

Abstract
This paper describes the terminal-selective Pt-catalyzed C(sp3)–H oxidation of aliphatic amines without the requirement for directing groups. CuCl2 is employed as a stoichiometric oxidant, and the reactions proceed in high yield at Pt loadings as low as 1 mol %. These transformations are conducted in the presence of sulfuric acid, which reacts with the amine substrates in situ to form ammonium salts. We propose that protonation of the amine serves at least three important roles: (i) it renders the substrates soluble in the aqueous reaction medium; (ii) it limits binding of the amine nitrogen to Pt or Cu; and (ii) it electronically deactivates the C–H bonds proximal to the nitrogen center. We demonstrate that this strategy is effective for the terminal-selective C(sp3)–H oxidation of a variety of primary, secondary and tertiary amines.
The development of methods for the selective functionalization of strong 1° C(sp3)–H bonds in the presence of weaker 3° and 2° C(sp3)–H bonds remains a major challenge in the burgeoning field of transition metal-catalyzed C–H functionalization.1,2,3 The Pt-catalyzed oxidation of alkanes offers a promising approach to tackle this challenge.4 In the 1970s, Shilov demonstrated that aqueous solutions of PtII and PtIV salts effect the C–H hydroxylation of alkanes.5 These transformations generally afford selective functionalization of 1° C-H bonds over 2° C–H bonds.4,6 However, despite the great promise of Shilov-type Pt catalysis, these transformations have found minimal application in organic synthesis over the past 40 years.7 This is due, in large part, to three key limitations: (1) the scope of substrates remains extremely narrow (predominantly due to the low water solubility of most organic molecules); (2) the selectivity remains modest (1° vs 2° C(sp3)–H bond selectivity generally ranges from 1.5 : 1 to 3 : 1); and (3) costly PtIV salts are typically used as the terminal oxidant (rendering the reactions impractical on even small scales).
We reasoned that all of these challenges could potentially be addressed in the context of the Pt-catalyzed C(sp3)–H oxidation of aliphatic amines. Aliphatic amines are a very important class of compounds that serve as the core structures of diverse organic materials, natural products, and bioactive molecules.8 Most existing methods for the C(sp3)–H functionalization of aliphatic amines involve either: (1) functionalization at the highly activated C–H site α-to nitrogen9 or (2) the use of the amine nitrogen as part of a directing group.7c,10,11 We sought to utilize Pt catalysis to achieve complementary reactivity: namely, terminal-selective C–H functionalization at sites remote to nitrogen.12
We hypothesized that terminal-selective Pt-catalyzed C–H oxidation would be enabled by protonation of the amine substrates (eq. 1). The formation of ammonium salts would address two of the existing challenges of Shilov catalysis. First, quaternization should render the substrates water-soluble. Second, the inductive electron withdrawing effect of the ammonium cation13 is expected to electronically deactivate C–H sites proximal to nitrogen, thereby enhancing selectivity for remote 1° C(sp3)–H bonds.14 In addition, we sought to leverage prior work by Sames7c and Sen7d demonstrating that CuII salts can be used as oxidants for Shilov-type reactions in place of costly K2PtCl6.15 Our protonation strategy is also crucial in this context, since unprotonated amines could potentially undergo undesired side reactions with the CuII oxidant.16
 |
(1) |
Our initial studies focused on the K2PtCl4-catalyzed C–H hydroxylation of dipropylamine, which was protonated in situ with H2SO4. We started with traditional Shilov conditions, using 10 mol % of K2PtCl4. 1 equiv of K2PtCl6 as the terminal oxidant (and limiting reagent), and 2 equiv of the amine H2SO4 salt at 120 °C. As shown in Table 1, entry 1, these conditions afforded the C(sp3)–H hydroxylation product with high terminal selectivity (>10 : 1 ratio of 1 to 1a) but only modest yield (36%) over 48 h. Changing the terminal oxidant to CuCl2 under otherwise analogous conditions provided an enhanced yield (66%), while maintaining high selectivity (>10 : 1; entry 2). Increasing the temperature to 150 °C afforded a similar yield (63%) over 30 h (entry 3). At this temperature, the catalyst loading could be dropped to 1 mol %, with minimal impact on the yield or selectivity (although this reaction now required 48 h; entry 5). Upon moving to 5 equiv of amine H2SO4 salt relative to Cu, the hydroxylated product was obtained in 97% yield (97 turnovers of Pt) and 8 : 1 selectivity for 1 over 1a (entry 6). To our knowledge, this result represents the highest combination of terminal selectivity and TON reported to date for Shilov-type Pt catalysis.4 Importantly, control reactions show that no product is formed in the absence of Cu or Pt (entries 7 and 8). Furthermore, <1% of the C–H hydroxylation product was observed under the standard conditions but in the absence of added acid (entry 9). This is consistent with our hypothesis that protonation of the amine is essential for this transformation.
