

Abstract
During neural development, growing axons extend to multiple synaptic partners by elaborating axonal branches. Axon branching is promoted by extracellular guidance cues like netrin-1 and results in dramatic increases to the surface area of the axonal plasma membrane. Netrin-1-dependent axon branching likely involves temporal and spatial control of plasma membrane expansion, the components of which are supplied through exocytic vesicle fusion. These fusion events are preceded by formation of SNARE complexes, comprising a v-SNARE, such as VAMP2 (vesicle-associated membrane protein 2), and plasma membrane t-SNAREs, syntaxin-1 and SNAP25 (synaptosomal-associated protein 25). Detailed herein isa multi-pronged approach used to examine the role of SNARE mediated exocytosis in axon branching. The strength of the combined approach is data acquisition at a range of spatial and temporal resolutions, spanning from the dynamics of single vesicle fusion events in individual neurons to SNARE complex formation and axon branching in populations of cultured neurons. This protocol takes advantage of established biochemical approaches to assay levels of endogenous SNARE complexes and Total Internal Reflection Fluorescence (TIRF) microscopy of cortical neurons expressing VAMP2 tagged with a pH-sensitive GFP (VAMP2-pHlourin) to identify netrin-1 dependent changes in exocytic activity in individual neurons. To elucidate the timing of netrin-1-dependent branching, time-lapse differential interference contrast (DIC) microscopy of single neurons over the order of hours is utilized. Fixed cell immunofluorescence paired with botulinum neurotoxins that cleave SNARE machinery and block exocytosis demonstrates that netrin-1 dependent axon branching requires SNARE-mediated exocytic activity.
Keywords: Neuroscience, Issue 109, TIRF Microscopy, DIC, Timelapse, Exocytosis, Axon Branching, Immunocytochemistry, Botulinum
Introduction
Recent estimates suggest that the human brain contains 1011 neurons with 1014 synaptic connections1, highlighting the importance of axon branching in vivo. Extracellular axon guidance cues such as netrin-1 guide axons to appropriate synaptic partners and stimulate axonal branching, thereby increasing synaptic capacity2-5. Netrin-1-dependent axonal arborization involves substantial plasma membrane expansion6, which we hypothesized requires delivery of additional membrane components via SNARE complex dependent exocytic vesicle fusion7.
Investigating the role of SNARE-mediated exocytosis in netrin-1 dependent axon branching is complicated by several factors. First, the heterogeneity of cortical neurons increases the sample size required to identify significant effects, complicating single cell techniques like imaging. Second, although biochemical techniques permit observation of changes that occur at the population level, they lack the temporal and spatial resolution necessary to localize plasma membrane expansion to the axon in the time frame of axon branching. Lastly, although axon branches form over hours, the cellular changes that contribute to axonal extension may begin within minutes and occur on the order of seconds, thus extending the temporal scope for experimental consideration.
We outline a multi-technique approach that addresses these diverse temporal and spatial scales of exocytosis and axon branching, and thus enhances our understanding of the fundamental cellular mechanisms. Utilizing these approaches provides evidence that supports a critical role for SNARE-mediated exocytosis in axon branching.
Protocol
Statement of research ethics: All experiments involving animals detailed herein are subject to the rules and regulations of the UNC Committee on Animal Care and to NIH standards for the care and use of laboratory animals.
1. Preparation and Plating of Dissociated Cortical Neurons
Euthanize timed-pregnant females by CO2 inhalation followed by cervical dislocation. Remove whole brains from the skulls of embryonic day 15.5 (E15.5) mice and microdissect cortices from each hemisphere. For detailed instructions regarding the microdissection of embryonic mouse cortex please see Viesselmann et al8.
Place no more than 4 cortices in 1 ml of dissecting media in a sterile 1.6 ml microtube. Add 120 μl of 10x trypsin and invert the tube 3 - 5 times to mix.
- Using a hemocytometer, count cells to seed cell culture dishes. Note: 1 cortical hemisphere provides approximately three million cells.
- For the SDS-resistant SNARE complex biochemistry assay, plate six million cells per 35 mm dish and utilize two dishes per condition. Note: This is simplified by using 6 well plates when comparing multiple conditions.
- For branching assays, plate neurons on nitric acid washed circular coverslips at a density of 250,000 cells per 35 mm dish, for DIC imaging of axon branching plate at a density of 150,000. These densities avoid overcrowding and simplify analysis.
- For live cell TIRF microscopy, resuspend two million neurons per transfection in nucleofection solution at RT.
