

Abstract
MyPro is a software pipeline for high-quality prokaryotic genome assembly and annotation. It was validated on 18 oral streptococcal strains to produce submission-ready, annotated draft genomes. MyPro installed as a virtual machine and supported by updated databases will enable biologists to perform quality prokaryotic genome assembly and annotation with ease.
Keywords: Bioinformatics, Prokaryote, Whole genome sequencing, Assembly, Annotation
The recent decrease in the cost of whole genome sequencing (WGS) technology has resulted in an increase in sequencing of various prokaryotic microbes. A typical genomics project requires processing of genome reads prior to data mining (Hasman et al., 2014; Rhoads et al., 2014). This process may include quality control checks and pre-processing measures, de novo assembly and/or reference-based assembly, automated annotation with or without manual improvement and improving genome quality by gap filling or other such methods. A variety of software is currently available for accomplishing genome assembly, annotation and improvement (Koren et al., 2014; Magoc et al., 2013; Seemann, 2014; Swain et al., 2012). However, the ability of a scientist or smaller laboratories without adequate bioinformatics training and support may limit execution of genome informatics tools to achieve meaningful results (Nocq et al., 2013).
Here, we introduce MyPro, a user-friendly genomics software pipeline for prokaryotic genomes that requires minimal programming skills. The pipeline consists of quality control and pre-processing tools, de novo assemblers, contig integration software, and tools for annotation and reference-based assembly.
To facilitate ease of use, MyPro is designed for automated de novo assembly, assembly integration and genome annotation (AutoRun.py). It consists of three main modules: Assemble, Integrate and Annotate (Fig. 1). Five commonly used assemblers including VelvetOptimiser 2.2.5 (Zerbino and Birney, 2008), Edena V3.131028 (Hernandez et al., 2008), Abyss 1.5.2 (Simpson et al., 2009), SOAPdenovo 2.01 (Luo et al., 2012) and SPAdes 3.1.1 (Bankevich et al., 2012) have been installed and configured on BioLinux 8 (Field et al., 2006), which enables the production of multiple assemblies within the Assemble module in MyPro (Assemble.py).
Fig. 1.
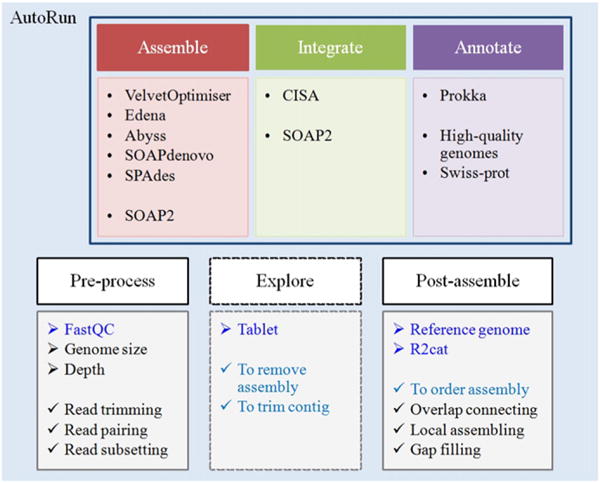
A schematic diagram shows modules, corresponding software programs and functions in the MyPro pipeline.
Most of the five assemblers use de Bruijn graph-based algorithms, which are highly dependent on the k-mer parameter. After inputting sequencing data in Fastq format, VelvetOptimiser is used first to perform a parameter sweep of k-mers ranging from 0.6 to 0.9 of read length. The optimal k-mer is automatically chosen for the highest N50 contig length and this k-mer is used for Abyss and SOAPdenovo assembly production. For paired reads, the VelvetOptimiser estimates insert size and its standard deviation, which are saved in MyPro for later use when desired. Depending on the read length of input reads, SPAdes assembly is obtained by setting k-mer lengths of 21, 33 and 55 for read lengths b150 bp, k-mer lengths of 21, 33, 55 and 77 for read lengths between 150 and 250 bp, and k-mer lengths of 21, 33, 55, 77, 99 and 127 for read lengths ≥250 bp. Edena uses an alternative approach to assemble, based on overlap layout, requiring a minimum overlap size (m) instead of k-mer for assembly. The value of m is set to half of the read length and is gradually increased to the read length. The optimal m is automatically chosen for the highest N50 contig length and the corresponding assembly is kept as Edena assembly.
Once multiple assemblies have been obtained, input reads are aligned using SOAP2 (Li et al., 2009) with the pre-calculated read information if desired (e.g., minimal and maximal insert size for paired reads). The alignment rate is used to evaluate the assemblies. To provide high-quality contigs and removing possible contaminants, the contigs with less than 100 aligned reads are removed and the basic assembly statistics, such as number of contigs, length of longest contig, N50 value and whole genome size are reported at the end of Assemble process. MyPro (Integrate.py) provides superior assembly by integrating the assemblies produced by the Assemble module using CISA 1.3 (Lin and Liao, 2013). In the automated pipeline, the worst assembly based on assembly statistics is removed prior to executing the Integrate module. Users are able to select among the five assemblies or add other assemblies into integration.
