

Abstract
The translational machinery, i.e., the polysome or polyribosome, is one of the biggest and most complex cytoplasmic machineries in cells. Polysomes, formed by ribosomes, mRNAs, several proteins and non-coding RNAs, represent integrated platforms where translational controls take place. However, while the ribosome has been widely studied, the organization of polysomes is still lacking comprehensive understanding. Thus much effort is required in order to elucidate polysome organization and any novel mechanism of translational control that may be embedded. Atomic force microscopy (AFM) is a type of scanning probe microscopy that allows the acquisition of 3D images at nanoscale resolution. Compared to electron microscopy (EM) techniques, one of the main advantages of AFM is that it can acquire thousands of images both in air and in solution, enabling the sample to be maintained under near physiological conditions without any need for staining and fixing procedures. Here, a detailed protocol for the accurate purification of polysomes from mouse brain and their deposition on mica substrates is described. This protocol enables polysome imaging in air and liquid with AFM and their reconstruction as three-dimensional objects. Complementary to cryo-electron microscopy (cryo-EM), the proposed method can be conveniently used for systematically analyzing polysomes and studying their organization.
Keywords: Neuroscience, Issue 109, Polysome, translational control, Atomic Force Microscopy, ribosome, brain, sucrose gradient, imaging, polysomal profile
Introduction
The synthesis of protein is the most energy consuming process in cells1,2. Hence, it is not surprising that protein abundances are mainly controlled at the translational rather than transcriptional level3,4,5. The polysome is the fundamental macromolecular component that converts the mRNA information into functional proteinaceous readouts. Polysomes are thus far recognized as macromolecular complexes where several translational controls converge6-13. Despite hundreds of studies on ribosome structure14- 16, the detailed molecular insights into the dynamics of translation and the topology of polysomes has encountered a limited interest. As a consequence, the organization of the native polysomal ribonucleoprotein complex and its potential effect on translation are still rather obscure issues. Polysomes may hide still unknown ordered and functional organizations, potentially mirroring what nucleosomes and epigenetic controls have represented for the transcription field. Indeed, the investigation of this intriguing hypothesis requires additional studies and new technical approaches. In this line, structural techniques and atomic force microscopy may fruitfully collaborate to unravel new mechanisms for controlling gene expression, similarly to what was achieved for the nucleosome17,18.
Since the discovery of the ribosome14-16, its structure has been extensively characterized in prokaryotes19,20, yeast21 and more recently in human22, providing the molecular description of the mechanisms at the base of protein synthesis. Polysomes were initially recognized on the membrane of the endoplasmic reticulum, forming typical 2D geometrical organizations14. As mentioned before, polysome assembly has not been the object of constant interest as the ribosome structure. In the past, polysomes have been studied essentially by transmission EM-based techniques. Only recently, cryo-EM techniques enabled the 3D reconstruction of purified polysomes from in vitro translation systems23-26, human cellular lysates27 or in cells28. These techniques offered more refined information about the ribosome-ribosome organization in polysomes23, 26-28 and a preliminary molecular description of the contact surfaces of adjacent ribosomes in wheat germ polysomes24. Thus, cryo-EM tomography allows the disclosure of ribosome-ribosome interactions with molecular detail, but it is burdened by extensive post-processing and reconstruction analyses that require heavy computational resources for data handling. Moreover, to obtain the molecular detail of ribosome-ribosome interactions, a highly resolved map of the ribosome is required, and this kind of reference ribosome is available only for a few species. Importantly, cryo-EM tomography is not able to detect free RNA. Therefore, new techniques are required to fully understand the organization of the translation machinery.
