

Abstract
Modifications to nucleotides in the genome can lead to mutations or are involved in regulation of gene expression, and therefore, finding the site of modification is a worthy goal. Robust methods for sequencing modification sites on commercial sequencers have not been developed beyond the epigenetic marks on cytosine. Herein, a method to sequence DNA modification sites was developed that utilizes DNA glycosylases found in the base excision repair pathway to excise the modification. This approach yields a gap at the modification site that is sealed by T4-DNA ligase yielding a product strand missing the modification. Upon sequencing, the modified nucleotide is reported as a deletion mutation, identifying its location. This approach was used to detect a uracil (U) or 8-oxo-7,8-dihydroguanine (OG) in codon 12 of the KRAS gene in synthetic oligodeoxynucleotides. Additionally, an OG modification site was placed in the VEGF promoter in a plasmid and sequenced. This method requires only commercially available materials and can be put into practice on any sequencing platform, allowing this method to have broad potential for finding modifications in DNA.
Graphical Abstract
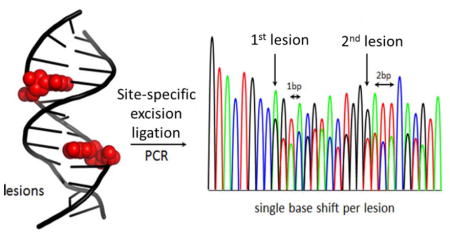
A diversity of chemical modifications to the genome has been identified, some of which play critical roles in epigenetic regulation of genes,1–3 while others cause mutations leading to diseases, such as cancer.4,5 Detection of a modified base in genomic DNA presents a significant challenge for currently available sequencing platforms. For the cytosine epigenetic markers, site-specific chemistry with bisulfite leading to an observable signal in sequencing experiments has been developed;2,6 however, this approach is not general and is not readily applicable to other modifications on other bases. Direct detection of modifications has been achieved using single-molecule real time (SMRT) sequencing technology7,8 or nanopore sequencing.9–14 These single-molecule methods and others need relatively large sample sizes to obtain suitable results. Therefore, amplification of genomic samples by PCR is typically required to significantly increase the amount of DNA for sequencing. One drawback to PCR amplification is the inability to retain the modification’s identity and location during the amplification step, due to replacement of the modified nucleotide by a standard nucleotide. To overcome this limitation, attempts for introduction of a marker nucleotide at the modification site have been pursued.15
One mutagenic modification found in the genome is the abasic site (AP). Methods have been developed in the Kool and Berdis laboratories for labeling AP with non-natural marker nucleotides during polymerase extension past the damage.16,17 This approach showed difficulty in extension past the site at which the marker nucleotide was inserted opposite an AP. The Sturla laboratory has extended this method by selectively incorporating a marker nucleotide opposite the alkylated lesion O6-benzylguanine followed by linear PCR amplification to generate a pool of labeled amplicons.18,19 We recently reported a method for introduction of a marker nucleotide at the site of lesions that are substrates for the base excision repair pathway.15 Our approach allowed exponential PCR amplification of the marker while retaining its location because it utilized the dNaM•d5SICS unnatural base pair developed by the Romesberg laboratory.20–23 In this method, one modified site was able to be sequenced using Sanger sequencing by observing an abrupt stop in the sequencing chromatogram at the marker site. Additionally, we were able to recognize more than one modification using a nanopore method, and recently, single nucleotide resolution sequencing for dNaM and d5SICS was demonstrated.24 Currently, other high throughput methods for sequencing this unnatural base pair have not been developed. Therefore, we sought an alternative method for sequencing modified sites that is readily adaptable to all currently available sequencing platforms.
This new method utilizes the selectivity of the base excision repair pathway to remove the modification to yield a gap in the DNA. Next, the gap is ligated to yield a new strand that is one or more nucleotides shorter than the reactant strand, in which the nucleotide missing is the modification. Development of this alternative method was achieved using the duplex DNA strands found in Table 1. These sequences centrally contain 30 nucleotides surrounding codon 12 of the KRAS gene that was flanked by two primer sequences allowing PCR amplification. In the center of codon 12 a uracil (U) was synthetically incorporated, representing the product of cytosine deamination, at the naturally occurring cytosine site (KRAS-U, Table S1). Also, an 8-oxo-7,8-dihydroguanine (OG), a guanine oxidation product, was incorporated at the naturally occurring G site (Supporting Information) for the method development.4,25
These lesions both cause mutations that have been observed at codon 12 of the KRAS gene in colon, breast, lung, and brain cancers.25 Further, the complementary strand contains 5′ and 3′ overhangs that were terminated with triethylene-glycol blocks that assured postlabeling with 32P only occurred on the strand to be analyzed by polyacrylamide gel electrophoresis (PAGE). These blocks also prevent interstrand ligation. Next, each step of this method was optimized to achieve the highest possible overall yields.