- Add 100 μl of cell suspension to microtube containing 10 μg of VAMP2-pHlourin expression plasmid and electroporate with a nucleofector according to manufacturer protocol9.
- After transfections immediately add 500 μl of trypsin quenching media, remove suspension from cuvette and plate cells at a density of 400,000 cells per 35 mm PDL-coated glass bottom dish.
Plate all cortical neurons in 2 ml of serum free media.
2. SNARE Complex Formation Assay
Note: SDS-resistant SNARE complexes were processed and analyzed as originally described10 with the modifications detailed below. For validated alternative antibodies to the ones used here, see the materials section.
Stimulate E15.5 cortical neurons 2 days in vitro (DIV) with 250 ng/ml netrin-1 or sham control for 1 hr prior to lysis.
Prior to processing samples, cool centrifuge to 4 °C, and set water bath temperature to 37 °C, and heat block to 100 °C.
- Remove cell dishes from incubator and place on ice.
- Aspirate media from cells, replace with ~ 2 ml ice cold Phosphate buffered saline (PBS) gently 2 times for 1 - 2 min per wash.
- For this assay, use 2 wells per condition.
- Aspirate PBS from the first well of a condition and replace with 250 μl homogenization buffer. Leave on ice for 5 min, and then using a cell lifter, homogenize cells on ice.
- Aspirate the PBS from the next well of the same condition and add the recently homogenized mixture to the well. This will increase protein concentrations. Repeat for all conditions.
- Upon completion, pipette homogenized solution to a pre-cooled 1.6 ml microtube on ice and add 20% TritonX-100 to reach a final concentration of 1% TritonX-100. Triturate mix 10 times with 1,000 μl pipette while minimizing bubbles.
Incubate tubes on ice for 2 min to solubilize proteins. Following incubation, centrifuge for 10 min at 6,010 rcf at 4 °C to pellet non-solubilized material. Move lysate supernatant into new chilled 1.6 ml tube on ice.
Perform protein concentration analysis using Bradford assay11 and dilute samples to a final protein concentration of 3 - 5 mg/ml with remaining homogenization buffer supplemented with 1% TritonX-100 and 5x sample buffer supplemented with B-Mercaptethanol (BME).
Divide each experimental sample evenly into 2 tubes. Incubate one sample at 37 °C for 30 min (SNARE complexes), and the other at 100 °C for 30 min (SNARE monomers). Invert tubes intermittently to keep samples in solution.
Freeze samples at -20 °C until ready to separate via SDS-PAGE. Prior to running samples via electrophoresis using standard techniques, reboil previously boiled samples for 5 min at 100 °C, but thaw 37 °C samples at RT.
Run duplicates of samples (30 - 40 μg of protein per sample) on both an 8% and a 15% gel; the 8% provides ideal separation of the complex, whereas the 15% is used to quantify SNARE protein monomers (SNAP-25 ~25kDa, VAMP2 ~18kDa, syntaxin-1 ~35kDa). Maintain power source at 70 V until the dye line is through the stacking gel and increase voltage to 90V. Run gels until the dye runs off.
To preserve monomers, transfer proteins to 0.2 μm nitrocellulose membrane on ice using cold transfer buffer with 20% Methanol (MeOH) added, at 70 V for 45 min.
Dry membrane in a covered dish for 2 hr to O/N at RT (O/N provides the best results).
Block SNARE complex membranes in 10% Bovine Serum Albumin (BSA) for 1 hr at RT.
- Prepare primary antibodies (recognizing specific SNARE complex components) at a dilution of 1:1,000 and probe O/N at 4 °C on a rotary shaker.
- Rinse blots in Tris Buffered Saline plus 0.1% Tween-20 (TBS-T) 3 times for 5 min each.
Prepare fluorescent secondary antibody solutions at a dilution of 1:20,000 in 1% BSA in TBS-T and probe for 1 hr, covered at RT. Repeat Step 2.12.1 after 1 hr.
Image blots on a fluorescent scanning machine equipped with software suite and quantify both the SNARE protein complex (immunoreactive bands above 40kDa) and monomer bands (immunoreactive bands at 25 kDa, 18 kDa, 35 kDa for SNAP-25, VAMP2 and syntaxin-1, respectively) using manufacturer's instructions for drawing rectangular ROIs.