Beside cryo-EM, AFM has been also employed as a useful tool for direct imaging of polysomes in lower eukaryotes29-33 and humans27. Compared to EM, AFM requires no sample fixation or labeling. In addition, measurements can be carried out in near physiological conditions and with the unique possibility of clearly identifying both ribosomes and naked RNA strands27. Imaging of single polysomes can be performed relatively quickly, obtaining thousands of images at nano-resolution with little post-processing effort compared to the extensive and heavy post-processing and reconstruction analyses required by cryo-EM microscopy. Consequently, AFM data handling and analysis does not need expensive workstations and high computing power. As such, this technique collects information on polysomal shapes, morphological characteristics (such as height, length and width), ribosome densities, the presence of free RNA and the number of ribosomes per polysome27 with a higher throughput than cryo-EM. In such way, AFM represents a powerful and complementary approach to EM techniques to portray polysomes27.
Here we present a complete pipeline from purification to data analysis where AFM is applied to image and analyze mouse brain polysomes. The proposed protocol focuses on purification issues and on the accurate deposition of polysomes on mica substrates that are used for AFM imaging. In addition to conventional particle analysis that can be easily performed with common software used by the AFM community, an ImageJ34 plugin, called RiboPick, is presented for counting the number of ribosomes per polysome27, 35.
Protocol
The practices used to obtain the mouse tissues were approved by the Body for the Protection of Animals (OPBA) of the University of Trento (Italy), protocol no. 04-2015, as per art.31 Legislative Decree no. 26/2014. All mice were maintained at the Model Organism Facility of the Centre for Integrative Biology (CIBIO), University of Trento, Italy.
Caution: To avoid any RNA degradation of the samples, prepare all buffers using DEPC-treated water for minimizing RNase contamination.
1. Preparation of Polysomes from Whole Brains
- Gathering brain tissues (15 min)
- Euthanize wild-type mouse strain C57BL/6 with CO2 asphyxiation for 5 min36. Carefully dissect the brain out from the skull36, place the tissue into a 1.5 ml tube and immediately place it in liquid nitrogen. Store at -80 °C until use.
- Preparation of lysate (30 min)
- Pulverize the whole brain tissue using a mortar and pestle under liquid nitrogen.
- Transfer about 25 mg powder to a cold microcentrifuge tube and immediately (to avoid the thawing of the pulverized tissue) add 0.8 ml of Lysis Buffer (see Table 1) and disrupt the cell by pipetting up and down 25 times quickly.
- Centrifuge the tube at 12,000 x g for 1 min at 4 °C to pellet cellular debris.
- Transfer the supernatant to a new microcentrifuge tube and keep the tube on ice for 15 min.
- Centrifuge the tube at 12,000 x g for 5 min at 4 °C to pellet nuclei and mitochondria.
- Transfer the supernatant to a new microcentrifuge tube.
- Store the supernatant at –80 °C for a maximum of 6 months or use it immediately.
- Sucrose gradient preparation and centrifugation (2 hr and 30 min)
- Wash ultracentrifuge tubes extensively with RNase-free water (diethylpyrocarbonate treated water (DEPC-water) or commercial) and 3% H2O2/DEPC H2O solution.
- Put the tubes on ice and add 5.5 ml of cold 50% sucrose solution at the bottom of each tube (see Table 1 for sucrose solutions). Carefully add the 15% sucrose solution drop by drop, staying close to the interphase in order to preserve a sharp interphase, until the tube is completely filled. When the tube is completely filled, close it with a rubber stopper to avoid air bubble formation.
- In a cold room gently lay down the tubes horizontally and keep it in this position for 2 hr. After this time, slowly straighten the tubes back into the vertical position and put them on ice. The gradients are now ready to be used. Alternatively, prepare the 15-50% sucrose gradient using a conventional gradient former.
- In a cold room remove carefully 1.0 ml from the top of the gradient and overlay the sucrose drop by drop with the cytosolic lysate (i.e., the supernatant obtained in step 1.2).
- Carefully lower the tubes into the buckets of swinging bucket rotor. Centrifuge the gradients for 100 min at 180,000 x g at 4 °C using an ultracentrifuge.
- After the centrifugation, leave the tubes in their buckets for 20 min at 4 °C to let the gradients stabilize.
