

Abstract
Poly(dimethyl siloxane) (PDMS) is extensively used for biomedical applications due to its low cost, ease of fabrication, high durability and flexibility, oxygen permeability, and self-healing properties. PDMS, however, has some significant drawbacks. PDMS endures unacceptably high levels of non-specific protein fouling when used with biological samples due to its superhydrophobic characteristics. Unfortunately, conventional surface modification methods do not work for PDMS due to its low glass transition temperature. This phenomenon has been well-known for years as “hydrophobic regeneration”. For the same reason, it is also very difficult to bring functionalities onto PDMS surfaces. Herein, we demonstrate how a superhydrophilic zwitterionic material, poly(carboxybetaine methacrylate) (pCBMA), can provide a highly stable coating with long term stabilty due to the sharp contrast in hydrophobicity between pCBMA and PDMS. This material is able to suppress nonspecific protein adsorption in complex media and functionalize desired biomolecules needed in applications, such as diagnostics, without sacrificing its nonfouling characteristics.
Keywords: Poly(dimethyl siloxane), surface modification, protein adsorption, zwitterions, functionalizable
Introduction
Due to its advantageous material properties, PDMS has been used to manufacture catheters,1 contact lenses,2 prosthetics,3 and many other devices. In recent years, PDMS has also found an exciting role in the manufacturing of microfluidic devices.4 Unfortunately, like many other hydrophobic materials, PDMS is susceptible to high levels of protein adsorption. For implants, this can lead to rejection from the body by the immune system, and when in contact with blood, protein fouling can result in thrombosis and clotting.5, 6
Currently, the easiest and most widely used method to reduce protein fouling of PDMS is to make the surface more hydrophilic by oxidizing the surface using UV-ozone,7 oxygen plasma,8 or an oxidative wet chemical method.9 These strategies introduce hydroxyl groups directly onto the PDMS surface to increase surface hydrophilicity, reduce non-specific protein fouling, and provide functionalizable groups. Due to its superhydrophobicity and low glass transition temperature, PDMS polymer chains will rearrange and undo most surface coatings.10-13 This is commonly referred to as hydrophobic regeneration (Figure 1a).8 Thus, these modifications often last only a few hours or sometimes a matter of minutes (Figure 1b).14 More effective methods introduce material coatings onto the surface, such as sol-gel coatings, layer-by-layer coatings, surfactant coatings, silane coatings, polymer attachment, and surface polymerizations.4, 15, 16 In recent years, poly(ethylene glycol) (PEG) has been the polymer most widely used to modify the surface of PDMS for interacting with biological samples. Despite their ubiquitous use, PEG coatings are unstable, which has been a well-known problem for years due to reconstruction that occurs at the interface between the PDMS and PEG polymers (Figure 1c).11, 13, 17 While PEG is often considered a hydrophilic polymer, this material is in fact amphiphilic, being soluble in such non-polar solvents such as dichloromethane and toluene, in addition to water.
Figure 1.
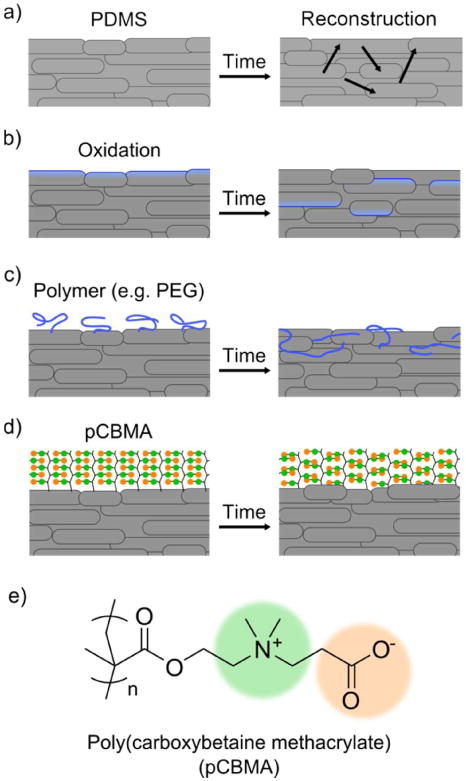
(a) Over time, PDMS chains will reconstruct, resulting in a surface that is constantly being renewed, known as hydrophobic regeneration. (b) The PDMS surface is made more hydrophilic by oxidation, but this surface will degrade over time. (c) The same problem occurs with polymers, particularly ones of amphiphilic nature like PEG. (d) pCBMA, due to its unique characteristics, forms a super stable nonfouling surface due to its zwitterionic nature. The structure of pCBMA is shown in (e).
