tsui L-C, Waye MMY, Wong JT-F, Zeng C, Zhang Q, Chee MS, Galver LM, Kruglyak S, Murray SS, Oliphant AR, Montpetit A, Hudson TJ, Chagnon F, Ferretti V, Leboeuf M, Phillips MS, Verner A, Kwok P-Y, Duan S, et al. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 146.McIver LJ, Fondon JW, III, Skinner MA, Garner HR. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics. 2011;97:193–199. doi: 10.1016/j.ygeno.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tae H, Kim D-Y, McCormick J, Settlage RE, Garner HR. Discretized Gaussian mixture for genotyping of microsatellite loci containing homopolymer runs. Bioinformatics. 2014;30:652–659. doi: 10.1093/bioinformatics/btt595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Castéra L, Krieger S, Rousselin A, Legros A, Baumann J-J, Bruet O, Brault B, Fouillet R, Goardon N, Letac O, Baert-Desurmont S, Tinat J, Bera O, Dugast C, Berthet P, Polycarpe F, Layet V, Hardouin A, Frébourg T, Vaur D. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet EJHG. 2014;22:1305–1313. doi: 10.1038/ejhg.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Austen B, Barone G, Reiman A, Byrd PJ, Baker C, Starczynski J, Nobbs MC, Murphy RP, Enright H, Chaila E, Quinn J, Stankovic T, Pratt G, Taylor AMR. Pathogenic ATM mutations occur rarely in a subset of multiple myeloma patients. Br J Haematol. 2008;142:925–933. doi: 10.1111/j.1365-2141.2008.07281.x. [DOI] [PubMed] [Google Scholar]
- 150.Ding H, Mao C, Li S-M, Liu Q, Lin L, Chen Q. Lack of association between ATM C.1066-6T > G mutation and breast cancer risk: a meta-analysis of 8,831 cases and 4,957 controls. Breast Cancer Res Treat. 2011;125:473–477. doi: 10.1007/s10549-010-0977-x. [DOI] [PubMed] [Google Scholar]
- 151.Chen J, Guo K, Kastan MB. Interactions of nucleolin and ribosomal protein L26 (RPL26) in translational control of human p53 mRNA. J Biol Chem. 2012;287:16467–16476. doi: 10.1074/jbc.M112.349274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Champion-Arnaud P, Reed R. The prespliceosome components SAP 49 and SAP 145 interact in a complex implicated in tethering U2 snRNP to the branch site. Genes Dev. 1994;8:1974–83. [DOI] [PubMed]
- 153.Li YI, Sanchez-Pulido L, Haerty W, Ponting CP. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015;25:1–13. doi: 10.1101/gr.181990.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dobričić J, Krivokuća A, Brotto K, Mališić E, Radulović S, Branković-Magić M. Serbian high-risk families: extensive results on BRCA mutation spectra and frequency. J Hum Genet. 2013;58:501–507. doi: 10.1038/jhg.2013.30. [DOI] [PubMed] [Google Scholar]
- 155.Nelson AC, Holt JT. Impact of RING and BRCT domain mutations on BRCA1 protein stability, localization and recruitment to DNA damage. Radiat Res. 2010;174:1–13. doi: 10.1667/RR1290.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Clark SL, Rodriguez AM, Snyder RR, Hankins GDV, Boehning D: Structure-Function Of The Tumor Suppressor BRCA1. Comput Struct Biotechnol J 2012, 1. [DOI] [PMC free article] [PubMed]
- 157.Leung CCY, Glover JNM. BRCT domains: easy as one, two, three. Cell Cycle Georget Tex. 2011;10:2461–2470. doi: 10.4161/cc.10.15.16312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Håkansson S, Johannsson O, Johansson U, Sellberg G, Loman N, Gerdes AM, Holmberg E, Dahl N, Pandis N, Kristoffersson U, Olsson H, Borg A. Moderate frequency of BRCA1 and BRCA2 germ-line mutations in Scandinavian familial breast cancer. Am J Hum Genet. 1997;60:1068–1078. [PMC free article] [PubMed] [Google Scholar]
- 159.Scottish/Northern Irish BRCAI/BRCA2 Consortium BRCA1 and BRCA2 mutations in Scotland and Northern Ireland. Br J Cancer. 2003;88:1256–1262. doi: 10.1038/sj.bjc.6600840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson J-Y, Moses R, West SC, de Winter JP, Ashworth A, Jones NJ, Mathew CG. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 2004;13:1241–1248. doi: 10.1093/hmg/ddh135. [DOI] [PubMed] [Google Scholar]
- 161.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 162.Oliveira C, Senz J, Kaurah P, Pinheiro H, Sanges R, Haegert A, Corso G, Schouten J, Fitzgerald R, Vogelsang H, Keller G, Dwerryhouse S, Grimmer D, Chin S-F, Yang H-K, Jackson CE, Seruca R, Roviello F, Stupka E, Caldas C, Huntsman D. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum Mol Genet. 2009;18:1545–1555. doi: 10.1093/hmg/ddp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Platinum Genomes [http://www.illumina.com/platinumgenomes/]. Accessed 31 July 2015.