Table 1.
Optimization of Pt-Catalyzed C–H Hydroxylation of Protonated Dipropylamine
 | ||||||
|---|---|---|---|---|---|---|
| entry | equiv amine |
oxidant | temp (° C) |
K2PtCl4 loading (mol %) |
yield of 1 + 1aa |
1 : 1aa |
| 1b | 2 | K2PtCl6 | 120 | 10 | 36% | >10 : 1 |
| 2b | 2 | CuCl2 | 120 | 10 | 66% | >10 : 1 |
| 3c | 2 | CuCl2 | 150 | 10 | 63% | >10 : 1 |
| 4c | 2 | CuCl2 | 150 | 1 | 40% | >10 : 1 |
| 5b | 2 | CuCl2 | 150 | 1 | 70% | 8 : 1 |
| 6c | 5 | CuCl2 | 150 | 1 | 97% | 8 : 1 |
| 7c | 5 | ---- | 150 | 1 | nde | ---- |
| 8 c | 5 | CuCl2 | 150 | ---- | nde | ---- |
| 9 c,d | 5 | CuCl2 | 150 | 1 | <1% | ---- |
Yield and ratio of products determined by 1H NMR. Reactions were conducted in sealed vials under an atmosphere of ambient air. Yields are calculated based on the oxidant (K2PtCl6 or CuCl2) as the limiting reagent.
48 h.
30 h.
No H2SO4 added.
Products 1 and 1a were not detected.
We next examined the Pt-catalyzed C–H hydroxylation of a series of N-alkyl pyrrolidine substrates in order to probe the impact of chain length on selectivity (Table 2). In all cases, the major product derived from C–H hydroxylation at the terminal position. As the terminal methyl group is moved closer to the amine nitrogen, the terminal selectivity increases sequentially from 2 : 1 in 5 to >20 : 1 in 2.17 In all cases, the site selectivity was determined by 1H NMR spectroscopic analysis of the crude reaction mixture (see Supporting Information for spectra). The amino alcohol products were then derivatized with pivaloyl chloride (PivCl) to facilitate product isolation and characterization. Using these conditions, the oxygenated amino ester products 3–5 were isolated in high yields.18
Table 2.
Pt-Catalyzed C–H Functionalization of N-Alkylpyrrolidines
 | |||
|---|---|---|---|
| substrate | major product | isolated yield | isolated selectivity (crude selectivity)a |
| 25%b | (β : α = >20 : 1) | ||
| 85% | γ : β = >20 : 1 (γ : β = 10 : 1) |
||
| 126% | δ : γ = 4 : 1 (δ : γ = 4 : 1) |
||
| 73% | ε : δ = 3 : 1 (ε : δ = 2 : 1) |
||
Crude selectivity determined by 1H NMR spectroscopic analysis prior to treatment with PivCl.
Yield determined by 1H NMR spectroscopic analysis prior to treatment with PivCl.