Herein, we modify the surface of PDMS with the zwitterionic polymer pCBMA (Figure 1d and 1e). When compared to PEG, zwitterionic polymers are chemically very different. Zwitterionic polymers are strongly hydrated via electrostatically induced hydration and have extremely poor solubility in non-aqueous solvents, making surface rearrangement with PDMS thermodynamically unfavourable. Furthermore, zwitterionic polymers have strong chain-chain interactions formed by ionic bridging,18 enhancing the coating's stability (Figure 1d). These properties make zwitterionic polymers ideal for the interface between biological media and PDMS.
Experimental Section
Materials
Copper(I) bromide (99.999%), Copper(II) bromide (>99.0%), 1,1,4,7,10,10-hexamethyltriethylene tetramine (HMTETA), d-Biotin, bovine plasma fibrinogen, and phosphate-buffered saline (PBS, 0.01 M phosphate, 0.138 M sodium chloride, 0.0027 M potassium chloride, pH 7.4) were purchased from Sigma Chemical Co. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-Hydroxysuccinimide (NHS), Sulfo-N-Hydroxysuccinimide (Sulfo-NHS), and fluorescein isothiocyanate (FITC) were purchased from Acros Organics. NeutrAvidin was purchased from Pierce Biotechnology, Inc.. Sylgard ® 184 Silicone Elastomer Kit was used from Dow Corning. 2-Carboxy-N,N,-dimethyl-N-(2′-(methacryloyloxy)ethyl) ethanaminium inner salt (carboxybetaine methacrylate, CBMA) was synthesized as previously reported.19 Synthesis of the atom transfer radical polymerization (ATRP) initiator trichlorosilane:10-Undecen-1-yl 2-Bromo-2-methylpropionate was synthesized as previously reported.20
PDMS preparation and initiator immobilization
Sylgard 184 Silicone Elastomer (Dow Corning) was prepared by thoroughly mixing 10 parts base to 1 part curing agent. The mixture was poured into a 20 cm petri dish and placed under vacuum to remove bubbles. The PDMS was cured at 60 degrees overnight. After curing, samples were cut and soxhlet extracted against acetone for one day, then dried under vacuum and mild heating. Clean samples of PDMS were treated with 3% ozone at 2 L/min for 1 hour. After oxidation, samples were exposed briefly to humid air and dried with nitrogen. For vapor deposition, the samples were placed on a clean glass cover slip in a vacuum desiccator with a small amount of ATRP trichlorosilane initiator. A strong vacuum was pulled on the desiccator, which was then sealed and placed at 80°C for 4 hours. After the chemical vapor deposition of the trichlorosilane, the PDMS samples were briefly dipped in a 1mM solution of NaOH (pH = 11) to crosslink the surface silanes and neutralize the HCl byproducts.