Treating the U-containing duplex (KRAS-U) with uracil-DNA glycosylase (UDG), a monofunctional base excision repair enzyme,26 yields an intact AP product (Figure 1A). To determine the yield of the reaction, an aliquot was treated with NaOH (200 mM) to affect strand scission followed by PAGE analysis (Figure 1b). This analysis determined the yield of the UDG reaction to be >99% (Figure 1c). The AP site was further reacted upon with APE1 to yield a gap at the modification site with ends (i.e., 3′-OH and 5′-phosphate) that are compatible with ligation (Figure 1a). The yield of this reaction was monitored by PAGE and found to be >99% (Figure 1b and c).
Figure 1.

Gap ligation approach for sequencing a single uracil lesion in the KRAS sequence. (a) The five-step scheme comprising excision, amplification, and Sanger sequencing of a lesion. (b) PAGE analysis monitoring and quantifying enzyme-catalyzed labeling reactions on the KRAS-U duplex. Reactions were monitored by 32P labeling of the lesion-containing strand, and the gels were quantified by phos-phorimager autoradiography. (c) Percent yield for each enzymatic step.
Lastly, the single-nucleotide gap was directly ligated with T4-DNA ligase to furnish two strands that differ in length by one nucleotide (Figure 1a). The shorter strand is the one with the modification removed, and the longer one is the complementary strand. The optimal reaction conditions for ligation were investigated, but the yield based on PAGE analysis could not be increased. The overall yield of this gap ligation labeling method was ~50% (Figure 1c). These steps were repeated with OG in the strand (Table S1) using Fpg as the DNA glycosylase for its removal. This additional demonstration gave similar yields as observed for gap ligating at a U lesion (Figure S6).
Because the newly synthesized strands only contain native DNA nucleotides, they were directly submitted to PCR amplification via standard protocols. Amplification of the sample yields two product amplicons that differ in length by one nucleotide (Figure 1a). Sanger sequencing was chosen for analysis because it has a much lower error rate (0.01%) compared to next-generation methods that offer high throughput but incur 10–1000-fold higher error rates.27 Upon sequencing, a unique and easily identifiable feature was observed that started at the modification site. The two different sequence lengths in solution caused two sequence readouts to be observed that were out of register by one nucleotide starting at the location of the modification (Figure 1a, bottom). This effectively caused a doubling of the sequencing peaks starting at the modification site that was easily identified.
Inspired by this positive result, studies were then conducted to determine if this method could identify more than one modification site per strand. The first test was conducted on KRAS-U-11 with two U lesions separated by nine nucleotides. Assays identified UDG could remove both Us in >99% yield; further, both sites were processed by APE1 to provide the desired gapped site in >99% yield (Figures 2a–c). Ligation of the two gap sites was again conducted with T4-DNA ligase to furnish full length product in ~45% yield (Figures 2b and c). The overall yield of this reaction was ~44% (Figure 2c). Submission of the processed KRAS-U-11 to Sanger sequencing could quickly identify the first site of U removal based on doubling of the sequencing peaks. The second U that was 9 nucleotides away was identified when the sequencing peaks became out of register by 2 nucleotides (Figure 2a). Ligation of the single-nucleotide gaps with a 9-mer duplex between them yielded the expected result. On the basis of this result, the position of two modifications in DNA within nearly one turn of the helix can be detected and sequenced.
Figure 2.

Gap ligation approach for sequencing two uracil lesions in the KRAS sequence positioned 9 nt apart. (a) The five-step scheme comprising excision, amplification, and Sanger sequencing of a lesion. (b) PAGE analysis monitoring and quantifying enzyme-catalyzed labeling reactions on the KRAS-U duplex with a U•G base pair. Reactions were monitored by 32P labeling of the lesion-containing strand, and the gels were quantified by phosphorimager autoradiography. (c) The percent yield for each enzymatic step.
