

Abstract
The zebrafish (Danio rerio) is a powerful model organism to study vertebrate development. Though many aspects of zebrafish embryonic development have been described at the morphological level, little is known about the molecular basis of cellular changes that occur as the organism develops. With recent advancements in microfluidics and multiplexing technologies, it is now possible to characterize gene expression in single cells. This allows for investigation of heterogeneity between individual cells of specific cell populations to identify and classify cell subtypes, characterize intermediate states that occur during cell differentiation, and explore differential cellular responses to stimuli. This study describes a protocol to isolate viable, single cells from zebrafish embryos for high throughput multiplexing assays. This method may be rapidly applied to any zebrafish embryonic cell type with fluorescent markers. An extension of this method may also be used in combination with high throughput sequencing technologies to fully characterize the transcriptome of single cells. As proof of principle, the relative abundance of cardiac differentiation markers was assessed in isolated, single cells derived from nkx2.5 positive cardiac progenitors. By evaluation of gene expression at the single cell level and at a single time point, the data support a model in which cardiac progenitors coexist with differentiating progeny. The method and work flow described here is broadly applicable to the zebrafish research community, requiring only a labeled transgenic fish line and access to microfluidics technologies.
Keywords: Developmental Biology, Issue 109, Single cell, zebrafish, multiplexing, cardiac progenitor, heart, development
Introduction
Most current studies of cell and molecular biology are based on population averages. However, important biological events may be masked by these traditional population-based analyses since minor populations can play major roles in biological processes and disease outcome. Understanding gene expression in heterogeneous populations at the single cell level can (and has) lead to relevant biological and clinical insights1,2. Of concern to embryonic development studies, in a larger population of cells, progenitor cells are often underrepresented, making it challenging to detect subtle changes in gene expression that ultimately initiate cell fate decisions3. Similarly, a single cell type may have different expression profiles in response to the microenvironment4. For example, resident endothelial cells in the same organ or in different organs (e.g., aorta or kidney) exhibit significant heterogeneity despite sharing common morphological and functional features5. In addition, cancer cells populating the same tumor can also have varying molecular profiles or mutations at the single cell level6.
In model systems, transcriptomics in single cells has successfully identified new cell populations, characterized intermediate states that occur during cell differentiation, and revealed differential cellular responses to stimuli7,8,9. Such insights would have been masked in conventional population-based studies. Zebrafish embryos are a tremendously under-utilized source of stem, progenitor, and differentiating cells for exploring questions of single cell heterogeneity and molecular regulation of cellular identities during development. Their highly stereotyped, ex vivo development and ease of genetic manipulation make them an excellent model system for this approach10,11. Specifically, a major limitation to interpretation of single cell gene expression data is that reliable identification of novel intermediate cell states during development requires very careful timing of tissue collection9. This is necessary to ensure that heterogeneity between captured cells represents heterogeneity within a tissue at a single time point rather than heterogeneity in gene expression presented by age-dependent cell differentiation. Compared to mice, zebrafish embryo development may be precisely synchronized across a large number of embryos12. Additionally, with large clutch sizes, zebrafish embryos can be used as an abundant source of stem and progenitor cells.
This protocol describes a method to isolate cells from zebrafish embryos and capture single cells using a commercially available integrated microfluidics circuit (IFC) chip and autoprep system for qRT-PCR gene expression analysis. This protocol can be rapidly transferrable to any high throughput multiplexing assays including whole transcriptome sequencing that allows more comprehensive analysis of cellular heterogeneity13. It also offers several advantages to traditional gene expression assays. The single cell isolation protocol yields high viability after FACS, which decreases the proportion of compromised cells that are included in downstream applications. By using an IFC, captured cells may be directly observed to evaluate capture rates and assess cell health morphologically. In addition, this protocol is broadly applicable to the zebrafish research community, requiring only a labeled transgenic fish line and access to microfluidic cell capture technologies.
As proof of principle, single cells derived from cardiac progenitors were isolated and captured on an IFC chip, and then the relative abundance of cardiac differentiation markers was measured by qRT-PCR. Gene expression analysis at the single cell level demonstrates that cardiac progenitors coexist with their differentiating progeny. The insight gained from single-cell profiling of cardiac progenitors may shed light on the heterogeneity in gene expression patterns among cardiac progenitor cells during vertebrate development, which may have been masked in traditional population-based analyses.
Protocol
This protocol requires the use of live, adult zebrafish to produce embryos. The embryos are harvested for tissue collection. It is essential to obtain approval from appropriate ethics review boards to conduct this experiment.
1. Obtain Staged Embryos
The day before the experiment, prepare healthy, adult zebrafish for breeding. Place one male and one female on opposite sides of a clear divider in a breeding tank.
Repeat 1.1 for as many breeding tanks as necessary for sufficient embryo production for the downstream application. Obtain embryos from both wild type fish and transgenic fish that express fluorescent proteins in the cell type of interest. NOTE: The number of embryos needed for downstream applications in Steps 2-8 depends on the relative abundance of the cells of interest at the time point of interest. Though this may vary by cell type, 200 embryos produce 2,000-5,000 sorted cells when the cells of interest represent <1.0% of the total cells at 24 hpf (hours post-fertilization).
The next morning, change the water in the breading tank by transferring fish to a fresh breeding tank and remove the divider. Tilt the tank at an angle to encourage breeding.
- Collect staged embryos.
- Every 15 min, collect embryos by transferring the adults to a fresh breeding tank and passing the eggs which are left behind through a tea strainer. NOTE: Zebrafish embryos develop synchronously when maintained at comparable densities and temperatures.
- Rinse the eggs with Egg Water (0.21 g/L Instant Ocean salts in 1 L double distilled water) and transfer to a petri dish. Transfer the petri dish to a humid incubator at 28.5 °C with air circulation.