- Sucrose gradient fractionation (2 hr)
- Carefully remove one ultracentrifuge tube from the ultracentrifuge rotor and mount it on the collector device of a Density Gradient Fractionation System. Collect 1 ml fractions monitoring the absorbance at 260 nm with a UV/VIS detector (See Figure 1 upper panel). Keep the collected fractions on ice.
- Prepare aliquots of 30-40 μl of the fractions of interest, keep them on ice before storing them at -80 °C till use. Do not use aliquots or sucrose fractions that underwent more than two freezing-thawing cycles (see Figure 1 lower panel).
2. Sample Preparation for Atomic Force Microscopy (3 hr)
Using tape, peel off the mica sheets.
Wash the mica sheets 3-4 times with DEPC-water and place it into a small Petri dish. Then dry the surface using air.
Cover the mica with 200 μl of 1 mM NiSO4 and incubate for 3 min at RT.
Remove the nickel solution and then dry the surface using air. Carry out all future steps at 4 °C by placing the Petri dish with the mica on ice.
Thaw an aliquot obtained in 1.4.2 on ice and gently add all of the sample drop by drop on the mica. Using a 100-200 μl tip, spread the sample on the entire surface of the mica. Incubate the sample on ice for 3 min.
Cover the mica sheet drop by drop with 200 μl cold Buffer-AFM (see Table 1) and incubate for 1 hr on ice.
- Imaging in liquid
- Remove carefully the Buffer-AFM and wash the mica sheets 3-4 times with 200 μl cold Buffer-AFM to remove excess sucrose. Then wash the mica sheets 3 times with cold Washing Solution (see Table 1), leaving the mica surface covered by some microliters of solution.
- Go to point 3 (Image acquisition).
- Imaging in air
- Carefully remove the Buffer-AFM and wash the mica 3-4 times with 200 μl cold Buffer-AFM to remove the excess sucrose. Then, wash the mica 3 times with cold Washing Solution (see Table 1) and drain the excess water using paper.
- Leave the sample to dry under the chemical hood with the top of the Petri dish partially open. After 2 hr, close the Petri dish and store at RT. Measure the sample after 2-3 hr as they are stable for years.
3. Image Acquisition (15 min per image after thermal stabilization)
Note: Polysomes immobilized on mica can be imaged in air or in liquid, using AC mode.
Attach the mica to the sample holder using double-sided tape.
Insert the sample holder in the AFM stage following manufacturer's directions. When imaging in liquid, if possible, try to maintain the sample at a temperature lower than 25 °C to increase polysome stability in time.
Select a cantilever suitable for AC imaging and mount it on the tip holder following manufacturer's directions. Here, use cantilevers with force constant between 2-20 N/m for air imaging and around 0.1 N/m for liquid imaging.
Adjust the laser spot on the cantilever and zero the quadrant detector signals.
Select an opportune driving frequency and drive the cantilever with an amplitude of 10-20 nm.
Approach the sample until the tip engages the surface.
Select a scan area of 2x2 μm, acquire at least 512x512 pixel images (pixel width < 4 nm), select a live background subtraction mode and a Z scale of 20-25 nm.
Inspect the image looking for the presence of round objects characterized by the height between 10 and 15 nm when acquiring in air and 25 and 30 nm when in liquid and the width in the range 25-30 nm. Adjust the setpoint and feedback parameters until sharp objects are visualized. The background should appear relatively flat in good samples, with some 2-4 nm height objects (see Figure 2A and B).
If the image looks good (as indicated at point 3.8), acquire several (at least ten) 2x2 micron scans at different sample areas.
(optional). If necessary, acquire high resolution images of selected polysomes (see Figure 2C).
Apply software corrections (plane subtraction and line-by-line correction algorithms) to correct the AFM images removing arbitrary tilt and drifting effects.