3. Imaging Exocytic Events via TIRF Microscopy
Note: This protocol requires specialized microscopy equipment including an environmental chamber to maintain temperature, humidity and CO2, an inverted TIRF microscope equipped with an epifluorescent illumination, a high magnification/ high numerical aperture (NA) TIRF objective, an automated XYZ stage, and a sensitive Charge Coupled Device (CCD) detector. This protocol uses a fully automated inverted microscope equipped with a 100x 1.49NA TIRF objective a solid state 491 nm laser and an Electron Multiplying CCD (EM-CCD). All equipment is controlled by imaging and laser control software. Prior to the beginning the imaging protocol power on the environmental chamber, stage, lamp, computer, and camera.
Select objective within imaging software. Once the objective is in place, fasten the objective heater around the collar.
Confirm that objective is lowered completely before securing the stage incubator in the stage slot. Pour distilled water into the stage incubator inlets evenly to prevent spill.
Turn on the incubation system. Open the valve to the CO2 tank and confirm the pressure is appropriate for the system per manufacturer's instructions. Allow chamber some time to reach 5% CO2 and 37 °C. Prior to adding the imaging dish, place an empty dish in the incubator to avoid water condensation on the objective.
Power on the laser source.
Open stage incubator and add immersion oil to the lens. Place sample in incubator and stabilize with stability arms or a dish weight. Raise the objective until the oil makes contact with the bottom of the sample. Switch to transmitted light illumination and find neuronal focal plane through the oculars.
Start laser software and connect to the laser control software. Set illumination to widefield and select the objective being used (100X 1.49 NA TIRF is recommended). Set refractive index of the sample (cells ~1.38). Adjust laser intensity by unchecking "TTL" for the 491 nm laser. Adjust the slider to 100 then bring it back down to a value between 20 - 40%. Recheck "TTL".
Focus on sample again in transmitted light illumination. Go to imaging software and select 491 nm laser illumination and open shutter. Fine adjust the focal point of the laser on the ceiling and center the point to the center of the closed field diaphragm with the condenser removed. Place condenser upside down on the optical bench, so as not to scratch lens.
Replace condenser and go to TIRF software and set penetration depth (PD) to 110 nm. Switch from widefield illumination to TIRF illumination mode for imaging.
- Find VAMP2-phluorin expressing cells through the oculars using widefield epifluorescence with epifluorescent light source.
- Adjust imaging parameters (exposure time, gain and laser power) to maximize signal to noise ratio and dynamic range using the minimal exposure time and laser intensity to reduce photobleaching and phototoxicity (for example: exposure between 50 - 100 msec, with a 15 - 30 gain at 30% maximum laser power).
- Set continuous autofocus per cell. Acquire a timelapse image set with acquisition occurring every 0.5 sec for 5 min. For the netrin-1 stimulated condition, add 500 ng/ml netrin-1 to dish of cells in a laminar flow hood and return the dish to the incubator for 1 hr prior to imaging.
- To quantify the frequency of exocytic events normalized per cell area and time, open image stacks in ImageJ by dragging the file into the window or going to File> Open> Filename (Figure 2A Inset 1).
- To remove stable fluorescent signals that do not represent vesicle fusion events, create an average z projection of the entire stack using Image>Stack>Z-Project>Projection type: Average Intensity. Subtract this mean image from each image in the timelapse using Process> Image Calculator> Image1: your stack Operation: Subtract Image2 newly created average z projection. This emphasizes exocytic events (Figure 2A Inset 2 - 3).
- Count exocytic fusion events by eye. Exocytic events are defined as the appearance of a diffraction-limited fluorescence signal, which rapidly diffuses as VAMP2-phluorin diffuses within the plasma membrane (Figure 2A Inset 6).
- Use the Threshold command to highlight the first image using Image>Adjust>Threshold> adjust slider until the whole cell is threshold highlighted (Figure 2A Inset 7 - 8).
- Under Analyze, choose SetMeasurements and check area and display label. Select the thresholded cellular area using the wand tracing tool and press 'm' to measure (Figure 2A Inset 7 - 8).
- Transfer both the exocytic fusion events count, the cell area measurements, and the elapsed imaging time to a spreadsheet to calculate number of exocytic events/mm2/time (Figure 2A Inset 9).
4. Differential Interference Contrast (DIC) Timelapse Microscopy of Axon Branching
Note: A complete protocol and demonstration for a general approach to DIC imaging is available12. While this protocol utilizes DIC, other transmitted light microscopy methods may be used for the same purposes (for example: phase contrast).
At 2 DIV, place glass bottom imaging dish containing untransfected neurons in pre-warmed environmental chamber to maintain a humid environment with 5% CO2 and 37 °C. Utilize a microscope equipped with a 60X Plan-Apochromat, 1.4 NA DIC objective lens and high NA condenser for best image quality and resolution.