Two hours after the last collection, sort fertilized, multicellular embryos into 10 cm petri dishes and reduce density to 50 embryos per dish. Select embryos from a single, 15 min time window of collection for downstream application. Incubate embryos at 28.5°C. NOTE: For example, collect embryos at 8:30, 8:45, 9:00, 9:15, 9:30, 9:45, 10:00 and 10:15 AM. Comparing across time points, if the largest number of fertilized embryos are from the clutches collected at 9:00, then use only these embryos for downstream applications.
2. Set Up for Single Cell Dissociation
Approximately 30 min prior to the time point of interest (18 hpf) remove embryos from their chorion manually with fine forceps.
Collect and label the following for each condition: two 2 ml microcentrifuge tubes, one 40 µm cell strainer, one 35 mm cell culture dish, and two FACS tubes topped with a 35 µm cell strainer.
Chill the following reagents on ice: Egg Water containing 0.21g/L Instant Ocean salts in 1L double distilled water; De-yolking Buffer containing 55 mM NaCl, 1.8 mM KCl, and 1.25 mM NaHCO3; and FACS Buffer containing Leibovitz's L-15 media supplemented with 5% heat-inactivated fetal bovine serum.
Bring 1 ml per sample of Cell Dissociation Reagent 1 to RT. Thaw 1 ml Cell Dissociation Reagent 2 per sample on ice. Bring to RT immediately before use.
3. Single Cell Dissociation
Using a wide bore glass pipette, transfer 100-300 embryos to a 2 ml micro centrifuge tube in a minimum volume of Egg Water.
Euthanize embryos by replacing water with 1 ml ice-cold Egg Water and submerging tube in ice for 20 min. CAUTION! It is critical to obtain approval from proper institutional animal care oversight committee for this euthanasia method (chilling on ice followed by cell dissociation as secondary euthanasia). Standard euthanasia typically requires that embryos <3 days post-fertilization are bleached after they are chilled, which is not appropriate for obtaining viable cells.
Wash the embryos two times with 1 ml ice-cold Egg Water. To wash, use a wide bore glass pipette to remove Egg Water and a P1000 to add Egg Water.
Remove the yolk by replacing the embryo water with 1 ml Deyolking Buffer and triturating 8-12 times with a P1000 tip, or until the yolk is dissolved and only the bodies of the embryos are visible.
Collect the tissue by centrifugation at 300 x g for 1 min. Use a pipette to gently remove the supernatant without disrupting the tissue pellet. Re-suspend in 1 ml Egg Water.
Repeat step 3.5 for a total of three washes, but on the final wash, re-suspend in 1 ml RT Cell Dissociation Reagent 1.
Incubate in Cell Dissociation Reagent 1 for 10 min at RT with tubes placed horizontally. Every 2-3 min, gently triturate with a P1000 pipette to prevent clumping. NOTE: Handle samples gently during trituration steps. Sometime a single, large clump will form in the tube from a tangle of embryo bodies. This will disperse with additional digestion aided by gentle trituration. DO NOT VORTEX.
Collect tissue by centrifugation at 300 x g for 3 min. Remove supernatant and resuspend in 1 ml Cell Dissociation Reagent 2.
- Incubate for 5-15 min at RT. Place the tubes horizontally. Every 2-3 min, gently triturate to prevent clumping. Every 5 min, assess digestion progress.
- To assess digestion progress, dilute 2 µl supernatant into 18 µl FACS Buffer and pipette as a droplet onto a cell culture dish.
- Place a coverslip over the sample and observe under a tissue culture microscope at 10X and 20X magnifications. NOTE: The preparation should appear as a mix of single cells, small clusters, large clusters, and occasional mostly-intact embryo body.
- Visually assess the proportion of the preparation that is single cells and small clusters. NOTE: Over-digestion will reduce viability; under-digestion will reduce single cell yield.
Collect the cell preparation by centrifugation at 300 x g for 5 min. Discard the supernatant and re-suspend in 1 ml cold FACS Buffer.
Moisten a 40 µm cell strainer with FACS Buffer. Pass the cell suspension through the 40 µm cell strainer onto a 35 mm cell culture dish. Wash the cell strainer one time with 1 ml FACS sorting buffer.
Transfer the flow-through to a 2 ml microcentrifuge tube. Collect cells by centrifugation at 300 x g for 5 min and re-suspend in 100 µl FACS Buffer.
Count cell yield using a hemocytometer. Further dilute samples to 5x106 cells per ml, or optimal concentration recommended for FACS machine of choice. Optional-When counting cell yield on the hemocytometer, counterstain dead cells with trypan blue to confirm viability of the cell preparation prior to FACS sorting. NOTE: Preparations that are too dilute will take excess amount of time to sort. Preparations that are too concentrated tend to clump into multimers and are suboptimal for sorting a pure population.
- Reserve 10-20% of each sample to use as unstained controls. For remaining samples, stain dead cells by adding a fluorescent live/dead (L/D) discrimination dye into each sample. DO NOT wash. NOTE: Any fluorescent dye that penetrates cells with compromised cell membranes and stains nucleic acids may be used as the L/D dye, provided that the fluorescence spectra is compatible with the FACS machine of choice and fluorophore labeling cells of interest.
- Do not wash the preparation after adding the dye as some cells will die in transit to FACS purification and should be excluded from the sorted population.
Moisten the 35 µm cell strainer capped FACS tube with 20 µl FACS buffer. Add cells to the strainer and collect by gravity. Store on ice.
4. FACS Enrichment
- Use FACS to enrich for single, live cells expressing the fluorescent marker.
- Set a gate to distinguish cells from debris on a scatter plot of forward scatter (FSC-A) amplitude vs side scatter amplitude (SSC-A), both with linear scaling. Caution! Zebrafish cells are typically smaller than mouse or human cells. This is reflected in a lower basal separation between cells and debris.