In the annotation module, we have significantly enhanced the limited core databases provided in Prokka (Seemann, 2014) to implement rapid annotation using the high-quality reference genome database (Tatusova et al., 2013) and an up-to-date Swiss-Prot database. MyPro (Annotate.py) annotates CISA assembly automatically, but users are able to annotate any other assembly placed on the specified folders (Assemble or Integrate).
In addition to the three main modules, MyPro provides functionalities for pre-process, exploration and post-assembly. In pre-process, with a specified genome size, MyPro (Preprocess.py) performs quality trimming and sub-samples input reads to a desired depth of coverage (default 100×). For assembly exploration, MyPro outputs aligned reads in BAM format, enabling visual inspection of assembly with Tablet (Milne et al., 2012) for quality assurance. If a closely related reference genome is available, r2cat (Husemann and Stoye, 2010) is employed for contig arrangement using the related reference to produce ordered and unmatched contigs. MyPro (Postassemble.py) utilizes both sets of contigs along with reads to post-assemble. Overlapping contigs (≥15 bp) at a reference-guided sequential order are merged. Reads unable to be aligned by SOAP2 on the merged contigs are assembled by Edena using a variety of m and those local assemblies are then used for bridging two contigs.
To validate MyPro, we performed genome assembly on three bacterial species chosen for extremes of GC-content: GC-poor Staphylococcus aureus (33% GC), GC-rich Rhodobacter sphaeroides 2.4.1 (69% GC) and Escherichia coli MG1655 (GC 51%). The publicly available sequencing reads were downloaded and analyzed by MyPro (see Supplementary data for details). The assembly results evaluated by QUAST 2.3 (Gurevich et al., 2013) demonstrate that MyPro produced high-quality annotated assemblies in terms of contiguity of assembly and annotation precision for coding sequences (Supplementary data). QUAST was also used to evaluate MyPro-produced assemblies for eight recently sequenced oral streptococcal strains that were post-assembled with available reference genomes (Table 1). For these strains, contigs less than 500 bp were discarded to maintain quality of data. Assemblies were also evaluated for ten other oral streptococcal strains de novo assembled by MyPro (AutoRun.py) (Table 2). For both oral streptococcal strains with available reference genomes and strains with no reference genome (Tables 1 and 2), MyPro consistently performed better than de novo assemblers and contig integration software (CISA) in terms of N50 value or number of contigs. We believe MyPro will be a useful bioinformatics tool for assembly, annotation and improvement of prokaryotic genomes for biomedical researchers. The software pipeline, components and user instructions are available for download at http://sourceforge.net/projects/sb2nhri/files/MyPro/.
Table 1.
N50 values and number of contigs (in parentheses) of 8 oral streptococcal genomes from various de novo assemblers, CISA and MyPro with CISA and post-assemble.
|
S. gordonii
|
S. salivarius
|
S. parasanguinis
|
S. cristatus
|
S. mitis
|
||||
|---|---|---|---|---|---|---|---|---|
| G9B | KB005 | MGH413 | CC5A | CR3 | OT25 | SK137 | SK145 | |
| Abyss | 343,271 (15) |
88,563 (58) |
404,146 (22) |
178,730 (43) |
193,676 (30) |
996,310 (8) |
191,221 (27) |
262,407 (29) |
| Edena | 335,948 (16) |
85,666 (43) |
260,686 (22) |
197,484 (33) |
496,588 (21) |
392,731 (12) |
247,103 (21) |
192,038 (19) |
| Velvet | 483,119 (16) |
88,921 (53) |
398,175 (17) |
191,762 (31) |
192,370 (25) |
427,010 (12) |
211,003 (24) |
257,188 (20) |
| SOAPdenovo | 78,330 (80) |
43,713 (108) |
28,220 (138) |
72,098 (70) |
26,261 (141) |
40,195 (89) |
124,486 (50) |
46,176 (79) |
| SPAdes | 334,420 (381) |
59,007 1093 |
141,257 (628) |
183,527 (270) |
89,833 (511) |
377,595 (295) |
310,465 (338) |
152,531 (671) |
| CISA | 344,471 (10) |
89,104 (37) |
404,146 (11) |
203,298 (25) |
496,588 (15) |
1,509,483 (5) |
372,102 (13) |
257,489 (16) |
| MyPro (CISA) |
556,432 (7) |
89,104 (37) |
404,146 (11) |
203,298 (22) |
496,588 (15) |
1,509,483 (5) |
446,044 (7) |
257,644 (19) |
| MyPro (post-assemble) |
1,555,780 (3) |
195,368 (22) |
2,007,789 (4) |
554,931 (8) |
540,612 (6) |
1,509,483 (4) |
1,089,748 (9) |
566,607 (12) |
Higher N50 value and lower number of contigs are indicative of superior genome assembly. Note that in comparison with other genome assembly software, MyPro consistently demonstrates values indicative of superior genome assembly (highlighted in bold).