Adjust the focal plane to find neurons through the oculars using transmitted light illumination.
- Using the multi area acquisition function on imaging software, find and save the XYZ locations of at least 6 cells. Position the stage so that neurons of interest fit within the field of view for best results.
- Stimulate with 250 ng/ml netrin-1 and press 'start multi area acquisition'. Sequentially acquire images at each position every 20 sec for 24 hr, pausing acquisition and refocusing as necessary.
- Review images in imaging software using Apps>Review Multi Dimensional Data>open file name. Identify stable axon branches (20 µm long) that form during the imaging session. Use the line draw tool to measure a stableaxon branch from the base at the axon to the tip to ensure the 20 µm length qualification is met.
- In a spreadsheet, record the frame number after netrin stimulation when membrane protrusions initiate in areas where branches later form as well as the frame number after netrin stimulation when nascent branches reach 20 µm in length.
- Multiply the number of frames between protrusion and branch formation by 20 sec to calculate the formation time per branch.
5. Toxin Manipulations and Fixed Cell Immunofluoresence
At 2 DIV, treat neurons in experimental conditions with 250 ng/ml netrin-1 and/or 10 nM Botulinum A toxin (BoNTA), which cleaves the SNARE protein SNAP25 and effectively inhibits SNARE mediated exocytosis13, plus 250 ng/ml netrin-1. Leave one set of untreated neurons as control condition. CAUTION: BoNTA requires the use of eye and respiratory protection when preparing and using solutions. Maintain all contaminated materials as biohazardous material and autoclave as soon as possible.
At 3 DIV (24 hr post treatment) aspirate media, and immediately replace with at least 1 ml of PHEM fix (see Table 1 for details) warmed to 37 °C. Incubate for 20 min in the dark at RT.
Rinse 3x with PBS (for future use seal with parafilm and store at 4 °C).
Place coverslips in dark, humidified staining chamber and permeabilize the cell membranes for 10 min in freshly diluted 0.2% TritonX-100 in PBS (made from 10% TritonX-100 stock).
Remove permeabilization solution and rinse with 50 µl of 1x with PBS. Block for 30 min in ~50 µl of 10% Bovine Serum Albumin in PBS (BSA-PBS) or 10% Boiled Donkey Serum in PBS at RT.
Rinse with 1x PBS again. Incubate coverslips in 50 µl of primary antibody (1:1,000 βIII tubulin, to confirm neuronal identity) in 1% BSA-PBS for 1 hr at RT, in the dark.
Perform 3x 5 min washes in PBS. Incubate coverslips in 50 µl of spectrally-distinct secondary antibody for tubulin (1:400), and fluorescent phalloidin (varies by excitation: 488 phalloidin at 1:400, 647 phalloidin at 1:100) in 1% BSA-PBS for 1 hr.
Wash 3x 5 min in 1x PBS and mount coverslips onto slides in mounting media.
Vacuum excess mounting media from the edges of the coverslip and seal with clear nail polish.
- Collect widefield epifluorescence images on an inverted microscope with a 40X 1.4NA objective, epifluorescent capabilities and EM-CCD.
- Manually analyze branching using ImageJ. Open image stacks in ImageJ by dragging the file into the window or going to File> Open> Filename (Figure 4A inset 1).
- Using line>segmented line, trace the axon, defined as the longest neurite extending from the soma(Figure 4A inset 2 - 3).
- Using Analyze>Tools>ROI Manager, save the axon tracing as a region of interest. Trace and save each axon branch, defined as a neurite ≥20 µm in length. Include only primary branches (outgrowths directly sprouting from the axon) in the analysis (Figure 4A inset 3 - 4).
- Press 'm' to measure the saved regions of interest, and transfer lengths in pixels to a spreadsheet to calculate the length in µm.
- Divide the number of axon branches ≥20 µm in length, by the length of the axon in µm divided by 100 to get number of axon branches per 100 µm length.
Representative Results
Utilizing in vitro biochemical techniques to assay the amount of SDS-resistant SNARE complexes in a population of neurons. Figure 1 shows the resulting western blot following completion of the SDS-resistant SNARE complex assay probed for SNAP-25, syntaxin1A and VAMP2.