- 166.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tavtigian SV, Greenblatt MS, Goldgar DE, Boffetta P, IARC Unclassified Genetic Variants Working Group Assessing pathogenicity: overview of results from the IARC Unclassified Genetic Variants Working Group. Hum Mutat. 2008;29:1261–1264. doi: 10.1002/humu.20903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro ANA, Iversen ES, Couch FJ, Goldgar DE. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Viner C, Dorman SN, Shirley BC, Rogan PK. Validation of predicted mRNA splicing mutations using high-throughput transcriptome data. F1000Res. 2014;3:8. doi: 10.12688/f1000research.3-8.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carleton KL. Quantification of transcript levels with quantitative RT-PCR. Methods Mol Biol Clifton NJ. 2011;772:279–295. doi: 10.1007/978-1-61779-228-1_17. [DOI] [PubMed] [Google Scholar]
- 171.ENIGMA BRCA1/2 Gene Variant Classification Criteria, v1.1 [http://enigmaconsortium.org/documents/ENIGMA_Rules_2015-03-26.pdf]. Accessed 1 June 2015.
- 172.Deveci M, Catalyürek UV, Toland AE. mrSNP: software to detect SNP effects on microRNA binding. BMC Bioinformatics. 2014;15:73. doi: 10.1186/1471-2105-15-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos I-L, Maniou S, Karathanou K, Kalfakakou D, Fevgas A, Dalamagas T, Hatzigeorgiou AG. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43(Database issue):D153–159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mucaki EJ, Ainsworth P, Rogan PK. Comprehensive prediction of mRNA splicing effects of BRCA1 and BRCA2 variants. Hum Mutat. 2011;32:735–742. doi: 10.1002/humu.21513. [DOI] [PubMed] [Google Scholar]
- 175.Bisio A, Nasti S, Jordan JJ, Gargiulo S, Pastorino L, Provenzani A, Quattrone A, Queirolo P, Bianchi-Scarrà G, Ghiorzo P, Inga A. Functional analysis of CDKN2A/p16INK4a 5’-UTR variants predisposing to melanoma. Hum Mol Genet. 2010;19:1479–1491. doi: 10.1093/hmg/ddq022. [DOI] [PubMed] [Google Scholar]
- 176.Berry JA, Cervantes-Sandoval I, Nicholas EP, Davis RL. Dopamine is required for learning and forgetting in Drosophila. Neuron. 2012;74:530–542. doi: 10.1016/j.neuron.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sribudiani Y, Metzger M, Osinga J, Rey A, Burns AJ, Thapar N, Hofstra RMW. Variants in RET associated with Hirschsprung’s disease affect binding of transcription factors and gene expression. Gastroenterology. 2011;140:572–582.e2. doi: 10.1053/j.gastro.2010.10.044. [DOI] [PubMed] [Google Scholar]
- 178.Knebelmann B, Forestier L, Drouot L, Quinones S, Chuet C, Benessy F, Saus J, Antignac C. Splice-mediated insertion of an Alu sequence in the COL4A3 mRNA causing autosomal recessive Alport syndrome. Hum Mol Genet. 1995;4:675–679. doi: 10.1093/hmg/4.4.675. [DOI] [PubMed] [Google Scholar]
- 179.Wallace MR, Andersen LB, Saulino AM, Gregory PE, Glover TW, Collins FS. A de novo Alu insertion results in neurofibromatosis type 1. Nature. 1991;353:864–866. doi: 10.1038/353864a0. [DOI] [PubMed] [Google Scholar]
- 180.Wiestner A, Tehrani M, Chiorazzi M, Wright G, Gibellini F, Nakayama K, Liu H, Rosenwald A, Muller-Hermelink HK, Ott G, Chan WC, Greiner TC, Weisenburger DD, Vose J, Armitage JO, Gascoyne RD, Connors JM, Campo E, Montserrat E, Bosch F, Smeland EB, Kvaloy S, Holte H, Delabie J, Fisher RI, Grogan TM, Miller TP, Wilson WH, Jaffe ES, Staudt LM. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood. 2007;109:4599–4606. doi: 10.1182/blood-2006-08-039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lévesque É, Bélanger A-S, Harvey M, Couture F, Jonker D, Innocenti F, Cecchin E, Toffoli G, Guillemette C. Refining the UGT1A Haplotype Associated with Irinotecan-Induced Hematological Toxicity in Metastatic Colorectal Cancer Patients Treated with 5-Fluorouracil/Irinotecan-Based Regimens. J Pharmacol Exp Ther. 2013;345:95–101. doi: 10.1124/jpet.112.202242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fujiwara Y, Minami H. An overview of the recent progress in irinotecan pharmacogenetics. Pharmacogenomics. 2010;11:391–406. doi: 10.2217/pgs.10.19. [DOI] [PubMed] [Google Scholar]