ATRP
Cu(I)Br (7.17 mg, 0.0500 mmol) and Cu(II)Br2 (2.79 mg, 0.0125 mmol) were placed in a small test tube under nitrogen protection and sealed with a rubber septum. 1.375 g (6 mmol) CBMA monomer and a PDMS substrate with immobilized initiator were placed in a large test tube, also under nitrogen protection and sealed with a rubber septum. Both test tubes were deoxygenated by five repetitions of a strong vacuum followed by nitrogen backfill. Deoxygenated water (deoxygenated by bubbling with N2 gas) was then added to both test tubes (2 mL to the small tube, 30 mL to the large tube). While stirring, 17 uL of HMTETA was added to the copper solution and stirred for 30 min for ligand complexation. To initiate polymerization, 1.2 mL of the copper catalyst solution was added to the CBMA monomer and PDMS substrate. Reaction time was controlled to adjust film thickness.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was used to measure fibrinogen adsorption on the modified and unmodified PDMS surfaces. Substrates were soaked in PBS for at least 30 minutes before the experiment. Next, the substrates were incubated in 1 mg/mL fibrinogen in PBS for 60 min. The substrates were then rinsed several times with fresh PBS. The samples were next incubated in PBS solution containing 5.5 μg/mL horseradish peroxidase (HRP)-conjugated anti-fibrinogen (USbiological) for 30 min. The substrates were again rinsed several times with PBS. Finally, the substrates were incubated in 1 mL of 0.1 M citrate-phosphate buffer (pH 5.0) containing 1 mg/mL chromogen of o-phenylenediamine (OPD) and 0.03% hydrogen peroxide for 60 minutes. Absorbance at 490 nm was measured using a spectrophotometer. Values of fibrinogen adsorption are represented as percentages of the adsorption levels from unmodified PDMS.
Bovine serum albumin and NeutrAvidin labeling with FITC
FITC- bovine serum albumin (FITC-BSA) and FITC-NeutrAvidin was prepared by protein conjugation with FITC. Briefly, 30 mg of protein was dissolved in 3 mL of 100 mM NaHCO3, pH 9. From a solution of 15 mg/mL FITC in DMSO, 300 μL was added to the protein solution to initiate labeling. After 2 hours, the FITC labeled conjugates were purified twice using 10 mL Bio-Gel P-6DG Bio-Rad disposable size exclusion columns.
Evaluation of surface serum binding
9.77 mg (40 mmol) d-Biotin was dissolved in 200 uL of DMSO. 7.67 mg EDC (40 mmol) and 8.69 mg Sulfo-NHS (40 mmol) were dissolved in 200 uL of water. The EDC/Sulfo-NHS solution was slowly added to the biotin while frequently vortexing. The coupling reaction was allowed to react for 20 minutes. Next, 5 uL of 2-mercaptoethanol was added to quench unreacted EDC and allowed to stand for 10 minutes. This solution was diluted 25× into 0.1 M phosphate, pH 7.4, containing the serum-exposed sample for 60 minutes. Samples were then washed several times with PBS to remove unbound biotin. Next, they were incubated in a solution of 1 mg/mL BSA and 0.01 mg/mL FITC-NeutrAvidin in PBS for 30 min. Samples were again washed with PBS and imaged using a fluorescence microscope.
Functionalization of proteins on pCBMA surfaces
57.4 mg EDC and 7.67 mg NHS were dissolved in 1.5 mL ultrapure (18.2 MΩ) water. pCBMA-coated PDMS was incubated in the activation solution for 10 minutes. The sample was removed, rinsed with water and dried with air. Next, 2 μL of FITC-BSA at 1 mg/mL in 10 mM phosphate, pH 7.4, was spotted on the activated pCBMA-PDMS surface and allowed to react for 30 min. The surface was then rinsed with PBS and incubated in 300 mM NaCl and 10 mM Na2CO3, pH 10.0, for 1 hour to deactivate any unreacted NHS ester groups still remaining on the surface. A similar procedure was followed, but without NHS in the activation solution, as a control. Another control was performed where FITC-BSA was not spotted on the activated surface, but instead the surface was deactivated first with basic solution. FITC-BSA was then spotted to test the effectiveness of deactivation. Samples were washed with PBS and imaged using a fluorescence microscope
Results and discussion
Surface polymerization and characterization
CBMA was introduced to the PDMS surface via surface initiated ATRP (SI-ATRP). This was done by first treating the surface with an ATRP trichlorosilane initiator using chemical vapor deposition. Polymerization of CBMA occurred in 100% water. This is worth noting because PDMS absorbs many organic solvents, deforming the material, which is important to avoid when dealing with medical devices. Polymer film thickness was measured as a function of reaction time using multiple-angle ellipsometry (Figure 2a), with a final film thickness around 30 nm. It is worth noting that this method for introducing pCBMA to the PDMS surface was done without needing to be exposed to organic solvents and only water.