TIRF microscopy at the basal cell membrane provides high resolution images of individual exocytic fusion events in single cells. Figure 2A demonstrates the image analysis methodology for identifying VAMP2-phluorin mediated exocytic events. The inset shows a single exocytic event as vesicle fusion occurs and as VAMP2-phluorin diffuses within the plasma membrane. Figure 2B shows an example of an exocytic event occurring over time (seconds) in a cortical neuron. Zoomed insets denote the soma, an axon branch and an axonal growth cone showing the spatial utility of this assay. Circles denote single exocytic fusion events, which can be seen via TIRF microscopy.
Timelapse DIC imaging of netrin stimulated axon branching reveals the timing of axon branch formation. Figure 3 depicts the formation of an axon branch in real time following netrin stimulation. White arrowheads denote the initial protrusion from a branch site. Black arrowheads denote a fully formed, stable branch of at least 20μm measuring from the main axon to the branch tip. Netrin dependent increases in axon branching occurs following netrin dependent increases in exocytic fusion.
Fixed cell immunocytochemistry combined with pharmacological inhibition of SNARE activity shows that SNARE mediated exocytosis is a requisite for cortical axon branching. Figure 4A, outlines a step by step process of branch tracings performed in ImageJ. Figure 4B shows representative images of cortical neurons at 3DIV. Conditions are as follows: untreated, stimulated with 250 ng/ml netrin, or treated with BoNTA and 250 ng/ml netrin for 24 hr.
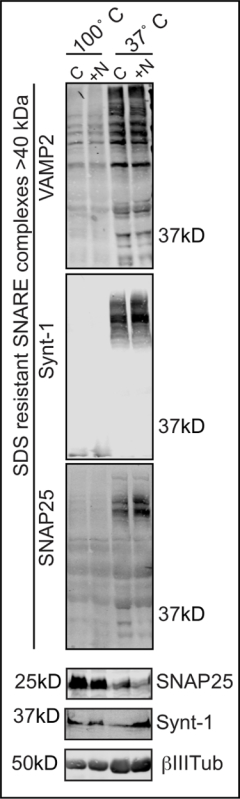 Figure 1. Using the SDS Resistance of SNARE Complexes as a Quantifiable Metric of SNARE Formation In Vitro. A representative western blot probed for SNARE complex members VAMP2, Syntaxin1A and SNAP-25 and the loading control BIII tubulin. Both SNARE proteins in complex and identifiable monomers are shown. Please click here to view a larger version of this figure.
Figure 1. Using the SDS Resistance of SNARE Complexes as a Quantifiable Metric of SNARE Formation In Vitro. A representative western blot probed for SNARE complex members VAMP2, Syntaxin1A and SNAP-25 and the loading control BIII tubulin. Both SNARE proteins in complex and identifiable monomers are shown. Please click here to view a larger version of this figure.
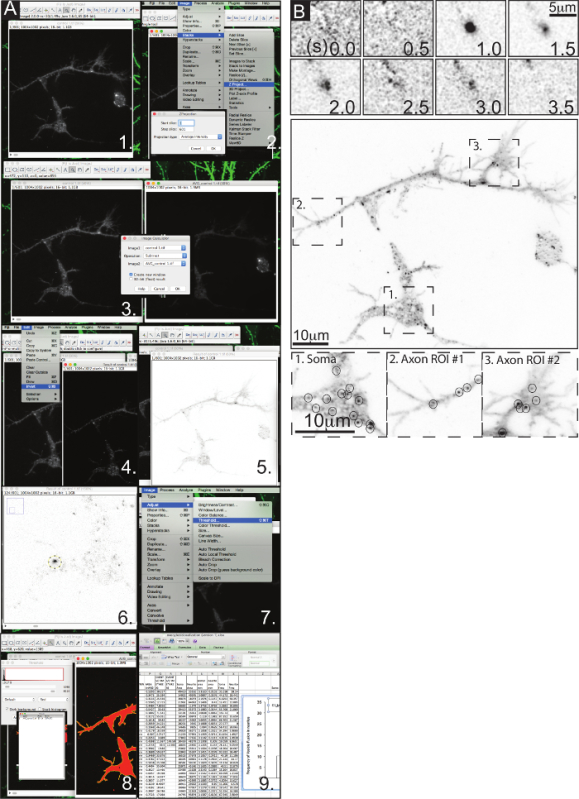 Figure 2. Live Cell Imaging and Quantification of Exocytic Vesicle Fusion Events in Cortical Neurons. (A) Step by step outline of the exocytic vesicle fusion image analysis as it was manually performed using ImageJ. (B) Inset panels show a single VAMP2-pHlourin vesicle fusion event as it occurs over time. The second panel featuring a TIRF microscopy image of a whole cortical neuron expressing VAMP2-pHlourin at 2DIV. Dotted line boxes denote the regions of interest as shown below: soma, axon branch, and an axonal growth cone. Circles with the regions of interest denote single vesicle fusion events. Please click here to view a larger version of this figure.