- From the gate set in 4.1.1, set a gate to enrich for single cells and exclude multimers using a scatter plot of forward scatter height (FSC-H) and low side scatter height (SSC-H), both with linear scaling. NOTE: Cells with disproportionately high FSC-H and low SSC-H are likely multimers and are excluded from sorting.
- From the gate set in 4.1.2, using single color controls, set a gate to include only live cells using a scatter plot of FSA-A with linear scaling and amplitude of the channel used to detect L/D stain with log scaling. Include L/D negative cells.
- From the gate set in 4.1.3, using single color controls, set a gate to include only live cells with positive fluorescence using a scatter plot of FSA-A with linear scaling and amplitude of the channel used to detect fluorescent protein cell marker with log scaling. Include positive cells.
- Set any compensation controls, if necessary, to account for interference between the L/D stain and fluorescent protein spectra.
- Set sort logic to cells that fall into all of the gates set in step 4.1.
- Using double labeled cells, verify gating for cell sorting. NOTE: Cells for sorting will be cells rather than debris, single cells rather than multimers, L/D negative and fluorescence positive cells.
Sort 2,000-4,000 cells from the population of interest into 5 µl of cold FACS Buffer in a microcentrifuge tube on ice. To minimize shearing and strain on cells during sorting, use the lowest pressures possible for the cell sorter. NOTE: The 5 µl droplet of FACS Buffer serves as a cushion for cells exiting the FACS machine. Collecting 2,000-4,000 cells directly into FACS Buffer eliminates the need for centrifugation prior to loading onto the IFC chips. This strategy is recommended because centrifugation can lead to formation of multimers if cells adhere to one another in the pellet.
- Assess the post-sort viability analysis.
- Transfer 1 µl sorted cells to a fresh FACS tube with 100 µl FACS sorting buffer and 1:1,000 dilution of L/D stain. Pass the cells through the FACS sorter with the gating strategy in Step 4.1. Use the proportion of L/D negative to positive to estimate viability of sorted cells.
5. Load Cells onto Microfluidics Chip
- Using a hemocytometer, measure both the concentration and size of sorted cells.
- Dilute sorted cells to at least 10 µl. Dilute an aliquot of 5 µl cells with 5 µl trypan blue. Load onto hemocytometer and apply coverslip.
- Collect bright field images of all cells in 4 x 4 grids of hemocytometer. Count the number of live cells and calculate live cells/ml. Live cells do not take up trypan blue. NOTE: Use any standard image analysis software to measure the diameter of all live cells. Calculate the average and standard deviation of the cell size, and select an IFC plate suitable for the cell size range of interest. If cell size range straddles an IFC plate cell size range, use multiple plates to capture the full range of cells of interest.
- Load cells on to IFC plate according to manufacturer's instructions (see Materials).
- Dilute cells to 1x106 cells/ml and perform buoyancy optimization as per manufacturer's instructions. NOTE: Buoyancy optimization ensures that cells neither sink to the bottom nor float to the top of the loading well for optimal loading onto IFC chip. The ratio of buffer to cells may vary by cell type, but typically ranges 6:4-7:3 of cells:buffer.
- Add cells to primed-IFC plate. Load plate into compatible fluidics machine, and run a cell loading script to push cells through microfluidics circuit and into capture lanes.
- Confirm that cells are lodged in capture sites on IFC plate.
- Remove IFC plate from fluidics machine. Mount the IFC plate on a microscope equipped with a plate adaptor.
- Collect snapshots of bright field and fluorescence for each capture site at 10X magnification. NOTE: Brightfield capture times may range from 10-50 ms, depending on lamp intensity. Fluorescence capture times may range from 250-750 ms, depending on fluorophore brightness. Avoid overexposing cells to prevent photodamage.
6. cDNA Synthesis
- Perform cell lysis in situ according to IFC plate manufacturer's instructions.
- Add lysis buffers to wells on IFC plate. Load plate onto compatible fluidics machine and run cell lysis script as per manufacturer's instructions.
- Perform reverse transcription according to IFC plate manufacturer's instructions.
- Add reverse transcription reagents to IFC plate. Load plate onto compatible fluidics machine. Run reverse transcription script. Sample cycling conditions: 1 cycle at 25°C for 10 min, 1 cycle at 42°C for 1 hr, then 1 cycle at 85°C for 5 min.
- Perform pre-amplification with gene-specific probes according to IFC plate manufacturer's instructions. NOTE: Due to their greater specificity with low-copy numbers, use fluorogenic-labeled probes rather than probes optimized for use with intercalator dyes.
- Pool primers and dilute to final 180 nM each. Add pooled primers to IFC plate. Load plate onto compatible fluidics machine. Run preamplification script.
Perform pre-amplification with the following cycle conditions: 1 cycle at 95°C for 10 min, then 18 cycles with denaturing at 95°C for 15 sec then annealing and extension at 60°C for 4 min. Store pre-amplified cDNA at 4°C until harvesting. Harvest cDNA from microfluidic plate according to manufacturer's instructions and store at -20˚C until use.
7. Select cDNA from Single Cells for Single Target qRT-PCR
- Dilute cDNA in 25 µl dilution reagent according to manufacturer's instructions. NOTE: Approximate final yield is 28 µl of pre-amplified cDNA per cell. Reserve an aliquot of diluent for use as a no-template control in qRT-PCR.
- Referring to bright field and fluorescent images recorded in step 5.3.2, score each capture site. Manually count and record the number of cells. Assess and record whether each cell is fluorescent. NOTE: Each individual site may contain 0, 1, or >1 cell, where >1 cell includes capture sites that contain multiple cells and/or unidentifiable debris. If the images are sufficient quality, a pixel value may also be assigned to quantify the intensity of the fluorescence signal.