In the next analysis, Us were placed closer than one turn of the helix to determine the threshold for modification spacing when sequencing via this method. These studies were conducted with the Us spaced 5 or 7 nucleotides apart (Figure S1 and S3). The Us were readily processed by UDG and APE1 for both U configurations, on the basis of gel analysis (Figure S4); however, when Sanger sequencing was performed following ligation, a new result was observed. When the Us were 5 nucleotides apart the sequencing chromatogram identified peaks out of register by 7 nucleotides (2 for the Us and 5 for the strand between), indicating that a 5-mer duplex was not stable and readily dissociates from the duplex (Figure S5a). In this case, the gap being ligated created a 7-nt bulge on the opposite strand. In contrast, when the Us were 7 nucleotides apart, two results were observed--one in which the 7-mer duplex remained intact giving the desired result, and the second was loss of the 7-mer duplex leading to sequencing peaks out of register by 9 nucleotides (Figures S4b and S5b). These observations identify the ability to sequence closely space lesions in DNA by this method, although the results will be complicated if the lesions are ~7 nucleotides apart, yielding a mixture of two ligation products. In the case of modifications ~5 nucleotides apart, the Sanger sequencing chromatogram will yield peaks out of register by the distance that separates the lesions.
In the final demonstration of this approach, an OG modification was sequenced in a plasmid. Site-specific incorporation of OG into the pBR322 plasmid in a G-rich region that would be prone to oxidation was achieved using a method developed in the Wang laboratory.28 The OG was placed at a possible hotspot for G oxidation in the VEGF promoter sequence.29 Site-selective removal of OG was achieved with Fpg and then processed by the approach developed above to yield the gap ligated product (Figure 3a). The yield for the individual reactions could not be determined. Upon Sanger sequencing, the sequencing chromatogram readily identified the location of OG by the observation of peak doubling (Figure 3b). Detection of the exact location is limited in tandem, single-nucleotide repeats because the method will not report which nucleotide of the repeat was removed during the DNA glycosylase step of this method. The plasmid-based sequencing experiment identifies this as a quick and robust method for locating modifications in plasmid DNA, which can be challenging by other methods. The results in the plasmid also provide convincing initial data supporting the utility of this method for detection of modifications in the genome.
Figure 3.

Sequencing of an 8-oxoguanine in a plasmid via the gap ligation approach. (a) The scheme comprising OG excision followed by amplification. (b) Sanger sequencing showing additional single base shift indicating the position of OG.
Nucleotide modifications in the genome can lead to mutations that are involved in cancer initiation, as is the case of benzo[a]pyrene adducts to DNA.30 Other modifications, such as methylation of C is the established epigenetic marker,2,6 which can be targeted by the base excision repair enzyme ROS1.31 Utilization of an approach, such as the one developed here, is broadly applicable and only limited by the selectivity of the BER DNA glycosylase. This approach does not require special unnatural nucleotides for labeling and can be readily adapted to any commercial sequencing platform. This method will be best suited for monitoring DNA modifications in single cells, in which the deletion will be observed in all amplicons after utilization of the method outlined herein.
Supplementary Material
Acknowledgments
This work was supported by a research grant from the National Institutes of Health (R01 GM093099). The oligonucleotides were provided by the DNA/Peptide core facility and sequencing was performed by the Sequencing core facility at the University of Utah; both are supported in part by a NCI Cancer Center Support Grant (P30 CA042014). The APE1 was a kind gift from Prof. Sheila David at the University of California Davis.
Footnotes
Notes
The authors declare no competing financial interests.
Complete methods, PAGE analysis, and Sanger sequencing chromatograms can all be found in the Supporting information. This material is available free of charge via the Inter-net at http://pubs.acs.org.