- 183.Palomaki GE, Bradley LA, Douglas MP, Kolor K, Dotson WD. Can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? An evidence-based review. Genet Med. 2009;11:21–34. doi: 10.1097/GIM.0b013e31818efd77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Biesecker LG. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: Lessons from the ClinSeqTM project. Genet Med Off J Am Coll Med Genet. 2012;14:393–398. doi: 10.1038/gim.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schneider TD. Information content of individual genetic sequences. J Theor Biol. 1997;189:427–441. doi: 10.1006/jtbi.1997.0540. [DOI] [PubMed] [Google Scholar]
- 186.Bonnet-Dupeyron M-N, Combes P, Santander P, Cailloux F, Boespflug-Tanguy O, Vaurs-Barrière C. PLP1 splicing abnormalities identified in Pelizaeus-Merzbacher disease and SPG2 fibroblasts are associated with different types of mutations. Hum Mutat. 2008;29:1028–1036. doi: 10.1002/humu.20758. [DOI] [PubMed] [Google Scholar]
- 187.Fei J. Splice site mutation-induced alteration of selective Regional activity correlates with the role of a gene in cardiomyopathy. J Clin Exp Cardiol. 2013;S12:004. [Google Scholar]
- 188.Khan SG, Metin A, Gozukara E, Inui H, Shahlavi T, Muniz-Medina V, Baker CC, Ueda T, Aiken JR, Schneider TD, Kraemer KH. Two essential splice lariat branchpoint sequences in one intron in a xeroderma pigmentosum DNA repair gene: mutations result in reduced XPC mRNA levels that correlate with cancer risk. Hum Mol Genet. 2004;13:343–352. doi: 10.1093/hmg/ddh026. [DOI] [PubMed] [Google Scholar]
- 189.von Kodolitsch Y, Berger J, Rogan PK. Predicting severity of haemophilia A and B splicing mutations by information analysis. Haemoph Off J World Fed Hemoph. 2006;12:258–262. doi: 10.1111/j.1365-2516.2006.01216.x. [DOI] [PubMed] [Google Scholar]
- 190.Martoni E, Urciuolo A, Sabatelli P, Fabris M, Bovolenta M, Neri M, Grumati P, D’Amico A, Pane M, Mercuri E, Bertini E, Merlini L, Bonaldo P, Ferlini A, Gualandi F. Identification and characterization of novel collagen VI non-canonical splicing mutations causing Ullrich congenital muscular dystrophy. Hum Mutat. 2009;30:E662–672. doi: 10.1002/humu.21022. [DOI] [PubMed] [Google Scholar]
- 191.Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, Lord GM, Gibbs JSR, Wilkins MR, Ohta-Ogo K, Nakamura K, Girerd B, Coulet F, Soubrier F, Humbert M, Morrell NW, Trembath RC, Machado RDl. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32:1385–9. [DOI] [PubMed]
- 192.Pink AE, Simpson MA, Desai N, Dafou D, Hills A, Mortimer P, Smith CH, Trembath RC, Barker JNW. Mutations in the γ-secretase genes NCSTN, PSENEN, and PSEN1 underlie rare forms of hidradenitis suppurativa (acne inversa) J Invest Dermatol. 2012;132:2459–2461. doi: 10.1038/jid.2012.162. [DOI] [PubMed] [Google Scholar]
- 193.Sanders DA, Ross-Innes CS, Beraldi D, Carroll JS, Balasubramanian S. Genome-wide mapping of FOXM1 binding reveals co-binding with estrogen receptor alpha in breast cancer cells. Genome Biol. 2013;14:R6. doi: 10.1186/gb-2013-14-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Suga Y, Tsuda T, Nagai M, Sakaguchi Y, Jitsukawa O, Yamamoto M, Hitomi K, Yamanishi K. Lamellar ichthyosis with pseudoexon activation in the transglutaminase 1 gene. J Dermatol. 2015;42:642–645. doi: 10.1111/1346-8138.12846. [DOI] [PubMed] [Google Scholar]