Figure 2. Live Cell Imaging and Quantification of Exocytic Vesicle Fusion Events in Cortical Neurons. (A) Step by step outline of the exocytic vesicle fusion image analysis as it was manually performed using ImageJ. (B) Inset panels show a single VAMP2-pHlourin vesicle fusion event as it occurs over time. The second panel featuring a TIRF microscopy image of a whole cortical neuron expressing VAMP2-pHlourin at 2DIV. Dotted line boxes denote the regions of interest as shown below: soma, axon branch, and an axonal growth cone. Circles with the regions of interest denote single vesicle fusion events. Please click here to view a larger version of this figure.
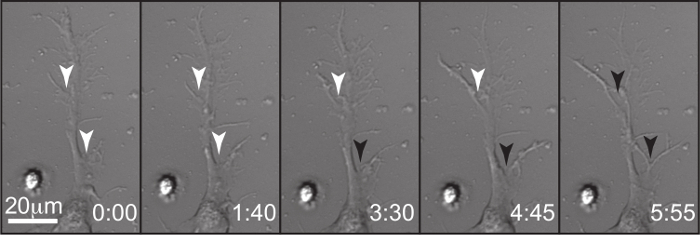 Figure 3. Long Term Live Cell DIC Imaging Reveals Timing of Netrin-1 Dependent Axon Branching. DIC live cell images of a cortical neuron showing the formation of axon branches in response to netrin stimulation. White arrowheads denote points of initial protrusion prior to the neurite reaching 20 μm in length. Black arrowheads denote bonafide branches (length ≥20 μm). Time denoted as hour:min. Please click here to view a larger version of this figure.
Figure 3. Long Term Live Cell DIC Imaging Reveals Timing of Netrin-1 Dependent Axon Branching. DIC live cell images of a cortical neuron showing the formation of axon branches in response to netrin stimulation. White arrowheads denote points of initial protrusion prior to the neurite reaching 20 μm in length. Black arrowheads denote bonafide branches (length ≥20 μm). Time denoted as hour:min. Please click here to view a larger version of this figure.
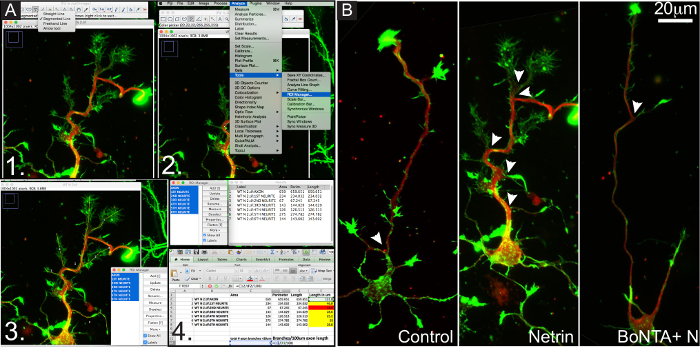 Figure 4. Fixed Cell Immunofluorescence Coupled with Toxin Inhibition of Exocytosis Shows that Exocytosis is Required for Netrin-1 Dependent Axon Branching. (A) Outline of the tracing and analysis steps for quantification of axon branching in a netrin stimulated cortical neuron at 3DIV. (B) Representative images of cortical neurons at 3DIV of each experimental condition: untreated, stimulated with 250 ng/mL netrin, or treated with 10 nM BoNTA toxin plus 250 ng/ml netrin. Green is F-Actin (phalloidin), red is βIIItubulin. Arrows denote points of axon branches ≥20 μm in length. Please click here to view a larger version of this figure.