Table 2.
N50 values and number of contigs (in parentheses) of 10 oral streptococcal genomes from various de novo assemblers and MyPro with CISA.
|
S. salivarius
|
S. sanguinis
|
S. mitis
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| UC3162 | I141 | VT517 | COL85/1862 | NCTC10712 | OP51 | SK141 | UC921A | UC5873 | UC6950A | |
| Abyss | 102,123 (44) | 205,597 (27) | 201,754 (34) | 258,394 (23) | 294,670 (13) | 325,986 (10) | 325,653 (11) | 224,590 (25) | 1,371,488 (26) | 140,468 (66) |
| Edena | 112,782 (36) | 79,082 (48) | 169,164 (47) | 190,728 (39) | 293,981 (16) | 321,991 (9) | 244,280 (13) | 224,608 (26) | 202,683 (43) | 137,264 (69) |
| Velvet | 93,598 (72) | 282,985 (23) | 202,625 (80) | 398,214 (33) | 293,951 (16) | 1,177,164 (15) | 1,229,524 (12) | 226,431 (22) | 1,380,504 (34) | 141,356 (88) |
| SOAPdenovo | 31,809 (166) | 79,987 (66) | 21,141 (307) | 36,934 (137) | 28,642 (143) | 84,194 (58) | 94,143 (54) | 41,314 (126) | 11,579 (376) | 28,278 (224) |
| SPAdes | 88,327 (69) | 292,810 (17) | 96,152 (91) | 335,392 (42) | 257,414 (20) | 79,761 (227) | 315,886 (13) | 223,855 (28) | 1,374,450 (27) | 95,372 (75) |
| MyPro (CISA) | 116,758 (25) | 331,769 (11) | 290,644 (25) | 722,753 (14) | 572,071 (10) | 1,183,284 (11) | 1,236,221 (6) | 226,881 (14) | 1,375,455 (12) | 142,219 (35) |
Higher N50 value and lower number of contigs are indicative of superior genome assembly. Note that in comparison with other genome assembly software, MyPro consistently demonstrates values indicative of superior genome assembly (highlighted in bold).
Supplementary Material
Acknowledgments
This research was supported by grants from the National Health Research Institutes (PH-104-PP-05 and PH-104-SP-03), Ministry of Science and Technology, Taiwan (103-2320-B-400-001) and the National Institute of Dental and Craniofacial Research, U.S.A. (R01DE022673).
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.mimet.2015.04.006.
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field D, Tiwari B, Booth T, Houten S, Swan D, Bertrand N, Thurston M. Open software for biologists: from famine to feast. Nat Biotechnol. 2006;24:801–804. doi: 10.1038/nbt0706-801. [DOI] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasman H, Saputra D, Sicheritz-Ponten T, Lund O, Svendsen CA, Frimodt-Møller N, Aarestrup FM. Rapid whole genome sequencing for the detection and characterization of microorganisms directly from clinical samples. J Clin Microbiol. 2014;52:139–146. doi: 10.1128/JCM.02452-13. http://dx.doi.org/10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez D, François P, Farinelli L, Østerås M, Schrenzel J. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802–809. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husemann P, Stoye J. r2cat: synteny plots and comparative assembly. Bioinformatics. 2010;26:570–571. doi: 10.1093/bioinformatics/btp690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Treangen TJ, Hill CM, Pop M, Phillippy AM. Automated ensemble assembly and validation of microbial genomes. BMC Bioinf. 2014;15:126. doi: 10.1186/1471-2105-15-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Lin SH, Liao YC. CISA: contig integrator for sequence assembly of bacterial genomes. PLoS One. 2013;8:e60843. doi: 10.1371/journal.pone.0060843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc T, Pabinger S, Canzar S, Liu X, Su Q, Puiu D, Tallon LJ, Salzberg SL. GAGE-B: an evaluation of genome assemblers for bacterial organisms. Bioinformatics. 2013;29:1718–1725. doi: 10.1093/bioinformatics/btt273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, Shaw PD, Marshall D. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2012 doi: 10.1093/bib/bbs012. bbs012. [DOI] [PubMed] [Google Scholar]
- Nocq J, Celton M, Gendron P, Lemieux S, Wilhelm BT. Harnessing virtual machines to simplify next-generation DNA sequencing analysis. Bioinformatics. 2013;29:2075–2083. doi: 10.1093/bioinformatics/btt352. [DOI] [PubMed] [Google Scholar]
- Rhoads DD, Sintchenko V, Rauch CA, Pantanowitz L. Clinical microbiology informatics. Clin Microbiol Rev. 2014;27:1025–1047. doi: 10.1128/CMR.00049-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain MT, Tsai IJ, Assefa SA, Newbold C, Berriman M, Otto TD. A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nat Protoc. 2012;7:1260–1284. doi: 10.1038/nprot.2012.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkv278. gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.