The observed increase in selectivity with shorter chain length correlates with a decreased rate of C–H functionalization at sites that are proximal to the ammonium cation. For example, the C–H hydroxylation of N-ethyl pyrrolidine to form 2 proceeded in modest 25% yield under conditions analogous to those used to obtain products 3–5. This is consistent with β-C(sp3)–H bonds being significantly less reactive than more remote C–H sites. In addition, a competition between N-propyl and N-butylpyrrolidine afforded a 10 : 7 : 1 : <1 ratio of products 4-OH : 3-OH : 4a-OH : 3a-OH (Scheme 1). Again, the selectivity for 4-OH over 3-OH as well as for 4a-OH over 3a-OH is consistent with higher reactivity of C–H sites that are more remote from nitrogen. Overall, these results support our hypothesis that quaternization of the amine deactivates the C(sp3)–H sites proximal to nitrogen via an inductive electron withdrawing effect.
Scheme 1.

Competition Between N-Propyl and N-Butylpyrrolidine
Our standard conditions proved effective for the C–H hydroxylation of a variety of other 2° and 3° aliphatic amine substrates (Table 3). In all cases, the site selectivity was determined by 1H NMR spectroscopic analysis of the crude reaction mixtures (see Supporting Information for spectra).17 The amino alcohol products were then derivatized with pivaloyl chloride to facilitate isolation and characterization. Notably, the oxidant is used as the limiting reagent in these transformations. As such, yields greater than 100% reflect regeneration of the Cu oxidant (presumably by ambient air).19
Table 3.
Substrate Scope of Pt-Catalyzed C-H Hydroxylation of Secondary and Tertiary Aminesa
| entry | major product | isolated yield | isolated selectivity (crude selectivity) |
|---|---|---|---|
| 1 | 87% | >20 : 1 (8 : 1) |
|
| 2 | 76% | 7 : 1 (5 : 1) |
|
| 3 | 47% | >10 : 1 (3 : 1) |
|
| 4 | 54% | >20 : 1 (8 : 1) |
|
| 5 | 46% | >20 : 1 (>20 : 1) |
|
| 6b | 65% | >10 : 1 (5 : 1) |
|
| 7c | 88% | >20 : 1 (>20 : 1) |
|
| 8d | 102% | >20 : 1 (7 : 1) |
|
| 9e | 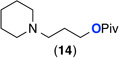 |
90% | >20 : 1 (14 : 1) |
| 10e | 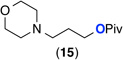 |
122% | >20 : 1 (10 : 1) |
| 11f | 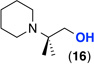 |
<5%g | ---- |
| 12f | 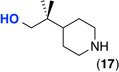 |
<5%g | ---- |
General conditions: 1 mol % K2PtCl4, 1 equiv CuCl2, 5 equiv of amine, 5.5 equiv H2SO4 (1.1 equiv relative to amine), 150 °C, 24 h.
Entry 6: Amine used as HCl salt, 10 mol % K2PtCl4.
Entry 7: 15 equiv amine, 16.5 equiv H2SO4, 48 h.
Entry 8: 0.5 mol % K2PtCl4, 15 equiv amine, 16.5 equiv H2SO4.
Entries 9–11: 5 mol % K2PtCl4.
Entries 11–12: amine used as HCl salt, 10 mol % K2PtCl4;
Yields estimated by 1H NMR analysis of crude reaction mixtures; <5% of C(sp3)–H hydroxylation or C(sp3)–H chlorination products were observed.
All of the examples in Table 3 display modest to excellent terminal selectivity.17 Notably, the derivatization and purification sequence generally results in an upgrade of selectivity for the terminal product. The terminal selectivity partially reflects the inherent steric preference of Pt for the activation of 1° over 2° or 3° C–H bonds.4,6 However, selectivity is highest in the formation of the products in entries 1, 4, 5, and 7–10, where the competing 2° C(sp3)–H sites are β or α to the quaternized nitrogen. Again, these results implicate significant inductive deactivation by the ammonium cation. Nitrogen heterocycles including pyrrolidine, piperidine, and morpholine are compatible with the reaction conditions, and minimal C–H oxidation of the ring (<5%) is observed in these systems. Although terminal β-C(sp3)–H sites are electronically deactivated, triethylamine does undergo high yielding β-C–H oxygenation under slightly modified reaction conditions (15 equiv of amine relative to CuCl2 over 48 h; entry 7). Minimal oxidation of tert-butyl groups is observed, even in the absence of competing terminal sites for C–H oxidation (e.g., entries 11, 12). Similar observations have been made by Hartwig in Ir-catalyzed alkane borylation2 and can be attributed to steric deactivation of the tert-butyl C–H bonds.