Figure 2.

(a) pCBMA film thickness as a function of reaction time, as measured by multiple-angle ellipsometry. Error bars represent standard deviation with samples measured in triplicate. AFM images of an (b) unmodified PDMS surface, (c) pCBMA modified PDMS surface in dry state, and (d) pCBMA modified PDMS surface in PBS.
The surface of the pCBMA modified PDMS was imaged using atomic force microscopy (AFM) to evaluate the structure of the modified surface in both wet and dry conditions. When compared to unmodified PDMS (Figure 2b), pCBMA polymer domain formations were visually obvious (Figure 2c). Under wet conditions (Figure 2d), the pCBMA film became hydrated and the dried polymer domains were no longer visible, leaving a smooth surface. This is a result of water penetration into the zwitterionic coating. The smooth hydrated film demonstrates the uniformity of the polymer film that is typical of SI-ATRP.
Long-term resistance to protein and serum fouling
Protein fouling was measured as a function of polymerization time using a fibrinogen adsorption ELISA, as shown in Figure 3a, with a minimum at reaction times from 8 to 24 hours. By controlling reaction time, protein adhesion can be kept at ∼2% compared to unmodified PDMS. Next, long-term stability was evaluated with samples that had undergone 18 hours of polymerization time. These were incubated in either wet conditions in PBS or dry conditions in a desiccator, both at room temperature. The results for fibrinogen adsorption relative to incubation time can be seen in Figure 3b for both conditions. Nearly no protein adhesion was seen for up to 31 days for samples incubated under dry conditions. After 31 days, the dry surface eventually became unstable and fouling increased at 74 days. When incubated in wet conditions, the nonfouling properties of pCBMA-coated PDMS remained ultralow, with <2% protein adsorption after 74 days. These results are significant, as most PDMS-based biomedical devices operate under aqueous conditions. To fully stress the performance of the coating, the pCBMA-coated PDMS surface was exposed to undiluted human serum. After washing, Sulfo-NHS activated biotin was used to non-specifically tag any adsorbed proteins that fouled. Next, FITC labelled NeutrAvidin was used to visualize any adsorbed protein. This was a particularly useful method we developed for detecting adsorption from protein mixtures on PDMS, as most traditional labelling methods using direct tagging with activated fluorescent tags will physically adsorb into PDMS. Our results showed that uncoated PDMS showed significant serum protein binding (Figure 3c), while little to no serum binding occurred on the pCBMA-coated PDMS sample (Figure 3d). With complete resistance to undiluted human serum, it is promising that pCBMA-PDMS coatings can be used on devices interacting with whole blood.
Figure 3.
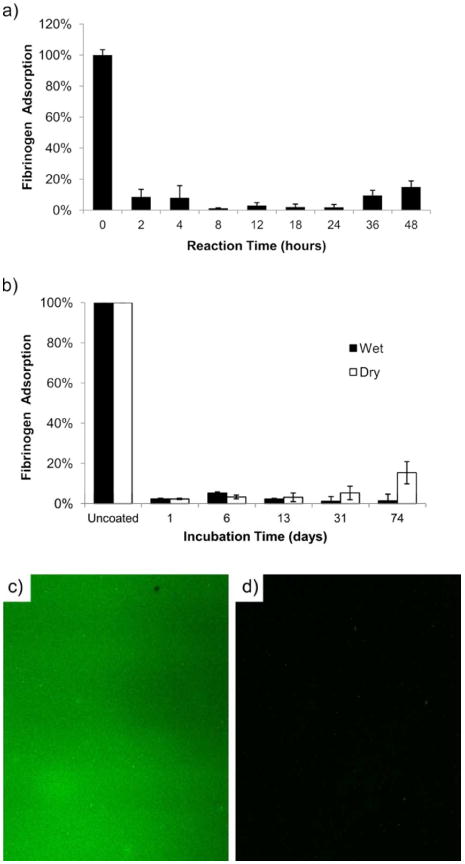
(a) Fibrinogen adsorption as a function of reaction time during polymerization. (b) Shows fibrinogen adsorption of pCBMA modified PDMS incubated under wet and dry conditions at room temperature up to 74 days after polymerization for 18 hours. Error bars represent standard deviation with samples measured in triplicate. (c) Bare PDMS and (d) pCBMA modified PDMS (18 hour polymerization) were exposed to 100% human serum. The surface was labeled by biotinylation and FITC-NeutrAvidin to visualize total surface binding.