- Select samples for qRT-PCR analysis.
- Select cDNA samples from capture sites identified in step 7.2 that contain exact 1 cell with positive fluorescence. Pool 1 µl aliquots of cDNA from each sample. NOTE: This is the "Pool" positive control used to determine whether genes of interest are expressed in any of the selected cells.
- Select cDNA from at least one capture site that contains exactly 0 cells as a negative control. Use diluent from step 7.1.2 as a no-template control.
- Run qRT-PCR assay.
- Use only probes used for pre-amplification in step 6.4.1. Load all samples in triplicate. Cycling conditions for a 10 µl reaction on a 384 well plate: 1 cycle 50°C 2 min, 1 cycle 95°C for 10 min, 40 cycles with denaturing at 95°C 15 sec then annealing and extension at 60°C for 1 min.
8. Data Analysis
Validate qRT-PCR products by standard gel electrophoresis. Confirm product is a single band at expected molecular weight (varies for each probe). NOTE: If a probe produces multiple bands or a single band at an inappropriate size, then specificity is questionable, and this excludes the gene from analysis.
Examine all amplification curves and account for any abnormalities14. Calculate the average CT value for each sample/gene combination15. NOTE: CT values for positive controls typically range from 10-30. CT values for negative controls typically range from 35-40 (no amplification). CT values 30-35 are considered very low expression and should be interpreted with caution14.
- Report data
- If samples fall in the CT=10-30, data may be also reported as fold change in transcript abundance relative to a control sample. NOTE: Fold change equals 2^(-ΔΔCT) where ΔCT is relative to a housekeeping gene by subtracting the average CT of the sample from the housekeeping gene and ΔΔCT is the differences in ΔCT between the sample and a positive control15.
Representative Results
As proof of principle, gene expression was assessed to explore differentiation dynamics during cardiac development. In zebrafish, cardiac progenitors arise from a mesodermal population of cells that migrate to the anterior lateral plate mesoderm where they fuse to form the linear heart tube. Prior to fusion, cardiac progenitors begin to express the transcription factor nkx2.5 (NK2 homeobox 5), which is thought to be the earliest specific marker of cardiac progenitors16,17. Here, a previously described BAC transgenic fish Tg(nkx2.5:ZsYellow)18, abbreviated nkx2.5:ZsY was used to examine cardiac differentiation markers in single cells at the 18 somite stage, 18 hr post fertilization (hpf)12. This was the earliest time point at which ZsYellow signal was visually detectable. As previously described for this transgenic line, ZsYellow labels cardiac progenitors, as well as a few extra-cardiac cells that give rise to the pharyngeal arch endothelial cells at 28 hpf19 (Figure 1A-B, data not shown). At 18 hpf, nkx2.5:ZsY embryos were dissociated into a single cell suspension then stained with Sytox Blue to exclude dead cells. Live, ZsYellow positive, Sytox Blue negative cells were FACS sorted using either a MoFloXDP or Sony SH800Z sorter equipped with 100 µm nozzle (Figure 1C-F). To assess purity of the sorted population and post-sort viability, an aliquot of sorted cells were stained with Sytox Blue and evaluated the percentage of events that fell within the original sorting gate (Figure 1G-J).
After sorting, cells were entered into an integrated microfluidic circuit (IFC) chip work flow (Figure 2A) according to manufacturer's instructions. A hemocytometer, combined with trypan blue exclusion was used to measure cell diameter, concentration, and viability (Figure 2B, and data not shown). Cell buoyancy was optimized (6.5:3.5 cells:buffer) according to manufacturer's instruction, and cells were loaded onto an IFC to capture 5-10 µm diameter cells. To assess capture efficiency, fluorescence and/or bright field signals were imaged at all capture sites, a tiling function in FIJI was used to stitch a single picture of the microfluidics plate. Each capture site contained 0, 1, or >1 individual cells (Figure 2C-F). As expected from FACS enrichment, captured cells expressed ZsYellow (Figure 2G). Capture efficiency exceeded 90% in 5 individual experiments using nkx2.5:ZsY sorted cells, and at least 70% of capture sites were occupied by a single cell (data not shown). Cells were lysed, RNA isolated, cDNA synthesized and specific target genes were amplified, all according to manufacturer's instructions.
A subset of cell capture sites were selected for qRT-PCR analysis as described in the protocol above. Specifically, elongation factor 1a (efl1a)20 and glyceraldehyde-3 phosphate (gapdh)21 were assayed as housekeeping genes expected to be expressed in every cell; GATA binding protein 4 (gata4)22 and NK2 homeobox 5 (nkx2.5) as early cardiac progenitor markers; ISL LIM homeobox 1 box 1 (isl1) as a second heart field marker23,24; and myosin light chain (myl7) and ventricular myosin heavy chain (vmhc) 25 to mark ventricular cardiomyocytes. Gene-specific probes were validated using cDNA from 48 hpf embryos (Figure 3). qRT-PCR was used to assess relative gene expression of these genes in 45 single cells from 18 hpf embryos, a no-template negative control, and a pooled population positive control containing cDNA from all 96 capture sites. Raw CT values for 40 representative cells and controls are shown in Table 1. Notably, from the pool of 46 cells examined, 6 cells were excluded from analysis because all gene expression CT values exceeded CT=35.0, and it is unknown whether these high CT values are attributable to true, very low expression levels or sample degradation. Since the range of CT values for the housekeeping gene, ef1a, was too broad to compare gene expression between samples, and many CT values exceeded 30, CT values were visualized as a heat map (Figure 3). Comparing across samples, substantial heterogeneity in gene expression was observed, and cells were classified cells as Type 1-5 based on expression pattern (Figure 3).