We next compared this C–H hydroxylation reaction to Ir-catalyzed C–H borylation, the current state-of-the-art method for terminal-selective alkane C–H functionalization.2 Hartwig has recently reported that the 3° amine substrate diethylbutylamine undergoes Ir-catalyzed C–H borylation to afford the terminal β- C(sp3)–H borylation product 18-BPin with 6 : 1 selectivity over the analogous terminal δ-C(sp3)–H borylation product 19-BPin (Scheme 2a).12 In this system, none of the 2° C(sp3)–H borylation product 19a-BPin was reported. In contrast, the Pt-catalyzed hydroxylation of this same substrate provides completely different site selectivity, affording a >20 : 1 preference for 19-OH over 18-OH, and a 3 : 1 ratio of 19-OH to 19a-OH (Scheme 2b). These results highlight the complementarity of the two methods. Notably, Pt catalysis has the additional advantages of compatibility with ambient air and moisture as well as applicability to 1°, 2° and 3° amine substrates.
Scheme 2.

Comparison of Selectivity of (a) Ir-Catalyzed C–H Borylation versus (b) Pt-Catalyzed C–H Oxygenation of Diethylbutylamine
A final set of investigations focused on probing the mechanism of this transformation. In particular, we noted that C–H hydroxylation products are formed exclusively under our reaction conditions, despite the presence of a high concentration of chloride. In other studies of Shilov-type alkane C–H oxidation, mixtures of C–H chlorination and C–H hydroxylation products have been reported under related conditions.5,6,20 When the Pt-catalyzed C–H oxidation of dipropylamine was monitored by 1H NMR spectroscopy, we observed rapid initial formation of an intermediate (A), which then fully converted to the C–H hydroxylation product over 24 h (Figure 1). Intermediate A was identified as the terminal C–H chlorination product based on in situ characterization by 1H NMR spectroscopy, as well as by the independent synthesis of this compound. We also confirmed that an authentic sample of A undergoes quantitative conversion to 1 over 24 h at 150 °C in H2O, both in the presence and in the absence of the Pt catalyst. This suggests that a significant quantity of the hydroxylated product 1 is formed from A via a nucleophilic substitution reaction.
Figure 1.

Formation and Decay of Intermediate A in the Pt-Catalyzed C–H Oxidation of Dibutylamine
The data in Figure 1 suggest the feasibility of selectively accessing C(sp3)–H chlorination products, particularly with substrates in which nucleophilic substitution with H2O is slow. As proof-of-principle, we subjected substrates 20 and 24 to our Pt-catalyzed C–H oxidation conditions for short reaction times (2 h). Analysis of the crude reaction mixtures by 1H NMR spectroscopy showed exclusive formation of the C–H chlorination products 21 and 25. While 21 and 25 proved challenging to isolate due to their high volatility, work up of these reactions with KHCO3 resulted in the formation of the corresponding oxazinones 23 and 26, which were isolated in 65% and 55% isolated yield, respectively (Scheme 3). In addition, the treatment of 21 with phenyl isothiocyanate afforded 22 in 73% yield.
Scheme 3.