- 195.Rogan PK, Svojanovsky S, Leeder JS. Information theory-based analysis of CYP2C19, CYP2D6 and CYP3A5 splicing mutations. Pharmacogenetics. 2003;13:207–218. doi: 10.1097/00008571-200304000-00005. [DOI] [PubMed] [Google Scholar]
- 196.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler and D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. [DOI] [PMC free article] [PubMed]
- 197.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32(Database issue):D23–26. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rogan P, Mucaki E: Population Fitness and Genetic Load of Single Nucleotide Polymorphisms Affecting mRNA splicing. ArXiv11070716 Q-Bio 2011.
- 199.Di L-J, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010;17:1406–1413. doi: 10.1038/nsmb.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wu X, Webster SR, Chen J. Characterization of tumor-associated Chk2 mutations. J Biol Chem. 2001;276:2971–2974. doi: 10.1074/jbc.M009727200. [DOI] [PubMed] [Google Scholar]
- 201.Durocher D, Henckel J, Fersht AR, Jackson SP. The FHA domain is a modular phosphopeptide recognition motif. Mol Cell. 1999;4:387–394. doi: 10.1016/S1097-2765(00)80340-8. [DOI] [PubMed] [Google Scholar]
- 202.Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286:2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- 203.Varley JM, Evans DG, Birch JM. Li-Fraumeni syndrome--a molecular and clinical review. Br J Cancer. 1997;76:1–14. doi: 10.1038/bjc.1997.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lee SB, Kim SH, Bell DW, Wahrer DC, Schiripo TA, Jorczak MM, Sgroi DC, Garber JE, Li FP, Nichols KE, Varley JM, Godwin AK, Shannon KM, Harlow E, Haber DA. Destabilization of CHK2 by a missense mutation associated with Li-Fraumeni Syndrome. Cancer Res. 2001;61:8062–8067. [PubMed] [Google Scholar]
- 205.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, Iglehart JD. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101:10137–10142. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Antoniou AC, Easton DF. Models of genetic susceptibility to breast cancer. Oncogene. 2006;25:5898–5905. doi: 10.1038/sj.onc.1209879. [DOI] [PubMed] [Google Scholar]
- 207.Peto J. Breast cancer susceptibility—A new look at an old model. Cancer Cell. 2002;1:411–412. doi: 10.1016/S1535-6108(02)00079-X. [DOI] [PubMed] [Google Scholar]
- 208.Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32:2001–2009. doi: 10.1200/JCO.2013.53.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kassahn KS, Scott HS, Caramins MC. Integrating massively parallel sequencing into diagnostic workflows and managing the annotation and clinical interpretation challenge. Hum Mutat. 2014;35:413–423. doi: 10.1002/humu.22525. [DOI] [PubMed] [Google Scholar]
- 210.Li M-X, Gui H-S, Kwan JSH, Bao S-Y, Sham PC. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012;40:e53. doi: 10.1093/nar/gkr1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kluska A, Balabas A, Paziewska A, Kulecka M, Nowakowska D, Mikula M, Ostrowski J. New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC Med Genomics. 2015;8:19. doi: 10.1186/s12920-015-0092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Caminsky NG, Mucaki EJ, Perri AM, Lu R, Knoll JHM, Rogan PK. Prioritizing variants in complete Hereditary Breast and Ovarian Cancer (HBOC) genes in patients lacking known BRCA mutations. Hum Mutat. 2016; [published online ahead of print March 18, 2016]. doi:10.1002/humu.22972. [DOI] [PubMed]