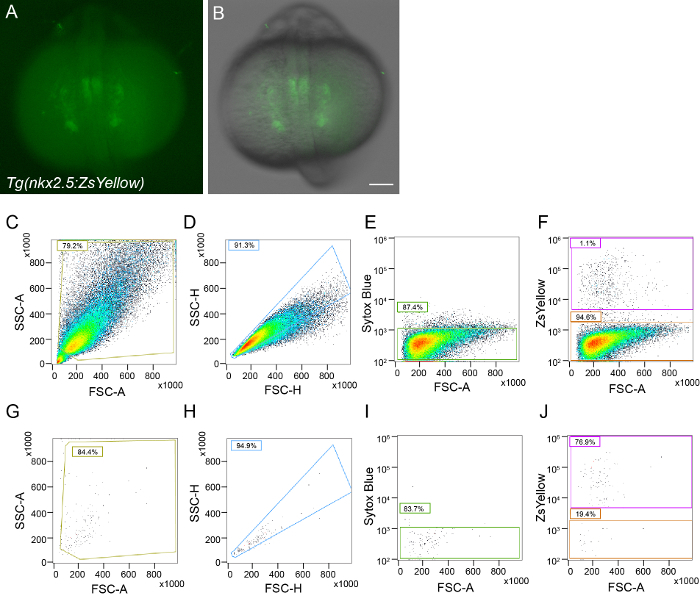 Figure 1. Single cell isolation of zebrafish nkx2.5:ZsY positive cells at 18 hpf. Whole mount images of representative Tg(nkx2.5:ZsYellow) embryo at 18 hpf with (A) ZsYellow fluorescence alone or (B) merged with bright field image. (C-F) Representative FACS gating strategy to enrich for ZsYellow positive cells and (G-J) post-sort analysis with (C,G) FSC/SSC size gating, (D,H) doublet discrimination, (E,I) Live/Dead gating, and (F,J) sorted population. Scale bar is 100 µm. Please click here to view a larger version of this figure.
Figure 1. Single cell isolation of zebrafish nkx2.5:ZsY positive cells at 18 hpf. Whole mount images of representative Tg(nkx2.5:ZsYellow) embryo at 18 hpf with (A) ZsYellow fluorescence alone or (B) merged with bright field image. (C-F) Representative FACS gating strategy to enrich for ZsYellow positive cells and (G-J) post-sort analysis with (C,G) FSC/SSC size gating, (D,H) doublet discrimination, (E,I) Live/Dead gating, and (F,J) sorted population. Scale bar is 100 µm. Please click here to view a larger version of this figure.
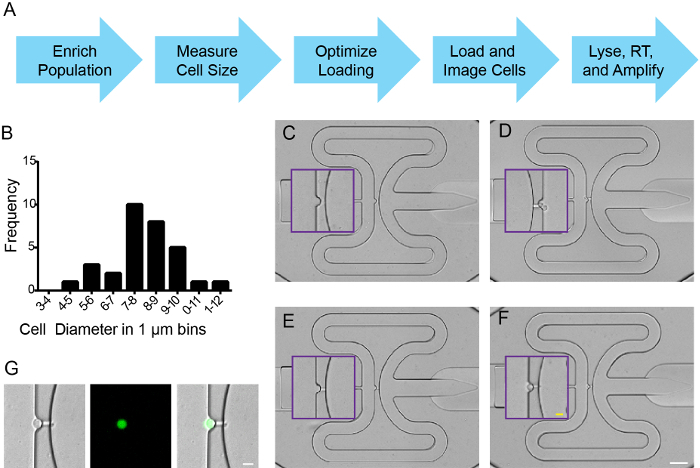 Figure 2. Single cell capture of zebrafish nkx2.5:ZsY positive cells. (A) Work flow. (B) Cell size distribution for sorted cells from Tg(nkx2.5:ZsYellow) embryos isolated at 18 hpf. (C-F) Representative cell capture events on IFC plate where (C) is an empty well, (D) contains two cells, (E) has a single cell lodged in the fluidics channel as a "channel capture", and (F) is a single captured cell. Purple boxes mark inset for magnified view of capture sites. (G) Single cell capture with brightfield, ZsYellow and merged images. (C-F) White scale bar is 50 µm; yellow scale bar is 10 µm. (G) Scale bar is 10 µm. Please click here to view a larger version of this figure.
Figure 2. Single cell capture of zebrafish nkx2.5:ZsY positive cells. (A) Work flow. (B) Cell size distribution for sorted cells from Tg(nkx2.5:ZsYellow) embryos isolated at 18 hpf. (C-F) Representative cell capture events on IFC plate where (C) is an empty well, (D) contains two cells, (E) has a single cell lodged in the fluidics channel as a "channel capture", and (F) is a single captured cell. Purple boxes mark inset for magnified view of capture sites. (G) Single cell capture with brightfield, ZsYellow and merged images. (C-F) White scale bar is 50 µm; yellow scale bar is 10 µm. (G) Scale bar is 10 µm. Please click here to view a larger version of this figure.