Pt-Catalyzed C(sp3)–H Chlorination and Subsequent Functionalization of 20 and 24
In conclusion, this report describes a Pt-catalyzed method for the terminal selective C(sp3)–H oxidation of protonated aliphatic amines using CuII as the terminal oxidant. These reactions proceed with some of the highest TONs and selectivities reported to date for Pt-catalyzed alkane oxidation. We propose that protonation of the amine is critical to the success of these transformations for several reasons. First, protonation renders the substrates water-soluble. Second, protonation prevents deactivation of the catalyst/oxidant by amine binding. Finally, the inductive electron withdrawing ammonium cation electronically deactivates proximal C–H bonds, resulting in high selectivity for terminal C(sp3)–H sites that are remote to nitrogen. Efforts to design second generation Pt catalysts that exhibit enhanced efficiencies and terminal selectivities in this transformation are underway in our lab and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge funding from NIH NIGMS (GM073836). ML thanks the NSF for a graduate fellowship. We also acknowledge Dr. Sarah J. Ryan for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental and spectral details for all new compounds and all reactions reported as well full details on selectivity determination from crude reactions. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Crabtree RH. Chem. Rev. 1985;85:245. [Google Scholar]; (b) Shilov AE, Shul’pin GB. Chem. Rev. 1997;97:2879. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]; (c) Crabtree RH. J. Chem. Soc.; Dalton Trans. 2001:2437. [Google Scholar]; (d) Labinger JA, Bercaw JE. Nature. 2002;417:507. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (e) Pérez PJ. Alkane C-H Activation by Single-Site Metal Catalysis. In: Pérez PJ, editor. Catalysis by Metal Complexes. Vol. 38. Dordrecht, The Netherlands: Springer; 2012. [Google Scholar]
- 2.Hartwig JF. Chem. Soc. Rev. 2011;40:1992. doi: 10.1039/c0cs00156b. [DOI] [PubMed] [Google Scholar]
- 3.(a) Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem. Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (c) Nam W. Acc. Chem. Res. 2007;40:522. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (d) White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lersch M, Tilset M. Chem. Rev. 2005;105:2471. doi: 10.1021/cr030710y. [DOI] [PubMed] [Google Scholar]; (b) Stahl SS, Labinger JA, Bercaw JE. Angew. Chem. Int. Ed. 1998;37:2180. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2180::AID-ANIE2180>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]; (c) Shilov AE, Shteinman AA. Coord. Chem. Rev. 1977;24:97. [Google Scholar]
- 5.(a) Gol’dshlger NF, Tyabin MB, Shilov AE, Shteinman AA. Zh. Fiz. Khim. 1969;43:2174. [Google Scholar]; (b) Gol’dshlger NF, Eskova VV, Shilov AE, Shteinman AA. Zh. Fiz. Khim. 1972;46:1353. [Google Scholar]
- 6.(a) Labinger JA, Herring AM, Lyon DK, Luinstra GA, Bercaw JE. Organometallics. 1993;12:895. [Google Scholar]; (b) Labinger JA, Herring AM, Bercaw JE. J. Am. Chem. Soc. 1990;112:5628. [Google Scholar]
- 7.For synthetic applications, see: Kao L-C, Sen A. J. Chem. Soc., Chem. Commun. 1991:1242. Basickes N, Sen A. Polyhedron. 1995;14:197–202. Dangel BD, Johnson JA, Sames D. J. Am. Chem. Soc. 2001;123:8149. doi: 10.1021/ja016280f. Lin M, Shen C, Garcia-Zayas EA, Sen A. J. Am. Chem. Soc. 2001;123:1000. doi: 10.1021/ja001926+. Lee JM, Chang S. Tetrahedron Lett. 2006;47:1375. Weinberg DR, Labinger JA, Bercaw JE. Organometallics. 2007;26:167. doi: 10.1021/om0606643. Chen GS, Labinger JA, Bercaw JE. Organometallics. 2009;28:4899.