Figure 4. Fixed Cell Immunofluorescence Coupled with Toxin Inhibition of Exocytosis Shows that Exocytosis is Required for Netrin-1 Dependent Axon Branching. (A) Outline of the tracing and analysis steps for quantification of axon branching in a netrin stimulated cortical neuron at 3DIV. (B) Representative images of cortical neurons at 3DIV of each experimental condition: untreated, stimulated with 250 ng/mL netrin, or treated with 10 nM BoNTA toxin plus 250 ng/ml netrin. Green is F-Actin (phalloidin), red is βIIItubulin. Arrows denote points of axon branches ≥20 μm in length. Please click here to view a larger version of this figure.
| Solution | Recipe |
| Homogenization buffer | 10 mM HEPES-NaOH pH 7.4,150 mM NaCI, 1 mM EGTA. Plus necessary protease and phosphatase inhibitors dependent upon cell type |
| 2x Sample buffer | 60 mM Tris-HCI pH 6.75, 5% (v/v) fI-mercaptoethanol, 2% (w/v) SDS, 10% (w/v) glycerol, 0.007% (w/v) bromophenol blue |
| PHEM fixative | 60 mM PIPES pH 7.0, 25mM HEPES pH 7.0, 10mM EGTA pH 8.0, 2 mM MgCl2, 0.12 M Sucrose, 4% PFA |
| Mounting Media | 20 mM TRIS pH 8.0, 0.5% N-Propyl-gallate, 90% high quality glycerol, MilliQ H2O |
| 10X SDS Running Buffer | Dissolve 30.0 g of Tris base, 144.0 g of glycine, and 10.0 g of SDS in 1,000 ml of H2O, pH 8.3. Dilute to 1x. |
| 10X Transfer Buffer | Dissolve 30.3 g Tris for 250 mM and 144.1 g Glycine for 1.92 M solution in 1 L H2O; (for 1 L 1x buffer add 100ml 10x solution to 200 ml MeOH and 700 ml cold DI H2O) |
| Serum Free Media (SFM) | 50 ml: 0.5 ml L-Glutamine, 1 ml B27, 48.5 ml Neurobasal Media |
| Trypsin Quenching Media (TQM) | 50 ml: 0.5 ml L-Glutamine, 2.5 ml FBS, 47 ml Neurobasal Media |
| TRIS Buffered Saline | 50 mM Tris-Cl, pH 7.5, 150 mM NaCl in 1 L H2O; (For TBS-T add 1 ml Tween-20) |
Table 1. Solutions
Discussion
Axon branching is a fundamental neurodevelopmental process and underpins the vast neuroconnectivity of the mammalian nervous system. Understanding the mechanisms involved in localized plasma membrane expansion is integral to our understanding of both normal and pathological neurodevelopment. The use of a multipronged approach incorporating both population level and single cell level methodologies enhances reproducibility and increases spatial and temporal resolution without compromising population level analysis. At the single cell level, utilizing both fixed and live cell approaches to examine netrin-dependent axon branching provides a three-fold benefit: the ability to collect a large sample size across multiple experiments, temporal resolution with DIC imaging, and spatiotemporal resolution of exocytic fusion using TIRF microscopy.
The SDS-resistant SNARE complex assay allows a measurement of SNARE activity that accounts for the heterogeneity of cortical neurons in culture. SNARE complexes are extremely stable; whereas most other protein complexes are dissociated by the addition of SDS. SNARE complexes are SDS resistant, though not temperature resistant. This property makes quantifying the amount of both the SNARE complex and SNARE monomers possible. Due to the necessity of running multiple SDS-PAGE gels at varying acrylamide percentages, seeding cells at a density no less than 6 million per well in order to acquire sufficient amounts of protein is important. As this assay requires common laboratory reagents, the ability to perform western blots and the antibodies for endogenous SNARE proteins, this is a relatively cost effective and simple method for producing quantifiable data on exocytic SNARE complex formation. Although omitted from this protocol, this method may be used in conjunction with a variety of pharmacological manipulations and multiple cell types. The main limitation to this method is the necessity for a relatively high number of cells. Those working in model systems where cell number and thus protein is limiting should take this into consideration.
TIRF microscopy-based imaging of exocytic events in single neurons allows the visualization of individual exocytic fusion events of VAMP2-pHluorin producing quantifiable data that can be categorized spatially or temporally. For example, exocytic events can be compared throughout the entire neuron or to specific cellular locations such as the soma, the axon or other neurites. Furthermore the frequency of fusion for single exocytic events at various cellular locations can be calculated from frame rate. This imaging protocol may be performed on any commercially available inverted TIRF system equipped with a stage incubator, an appropriate laser source, and an EMCCD camera, although adjustments to laser power and exposure time are necessary on a per cell basis. This protocol can be used in conjunction with any cell type that is suitable for transfection, although dividing cell lines must be seeded appropriately to allow for the isolation of single cells for imaging. Careful consideration of post processing for images is recommended, as the high level of background noise in live cells can make identifying fusion events difficult. Utilizing the z-projection function in ImageJ, we created average z projections of image stacks then subtracted the resulting image from the original stack. This process significantly decreased background, increasing the signal-to ratio, and simplifying identification of fusion events.