Functionalization
While the nonfouling capacity and long term stability of pCBMA-modified PDMS are considerable, the unique structure of pCBMA allows for covalent immobilization of biomolecules such as RGD-containing peptides for cell adhesion or antibodies for biomarker detection21, 22 without sacrificing the nonfouling characteristics of the surface. We chose FITC labelled BSA (FITC-BSA) as a model biomolecule to demonstrate functionality. This was done first by activating the exposed carboxylic acid groups with reactive N-hydroxysuccinimide (NHS) esters using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC). The activated NHS groups are able to react with biomolecules containing accessible primary amines. Once the proteins are covalently bound to the surface, the surface can be deactivated under slightly basic conditions (pH ∼ 10) to hydrolyze unreacted NHS groups back to carboxylic acids, thereby restoring the zwitterionic nonfouling background. In Figure 4a, a pCBMA-PDMS surface is shown that was exposed to FITC-BSA without any activation, showing no visible protein adhesion. Figure 4b shows the pCBMA-PDMS surface activated with EDC/NHS, which was then exposed to FITC-BSA. Figure 4c shows a pCBMA-PDMS surface that was activated with EDC/NHS, but then deactivated with 10 mM sodium carbonate, pH 10.0, before being exposed to FITC-BSA. This confirms that the pCBMA surface can controllably immobilize desired biomolecules without affecting its nonfouling properties.
Figure 4.

(a) Unactivated pCBMA-PDMS surface exposed to FITC-BSA showed no protein binding. (b) NHS/EDC activated pCBMA-PDMS surface exhibited high levels of controlled protein attachment. (c) NHS/EDC activated pCBMA-PDMS surface followed by deactivation demonstrated no protein binding. This was to show that the surface does not lose its nonfouling characteristics during activation.
Conclusions
By employing a superhydrophilic zwitterionic polymer, we are able to modify zwitterionic PDMS and to suppress surface reconstruction for long-term stability. We have been able to introduce excellent nonfouling properties in undiluted blood serum and provide the ability to specifically functionalize desired biomolecules. Results show that a thin and uniform pCBMA layer can be introduced onto PDMS to create a nonfouling surface that will last up to 31 days in dry air, and >74 days in aqueous conditions (saline). It is worth noting that, due to the swelling properties of PDMS to non-aqueous solvents, surface modification was accomplished without exposure to any solvents except water. This avoids any deformation which greatly increases amenability to biomedical applications.23 Due to long-term stability, nonfouling properties, and functionality, the proposed surface modification method based on pCBMA will enable a wider range of biomedical applications that require specific binding or targeting.
Acknowledgments
This work has been supported by the Office of Naval Research (N000140910137 and N000141010600). A. J. Keefe acknowledges partial financial support through an NCI training grant (T32CA138312).