- 8.Vitaku E, Smith DT, Njardarson JT. J. Med. Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 9.For examples of the α-C–H oxygenation of amines, see: Kim S, Ginsbach JW, Lee JY, Peterson RL, Liu JJ, Siegler MA, Sarjeant AA, Solomon EI, Karlin KD. J. Am. Chem. Soc. 2015;137:2867. doi: 10.1021/ja508371q. Genovino J, Lütz S, Sames D, Touré BB. J. Am. Chem. Soc. 2013;135:12346. doi: 10.1021/ja405471h. Ling Z, Yun L, Liu L, Fu X. Chem. Commun. 2013;49:4214. doi: 10.1039/c2cc37263k. Liu P, Liu Y, Wong EL-M, Xiang S, Che C-M. Chem. Sci. 2011;2:2187. Park J, Morimoto Y, Lee Y-M, You Y, Nam W, Fukuzumi S. Inorg. Chem. 2011;50:11612. doi: 10.1021/ic201545a. Murahashi S, Naota T, Yonemura K. J. Am. Chem. Soc. 1988;110:8256.
- 10.(a) McNally A, Haffemayer B, Collins BSL, Gaunt MJ. Nature. 2014;510:129. doi: 10.1038/nature13389. [DOI] [PubMed] [Google Scholar]; (b) Smalley AP, Gaunt MJ. J. Am. Chem. Soc. 2015;137:10632. doi: 10.1021/jacs.5b05529. [DOI] [PubMed] [Google Scholar]
- 11.For examples of protected amines as directing groups for C(sp3)–H oxidation, see: Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. He G, Zhao Y, Zhang S, Lu C, Chen G. J. Am. Chem. Soc. 2012;134:7313. doi: 10.1021/ja3023972. Chan KSL, Wasa M, Chu L, Laforteza BN, Miura M, Yu JQ. Nature Chem. 2013;6:146. doi: 10.1038/nchem.1836.
- 12.For a complementary strategy involving terminal-selective Ir-catalyzed C–H borylation of aliphatic amines, see: Li Q, Liskey CW, Hartwig JF. J. Am. Chem. Soc. 2014;136:8755. doi: 10.1021/ja503676d.
- 13.Isaacs NS. Physical Organic Chemistry. 2nd. London: Longman Group Limited; 1995. pp. 146–192. [Google Scholar]
- 14.Inductive EWGs are known to deactivate adjacent C–H bonds towards Pt-catalyzed C(sp3)–H activation. For example, see ref. 7d as well as Periana RA, Taube DJ, Gamble S, Taube H, Satoh T, Fujii H. Science. 1998;280:560. doi: 10.1126/science.280.5363.560.
- 15.For a later example of CuCl2as an oxidant in Shilov chemistry, see ref 7e and: Wang Z, Sugiarti S, Morales CM, Jensen CM, Morales-Morales DM. Inorg. Chim. Acta. 2006;359:1923.
- 16.(a) Maeda Y, Nishimura T, Uemura S. Bull. Chem. Soc. Jpn. 2003;76:2399. [Google Scholar]; (b) Allen SE, Walvoord RR, Padilla-Salinas R, Kozlowski MC. Chem Rev. 2013;113:6234. doi: 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The only internal chain oxidation products detected in more than trace quantities were at the site that is one carbon away from the terminal position. Additionally, only small amounts of products derived from C–H functionalization on the pyrrolidine ring were observed, except with N-ethylpyrrolidine. In the latter case, we estimate that ~15% of ring oxidation products are formed.
- 18.The yields are calculated based on CuCl2•2H2O as the limiting reagent. Since Cu is a 1e− oxidant, 100% yield = 0.5 × mmol CuCl2•2H2O added to the reaction). Yields above 100% reflect regeneration of Cu by ambient air.
- 19.The NMR yield of the C–H hydroxylation to form 4 under our standard conditions was 142%. When this same reaction was conducted under an atmosphere of N2(rather than air), a significantly lower yield of 41% was obtained. Furthermore, when the reaction under air was conducted in a small vessel (4 mL vs 10 mL sealed vial), the yield decreased to 47%. All of these pieces of data are consistent with the proposal that the O2(in air) is turning over the Cu.
- 20.(a) Luinistra GA, Wang L, Stahl SS, Labinger JA, Bercaw JE. J. Organomet. Chem. 1995;504:75. [Google Scholar]; (b) DeVries N, Roe DC, Thorn DL. J. Mol. Catal. A. 2002;189:17. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.