Time lapse DIC imaging is a simple and effective method for long-term spatiotemporal examination of neuronal morphogenesis in vitro. As transmitted light illumination has little negative impact on neurons, this approach can, in theory, be utilized for long timecourses on the order of days. Utilizing the multi area acquisition function increases the number of cells that can be simultaneously imaged, but this number will be constrained by the chosen frame rate and the distance between regions of interest. Thus, balancing the desired number of cells and required frame rate for the phenomena of interest to optimize the experimental throughput is necessary.
In both live cell imaging protocols, the use of a stage incubator, which maintains CO2, humidity and temperature, allows for acute stimulation or pharmacological manipulation in real time though we did not take advantage of this possibility. Thus the live cell imaging approaches outlined here allow an effective means by which the spatial and temporal aspects of netrin dependent axon branching, and other cellular phenomena can be examined in real time.
Our SNARE complex biochemistry assays and live cell imaging produced quantifiable measures of population level and single cell level changes. Fixed cell immunocytochemistry, coupled with pharmacological manipulations of exocytic activity allowed us to both retrieve temporal information about axon branching and connect branching to the requirement of exocytosis. Dependent on which pharmacological manipulation is desired, it may be necessary to perform a concentration assay, to identify the most effective but least cytotoxic dose possible for cells of interest. These assays require relatively few cells and only three DIV, making them both cost and time effective.
Together, the assays outlined above are powerful tools in elucidating the roles of specific proteins in exocytic fusion and axon branching. Using this approach coupled with a genetic knockout model of the E3 ubiquitin ligase TRIM9, we showed that TRIM9 acts as a regulator of exocytosis through an interaction with SNAP25, and thus constrains axon branching7. In sum, our multi-pronged strategy to examine membrane addition during axon branching is likely to be instrumental in dissecting detailed molecular and cellular mechanisms governing plasma membrane expansion necessary to neuronal development and cell shape change.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the National Institutes of Health: RO1-GM108970 (SLG) and F31-NS087837 (CW).
References
- Drachman DA. Do we have brain to spare? Neurology. 2005;64(12):2004–2005. doi: 10.1212/01.WNL.0000166914.38327.BB. [DOI] [PubMed] [Google Scholar]
- Serafini T, et al. Netrin-1 Is Required for Commissural Axon Guidance in the Developing Vertebrate Nervous System. Cell. 1996;87(6):1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- Métin C, Deléglise D, Serafini T, Kennedy TE, Tessier-Lavigne M. A role for netrin-1 in the guidance of cortical efferents. Development. 1997;124(24):5063–5074. doi: 10.1242/dev.124.24.5063. [DOI] [PubMed] [Google Scholar]
- Kennedy TE, Serafini T, de la Torre JR, Tessier-Lavigne M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 1994;78(3):425–435. doi: 10.1016/0092-8674(94)90421-9. [DOI] [PubMed] [Google Scholar]
- Sun KLW, Correia JP, Kennedy TE. Netrins: versatile extracellular cues with diverse functions. Development. 2011;138(11):2153–2169. doi: 10.1242/dev.044529. [DOI] [PubMed] [Google Scholar]
- Pfenninger KH. Plasma membrane expansion: a neuron's Herculean task. Nature Reviews Neuroscience. 2009;10(4):251–261. doi: 10.1038/nrn2593. [DOI] [PubMed] [Google Scholar]
- Winkle CC, McClain LM, Valtschanoff JG, Park CS, Maglione C, Gupton SL. A novel Netrin-1-sensitive mechanism promotes local SNARE-mediated exocytosis during axon branching. The Journal of Cell Biology. 2014;205(2):217–232. doi: 10.1083/jcb.201311003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viesselmann C, Ballweg J, Lumbard D, Dent EW. Nucleofection and Primary Culture of Embryonic Mouse Hippocampal and Cortical Neurons. Journal of Visualized Experiments. 2011. p. e2373. [DOI] [PMC free article] [PubMed]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Hayashi T, et al. Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. The EMBO Journal. 1994;13(21):5051–5061. doi: 10.1002/j.1460-2075.1994.tb06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Centonze Frohlich V. Phase Contrast and Differential Interference Contrast (DIC) Microscopy. Journal of Visualized Experiments. 2008. [DOI] [PMC free article] [PubMed]
- Blasi J, et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 1993;365(6442):160–163. doi: 10.1038/365160a0. [DOI] [PubMed] [Google Scholar]