References
- 1.Park JH, Lee KB, Kwon IC, Bae YH. Journal of Biomaterials Science-Polymer Edition. 2001;12:629–645. doi: 10.1163/156856201316883458. [DOI] [PubMed] [Google Scholar]
- 2.Liu L, Sheardown H. Biomaterials. 2005;26:233–244. doi: 10.1016/j.biomaterials.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 3.Eleni PN, Katsavou I, Krokida MK, Polyzois GL, Gettleman L. Dental Materials. 2009;25:1493–1502. doi: 10.1016/j.dental.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 4.Zhou JW, Ellis AV, Voelcker NH. Electrophoresis. 2010;31:2–16. doi: 10.1002/elps.200900475. [DOI] [PubMed] [Google Scholar]
- 5.Ratner BD, Bryant SJ. Annual Review of Biomedical Engineering. 2004;6:41–75. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 6.Tsai WB, Grunkemeier JM, Horbett TA. Journal of Biomedical Materials Research. 1999;44:130–139. doi: 10.1002/(sici)1097-4636(199902)44:2<130::aid-jbm2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 7.Olah A, Hillborg H, Vancso GJ. Applied Surface Science. 2005;239:410–423. [Google Scholar]
- 8.Bodas D, Khan-Malek C. Sensors and Actuators B-Chemical. 2007;123:368–373. [Google Scholar]
- 9.Bodas D, Khan-Malek C. Microelectronic Engineering. 2006;83:1277–1279. [Google Scholar]
- 10.Delamarche E, Donzel C, Kamounah FS, Wolf H, Geissler M, Stutz R, Schmidt-Winkel P, Michel B, Mathieu HJ, Schaumburg K. Langmuir. 2003;19:8749–8758. [Google Scholar]
- 11.Demming S, Lesche C, Schmolke H, Klages CP, Buttgenbach S. Physica Status Solidi a-Applications and Materials Science. 2011;208:1301–1307. [Google Scholar]
- 12.Hellmich W, Regtmeier J, Duong TT, Ros R, Anselmetti D, Ros A. Langmuir. 2005;21:7551–7557. doi: 10.1021/la0510432. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZW, Feng XJ, Luo QM, Liu BF. Electrophoresis. 2009;30:3174–3180. doi: 10.1002/elps.200900132. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Chaudhury MK, Owen MJ, Orbeck T. Journal of Colloid and Interface Science. 2001;244:200–207. [Google Scholar]
- 15.Lopez AI, Kumar A, Planas MR, Li Y, Nguyen TV, Cai C. Biomaterials. 2011;32:4336–4346. doi: 10.1016/j.biomaterials.2011.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauer WAC, Fischlechner M, Abell C, Huck WTS. Lab on a Chip. 2010;10:1814–1819. doi: 10.1039/c004046k. [DOI] [PubMed] [Google Scholar]
- 17.Sharma V, Dhayal M, Govind, Shivaprasad SM, Jain SC. Vacuum. 2007;81:1094–1100. [Google Scholar]
- 18.Kathmann EE, White LA, McCormick CL. Polymer. 1997;38:879–886. [Google Scholar]
- 19.Zhang Z, Chao T, Chen SF, Jiang SY. Langmuir. 2006;22:10072–10077. doi: 10.1021/la062175d. [DOI] [PubMed] [Google Scholar]
- 20.Matyjaszewski K, Miller PJ, Shukla N, Immaraporn B, Gelman A, Luokala BB, Siclovan TM, Kickelbick G, Vallant T, Hoffmann H, Pakula T. Macromolecules. 1999;32:8716–8724. [Google Scholar]
- 21.Ebara M, Hoffman JM, Stayton PS, Hoffman AS. Radiation Physics and Chemistry. 2007;76:1409–1413. [Google Scholar]
- 22.Sui GD, Wang JY, Lee CC, Lu WX, Lee SP, Leyton JV, Wu AM, Tseng HR. Analytical Chemistry. 2006;78:5543–5551. doi: 10.1021/ac060605z. [DOI] [PubMed] [Google Scholar]
- 23.Lee JN, Park C, Whitesides GM. Analytical Chemistry. 2003;75:6544–6554. doi: 10.1021/ac0346712. [DOI] [PubMed] [Google Scholar]