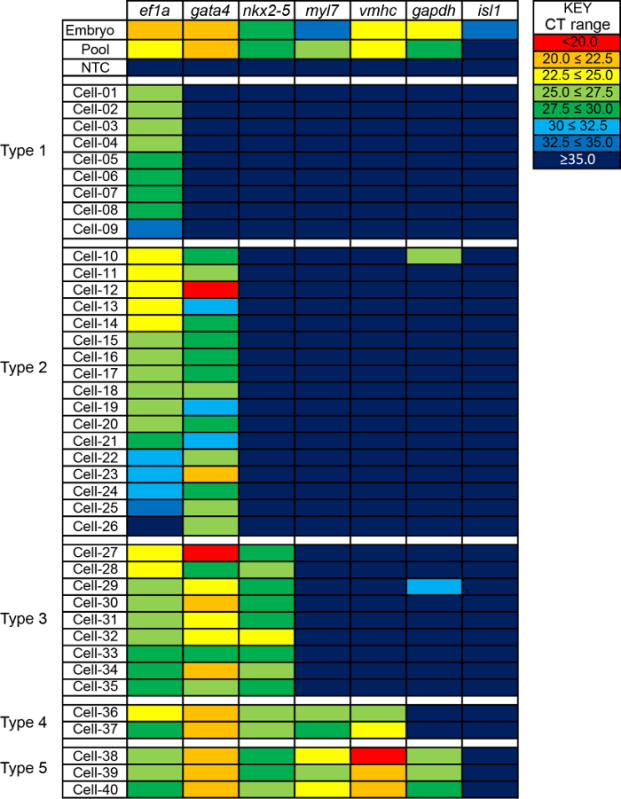 Figure 3. Gene expression analysis of capture single zebrafish nkx2.5:ZsY positive cells. Gene expression by qRT-PCR in positive controls (embryos at 2 days-post-fertilization, pooled cDNA from single cells), negative control (no-template) and 40 single cells (Cell 01-40). Raw CT values were color coded based as described in the Key. Ef1a = elongation factor 1a, gata4 = GATA binding protein 4, nkx2.5 = NK2 homeobox 5, myl7 = myosin light chain 7, vmhc = ventricular myosin heavy chain, gapdh = glyceraldehyde-3 phosphate, and isl1 = ISL LIM homeobox 1. Please click here to view a larger version of this figure.
Figure 3. Gene expression analysis of capture single zebrafish nkx2.5:ZsY positive cells. Gene expression by qRT-PCR in positive controls (embryos at 2 days-post-fertilization, pooled cDNA from single cells), negative control (no-template) and 40 single cells (Cell 01-40). Raw CT values were color coded based as described in the Key. Ef1a = elongation factor 1a, gata4 = GATA binding protein 4, nkx2.5 = NK2 homeobox 5, myl7 = myosin light chain 7, vmhc = ventricular myosin heavy chain, gapdh = glyceraldehyde-3 phosphate, and isl1 = ISL LIM homeobox 1. Please click here to view a larger version of this figure.
| Cycle Threshold (CT) | ||||||||
| ef1a | gata4 | nkx25 | myl7 | vmhc | gapdh | isl1 | ||
| Embryo | 20.69 | 20.92 | 28.59 | 32.87 | 26.30 | 27.00 | 33.57 | |
| Pool | 26.4 | 22.4 | 27.7 | 27.4 | 24.5 | 29.2 | 40.0 | |
| NTC | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Type 1 | Cell-01 | 25.1 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 |
| Cell-02 | 25.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-03 | 26.3 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-04 | 27.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-05 | 28.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-06 | 28.2 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-07 | 29.3 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-08 | 30.7 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-09 | 34.3 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Type 2 | Cell-10 | 23.5 | 28.0 | 40.0 | 40.0 | 40.0 | 26.3 | 40.0 |
| Cell-11 | 23.5 | 27.2 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-12 | 23.7 | 17.9 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-13 | 24.1 | 32.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-14 | 24.9 | 27.5 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-15 | 25.9 | 28.6 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-16 | 26.0 | 28.1 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-17 | 27.0 | 27.7 | 40.0 | 40.0 | 40.0 | 38.5 | 40.0 | |
| Cell-18 | 27.0 | 27.4 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-19 | 27.0 | 30.2 | 40.0 | 40.0 | 40.0 | 38.2 | 40.0 | |
| Cell-20 | 27.0 | 28.2 | 40.0 | 40.0 | 40.0 | 39.2 | 40.0 | |
| Cell-21 | 27.7 | 30.3 | 40.0 | 40.0 | 40.0 | 38.6 | 40.0 | |
| Cell-22 | 30.4 | 26.2 | 40.0 | 40.0 | 40.0 | 38.3 | 40.0 | |
| Cell-23 | 31.3 | 22.4 | 40.0 | 40.0 | 40.0 | 39.7 | 40.0 | |
| Cell-24 | 31.5 | 28.8 | 40.0 | 40.0 | 40.0 | 39.5 | 40.0 | |
| Cell-25 | 33.8 | 27.4 | 40.0 | 40.0 | 40.0 | 37.3 | 40.0 | |
| Cell-26 | 40.0 | 27.4 | 40.0 | 40.0 | 40.0 | 36.6 | 40.0 | |
| Type 3 | Cell-27 | 24.7 | 19.9 | 28.8 | 40.0 | 40.0 | 40.0 | 40.0 |
| Cell-28 | 24.8 | 28.4 | 26.3 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-29 | 25.1 | 23.5 | 27.6 | 40.0 | 40.0 | 32.5 | 40.0 | |
| Cell-30 | 26.3 | 21.4 | 27.5 | 40.0 | 40.0 | 39.9 | 40.0 | |
| Cell-31 | 26.9 | 24.8 | 28.2 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-32 | 27.0 | 22.5 | 23.8 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-33 | 27.8 | 27.6 | 27.7 | 40.0 | 40.0 | 40.0 | 40.0 | |
| Cell-34 | 28.1 | 21.9 | 26.6 | 40.0 | 40.0 | 37.7 | 40.0 | |
| Cell-35 | 29.3 | 26.5 | 28.1 | 40.0 | 40.0 | 38.3 | 40.0 | |
| Type 4 | Cell-36 | 24.8 | 20.3 | 26.0 | 27.4 | 26.6 | 40.0 | 40.0 |
| Cell-37 | 28.7 | 22.3 | 25.9 | 28.9 | 22.9 | 38.5 | 40.0 | |
| Type 5 | Cell-38 | 25.5 | 20.0 | 29.3 | 23.0 | 19.9 | 25.9 | 40.0 |
| Cell-39 | 25.9 | 21.8 | 28.6 | 25.1 | 21.7 | 25.7 | 40.0 | |
| Cell-40 | 28.9 | 22.0 | 27.0 | 23.0 | 21.3 | 27.6 | 40.0 |
Table 1. Raw CT values.