References
- 1.Song CX, Yi C, He C. Nat Biotechnol. 2012;30:1107–1116. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Booth MJ, Raiber EA, Balasubramanian S. Chem Rev. 2015;115:2240–2254. doi: 10.1021/cr5002904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner M, Steinbacher J, Kraus TF, Michalakis S, Hackner B, Pfaffeneder T, Perera A, Muller M, Giese A, Kretzschmar HA, Carell T. Angew Chem Int Ed Engl. 2015;54:12511–12514. doi: 10.1002/anie.201502722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burmer GC, Loeb LA. Proc Natl Acad Sci U S A. 1989;86:2403–2407. doi: 10.1073/pnas.86.7.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irani K, Xia Y, Zweier J, Sollott S, Der C, Fearon E, Sundaresan M, Finkel T, Goldschmidt-Clermont P. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 6.Zheng G, Fu Y, He C. Chem Rev. 2014;114:4602–4620. doi: 10.1021/cr400432d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 8.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. Nat Methods. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schibel AEP, An N, Jin Q, Fleming AM, Burrows CJ, White HS. J Am Chem Soc. 2010;132:17992–17995. doi: 10.1021/ja109501x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An N, Fleming AM, White HS, Burrows CJ. Proc Natl Acad Sci U S A. 2012;109:11504–11509. doi: 10.1073/pnas.1201669109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wanunu M. Phys Life Rev. 2012;9:125–158. doi: 10.1016/j.plrev.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wescoe ZL, Schreiber J, Akeson M. J Am Chem Soc. 2014;136:16582–16587. doi: 10.1021/ja508527b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Adey A, Nova IC, Craig JM, Langford KW, Samson JM, Daza R, Doering K, Shendure J, Gundlach JH. Nat Biotechnol. 2014;32:829–833. doi: 10.1038/nbt.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayley H. Clin Chem. 2015;61:25–31. doi: 10.1373/clinchem.2014.223016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riedl J, Ding Y, Fleming AM, Burrows CJ. Nat Commun. 2015;6:8807. doi: 10.1038/ncomms9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matray TJ, Kool ET. Nature. 1999;399:704–708. doi: 10.1038/21453. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Donnelly A, Lee I, Berdis AJ. Biochemistry. 2006;45:13293–13303. doi: 10.1021/bi060418v. [DOI] [PubMed] [Google Scholar]
- 18.Wyss LA, Nilforoushan A, Eichenseher F, Suter U, Blatter N, Marx A, Sturla SJ. J Am Chem Soc. 2015;137:30–33. doi: 10.1021/ja5100542. [DOI] [PubMed] [Google Scholar]
- 19.Gahlon HL, Boby ML, Sturla SJ. ACS Chem Biol. 2014;9:2807–2814. doi: 10.1021/cb500415q. [DOI] [PubMed] [Google Scholar]
- 20.Malyshev DA, Dhami K, Quach HT, Lavergne T, Ordoukhanian P, Torkamani A, Romesberg FE. Proc Natl Acad Sci U S A. 2012;109:12005–12010. doi: 10.1073/pnas.1205176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo YJ, Malyshev DA, Lavergne T, Ordoukhanian P, Romesberg FE. J Am Chem Soc. 2011;133:19878–19888. doi: 10.1021/ja207907d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malyshev DA, Seo YJ, Ordoukhanian P, Romesberg FE. J Am Chem Soc. 2009;131:14620–14621. doi: 10.1021/ja906186f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Correa IR, Jr, Romesberg FE. Nature. 2014;509:385–388. doi: 10.1038/nature13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Craig JM, Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Nova IC, Doering K, Tickman BI, Svet MT, Gundlach JH. PLoS ONE. 2015;10:e0143253. doi: 10.1371/journal.pone.0143253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capella G, Cronauer-Mitra S, Pienado MA, Perucho M. Environ Health Perspect. 1991;93:125–131. doi: 10.1289/ehp.9193125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.David S, Williams S. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 27.Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. J Biomed Biotechnol. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.You C, Dai X, Yuan B, Wang J, Brooks PJ, Niedernhofer LJ, Wang Y. Nat Chem Biol. 2012;8:817–822. doi: 10.1038/nchembio.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fleming AM, Zhou J, Wallace SS, Burrows CJ. ACS Cent Sci. 2015;1:226–233. doi: 10.1021/acscentsci.5b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeifer G, Besaratinia A. Human Genetics. 2009;125:493–506. doi: 10.1007/s00439-009-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ponferrada-Maran MI, Roldan-Arjona T, Ariza RR. Nucleic Acids Res. 2009;37:4264–4274. doi: 10.1093/nar/gkp390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.