Discussion
The method described herein uses expression of a fluorescent protein under control of a cell-type specific promoter to enrich a population of cardiac progenitor cells from zebrafish embryos for use in microfluidic assisted single cell capture system to assess expression of a subset of cardiac genes in single cells. Provided that FACS laser excitation and emission capabilities are compatible with the fluorophore(s) of choice, this method can be used for any fluorescent reporter line. Many zebrafish reporter lines are already in existence, and transgenic fish carrying novel reporters can be generated in as little as 3 months. Additionally, this work flow may be adapted to produce cDNA from the whole transcriptome for single cell-RNA sequencing. To do so, steps 5-7 will need to be modified slightly for use with chemistries optimized for preparing cDNA for generating libraries suitable for RNA sequencing. However, it is advisable to validate population heterogeneity by using the method described here prior to pursing high throughput sequencing applications.
It is important to note that there are some limitations to the described method. First, use of integrated microfluidic circuit (IFC) plates requires specialized, costly equipment. However, microfluidic chips offer substantial advantages over traditional 96 well and 384 well plate formats for assaying single cells. By flowing cells and reagents through micrometer scale lanes, single cells are positioned into single capture sites where they can be directly observed to confirm that they are single cells in good health. RNA extraction and cDNA synthesis occur in situ on the IFC chip using very low volumes for each cell. Integrated fluidic chips, at the time of this writing, are commercially available through only one source. Though non-commercial fabrication has been performed by several groups, it is outside of the capacity of most laboratories. Steps 5-7 may need to be modified for platforms produced by other companies or in-house fabrications. Second, though commercially available IFC plates can accommodate up to 96 genes, the genes selected are limited by the availability of fluorogenic-labeled gene probes validated for use in zebrafish. Third, zebrafish cells are typically smaller than mammalian cells, and this small size presents challenges to sample processing. A small change in cell diameter translates into a large change in cell volume, reducing available material and decreasing the likelihood of detecting low expressing genes.
There are several additional considerations for this protocol. In step 1, it is important to only use embryos fertilized within a short time window. Keeping temperature constant and without limiting oxygen conditions, zebrafish embryos develop synchronously. The ultimate readout of this protocol (relative gene expression from single cells) cannot discriminate the source of heterogeneity between individual cells. For this reason, interpretation of intercellular heterogeneity relies on carefully staged embryos and synchronous development. In steps 2-3, there is a tradeoff between dissociation and viability. Though step 4 selects for viable cells, and step 3 contains several filtering steps that help exclude multimers, over-digestion will reduce the viable material available for FACS and potentially reduce sorted cell yield. Steps to troubleshoot low viability include reducing digestion time, reducing trituration intensity, and optimizing the number of embryos in the starting material. FACS enrichment of the cell population of interest (step 4) is arguably the most critical step in this protocol. Stringent sorting criteria are essential to generate a single cell preparation containing only live cells expressing the fluorescent protein of interest. Contaminating cells from other populations could lead to false representation of intercellular heterogeneity. Notably, FACS enrichment of a population of interest is not restricted to the two color strategy shown in this protocol. More complex strategies can produce more refined cell populations and are a rapid way to extend the methodology reported in the proof of principle study. In step 5, cells may be directly observed to confirm the number of cells captured, cell health, and expression of fluorescence protein. But, depending on cell size and brightness, the fluorophore of interest may not be visually detectable and is not a reliable readout. Successful completion of steps 6-8 requires careful adherence to manufacturer's protocols.
As proof of principle, expression levels of a subset of known cardiac markers were assessed in single derived from a previously described BAC transgenic zebrafish line nkx2.5:ZsY to examine cardiac differentiation markers in individual nkx2.5:ZsY positive cells at 18 hr post fertilization (hpf). To explore the heterogeneity of differentiation markers expressed in captured nkx2.5:ZsYexpressing cells, a suite of genes were assayed including housekeeping genes (ef1a, gapdh), transcription factors known to be turned on early in cardiac specification (gata4, nkx2.5), a second heart field progenitor marker (isl1), and genes known to be turned on later during cardiac differentiation (myl7, vmhc). The relative abundance of each gene was measured by qRT-PCR by calculating cycle threshold (CT). The lowest CT value calculated was 17.9 and the highest was 40, representing no amplification (Table 1). CT values of >35.0 were considered below detection.
Of the 46 cells in the data set, 6 were excluded due to failed amplification of any genes. The remaining cells were sub-categorized cells into 5 types based on expression of gata4, nkx2.5, myl7, and vmhc, and were sorted by ef1a value within groups. Though gapdh was originally included as a possible housekeeping gene, expression was highly variable across cells, making it unsuitable as a housekeeping gene. Gapdh expression likely better represents changes to energy metabolism associated with cardiac differentiation in this cell type. Interestingly, there was one cell in which ef1a was un-detectable but another gene was detectable (gata1 in cells 26). This suggests that while ef1a is generally an adequate housekeeping gene to differentiate between successful and unsuccessful single cell reverse transcription reactions, it may not be ideal for all applications. The lack of ef1a expression in cell 26 could be due to single cell transcriptional dynamics. Recent single-cells studies demonstrate that gene transcription is fundamentally stochastic, alternating between phases of rapid and negligible transcriptional activity26. This transcriptional bursting model suggests that use of a housekeeping gene for quantitative comparisons between single cells may be altogether inappropriate.
In the Type 1 group, ef1a is the only gene detectable. These cell may comprise either nkx2.5:ZsY negative cells or cells with very low expression of the other genes of interest. Type 2 cells, with the exception of Cell-26, expressed both ef1a and gata4 with no correlation in the relative expression levels of these genes. Notably, nkx2.5 expression in Type 1 and Type 2 cells may be due to sub-optimal FACS stringency, transcriptional bursting, transgene leakiness, or sub-threshold expression levels. Type 3 cells expressed detectable levels of ef1a, gata4, and nkx-2.5. These cells are likely cardiac progenitor cells and may include differentiating atrial cardiomyocytes. Type 4 cells expressed detectable levels of ef1a, gata4, nkx2.5, myl7 and vmhc and are likely cells differentiating into ventricular cardiomyocytes. Type 5 cells expressed ef1a, gata4, nkx2.5, myl7, vmhc, and gapdh. The addition of detectable levels of gapdh suggests that these cells have the capacity for enhanced glycolytic metabolism found in differentiated cardiomyocytes. It was intriguing that, despite sorting for ZsYellow positive cells from Tg(nkx2.5:ZsYellow), embryos, 21/40 cells had sub-threshold levels of nkx2.5 in our qRT-PCR analysis. This could be due to transcriptional bursting or could reflect differences in transcript processing between endogenous nkx2.5 and ZsYellow. Importantly, the second heart field marker isl1 was undetectable in any cells or cDNA pooled from all of our capture sites. In sum, comparing across samples, substantial heterogeneity was evident at the single cell level, suggesting that at 18 hpf, nkx2.5:ZsY+ cells comprise cardiac progenitors as well as progeny at different stages of differentiation.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. C. Geoffrey Burns for fish stock. The authors are grateful to UNC Flow Cytometry Core Facility, UNC-CGIBD AAC core for resources enabling this project, and the ZAC facility for animal care. L.S. is supported by NIH T32 grant HL069768-13 (PI, Nobuyo Maeda). N.F. is supported by NSF Graduate Research Fellowship NSF-DGE-1144081. This study was supported by NIH P30DK034987 grant (to UNC Advanced Analytics Core), American Heart Association Scientist Development Grant 13SDG17060010 and Ellison Medical Foundation New Scholar Grant AG-NS-1064-13 (to Dr. Qian), and NIH R00 HL109079 grant (to Dr. Liu).
References
- Speicher MR. Single-cell analysis: toward the clinic. Genome medicine. 2013;5 doi: 10.1186/gm478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay IC, Voet T. Single cell genomics: advances and future perspectives. PLoS genetics. 2014;10:e1004126. doi: 10.1371/journal.pgen.1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco E, et al. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5643–5650. doi: 10.1073/pnas.1408993111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Marshall D. Microfluidics for single cell analysis. Current opinion in biotechnology. 2012;23:110–119. doi: 10.1016/j.copbio.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:1193–1202. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- Diaz-Cano SJ. Tumor heterogeneity: mechanisms and bases for a reliable application of molecular marker design. International journal of molecular sciences. 2012;13:1951–2011. doi: 10.3390/ijms13021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, et al. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell stem cell. 2013;13:492–505. doi: 10.1016/j.stem.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, et al. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Developmental cell. 2010;18:675–685. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Treutlein B, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB. Genetics and early development of zebrafish. Trends in genetics : TIG. 1989;5:283–288. doi: 10.1016/0168-9525(89)90103-0. [DOI] [PubMed] [Google Scholar]
- Liu J, Stainier DY. Zebrafish in the study of early cardiac development. Circulation research. 2012;110:870–874. doi: 10.1161/CIRCRESAHA.111.246504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PloS one. 2014;9:e78644. doi: 10.1371/journal.pone.0078644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, McMurray HR, Land H, Almudevar A. On non-detects in qPCR data. Bioinformatics. 2014;30:2310–2316. doi: 10.1093/bioinformatics/btu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Lyons I, et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes & development. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- Chen JN, Fishman MC. Zebrafish tinman homolog demarcates the heart field and initiates myocardial differentiation. Development. 1996;122:3809–3816. doi: 10.1242/dev.122.12.3809. [DOI] [PubMed] [Google Scholar]
- Zhou Y, et al. Latent TGF-beta binding protein 3 identifies a second heart field in zebrafish. Nature. 2011;474:645–648. doi: 10.1038/nature10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paffett-Lugassy N, et al. Heart field origin of great vessel precursors relies on nkx2.5-mediated vasculogenesis. Nature cell biology. 2013;15:1362–1369. doi: 10.1038/ncb2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurley AT, Callard GV. Characterization of housekeeping genes in zebrafish: male-female differences and effects of tissue type, developmental stage and chemical treatment. BMC molecular biology. 2008;9:102. doi: 10.1186/1471-2199-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R, Dodd A, Lai D, McNabb WC, Love DR. Validation of zebrafish (Danio rerio) reference genes for quantitative real-time RT-PCR normalization. Acta biochimica et biophysica Sinica. 2007;39:384–390. doi: 10.1111/j.1745-7270.2007.00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbedzija GN, Chen JN, Fishman MC. Regulation in the heart field of zebrafish. Development. 1998;125:1095–1101. doi: 10.1242/dev.125.6.1095. [DOI] [PubMed] [Google Scholar]
- de Pater E, et al. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development. 2009;136:1633–1641. doi: 10.1242/dev.030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hami D, Grimes AC, Tsai HJ, Kirby ML. Zebrafish cardiac development requires a conserved secondary heart field. Development. 2011;138:2389–2398. doi: 10.1242/dev.061473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelon D, Horne SA, Stainier DY. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Developmental biology. 1999;214:23–37. doi: 10.1006/dbio.1999.9406. [DOI] [PubMed] [Google Scholar]
- Molina N, et al. Stimulus-induced modulation of transcriptional bursting in a single mammalian gene. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:20563–20568. doi: 10.1073/pnas.1312310110. [DOI] [PMC free article] [PubMed] [Google Scholar]