4. Data Analysis (30 min per image)
Export images in ImageJ (optional: apply a scale factor) preferentially using a lossless compression format, e.g., the TIFF format (see Figure 3A).
- Use the ImageJ macro toolset RiboPick.ijm (to be copied in the ImageJ macro/toolset subdirectory) to count ribosomes in polysomes (see Figure 3B) and compute statistical properties of the sample (see Figure 3C).
- Start loading an image using the <Read image> tool that initializes the program.
- Pick the ribosome centers with the standard ImageJ <Point selection> tool (shift + left click allows the multi-selection of ribosomes). Mark the selected ribosomes with the <Mark ribosomes> tool. Ribosome coordinates will appear in a custom text window.
- Add more ribosomes to the same polysome repeating the procedure indicated at the point 4.2.2.
- Remove wrongly marked ribosomes using the <Undo last pick> tool (starts removing from the last added ribosome to the first one — in the current polysome only).
- When the polysome is completed, use the <New polysome> tool. As the polysome number is added as an overlay to the image, the text window is updated.
- Use the <Save results and close> to write the ribosome coordinates file (the default units of the image are used) and a PNG image that summarizes the picked ribosomes and polysomes. The original image is closed without being saved.
Representative Results
Sucrose gradient polysomal profiling of whole mouse brain
It is possible to purify polysomes from a cellular lysate by polysomal profiling, which separates macromolecules in accordance to their weight and size. With polysomal profiling, cytoplasmic lysates obtained from cultured cells or tissues, such as in this example, are loaded onto a linear sucrose gradient and processed by ultracentrifugation to separate free RNA from 40S and 60S subunits, 80S ribosomes and polysomes according to their sedimentation coefficients (Figure 1 upper panel). The absorbance of the sample at 254 nm and the fraction collection enable the isolation of the sucrose fraction enriched in ribosomes or polysomes with different number of ribosomes per transcript. The fractions can be collected, aliquoted for AFM imaging and stored at -80 °C (Figure 1 lower panel).
Native polysomes from mouse brain observed by AFM in air reveal tight ribosome interactions
Image acquisition is performed using AC mode (also known as tapping mode). This modality is especially suitable for imaging soft samples (such as the polysomes) because the tip-sample interaction and the corresponding shear forces are greatly reduced. In this way both the tip and the sample are preserved. AFM imaging allows the acquisition of data at high resolution, with exact values that depend on various factors, such as the measuring conditions, the kind of the tip, and the properties of the sample. In the conditions described in this paper a lateral resolution of about 4-6 nm and a vertical resolution of 0.1-0.2 nm are attainable. After the deposition of sucrose aliquots on mica, AFM provides descriptions of single polysomes that appear as clusters of tightly packed ribosomes (Figure 2A, C and C). By cross section analysis (Figure 2D), the height of ribosomal peaks is around 14 nm in accordance to what was previously observed for human ribosomes in polysomes after air drying27. The center to center distance of ribosomes identified by the line profile is 23 nm, which is in agreement with the dimension of ribosomes in solution37, 38.
The number of ribosomes per polysome from a single fraction shows a mono-dispersed distribution
Figure 3A reports a typical AFM image obtained by acquiring a sample absorbed on mica from a fraction of the sucrose gradient. The inset shows a digital zoom of a single polysome, with a magnification factor suitable for the identification of ribosomes in the polysome. Panel B shows the same image after the ribosome identification with the RiboPick macro. Red circles (see the inset) mark the ribosomes, and a progressive number (cyan) is used to help the association of the ribosome coordinates with a specific polysome. The frequency distribution of the number of ribosomes per polysome was analyzed for fraction #10 (See Figure1, lower panel, corresponding to Medium Molecular Weight polysomes (MMW)), which clearly shows a single peak (Panel C, grey bars). The experimental distribution was fitted with a Gaussian curve (black line) that was centered at 5.8 ribosomes/polysome, with a standard deviation of 1.3 ribosomes/polysome.
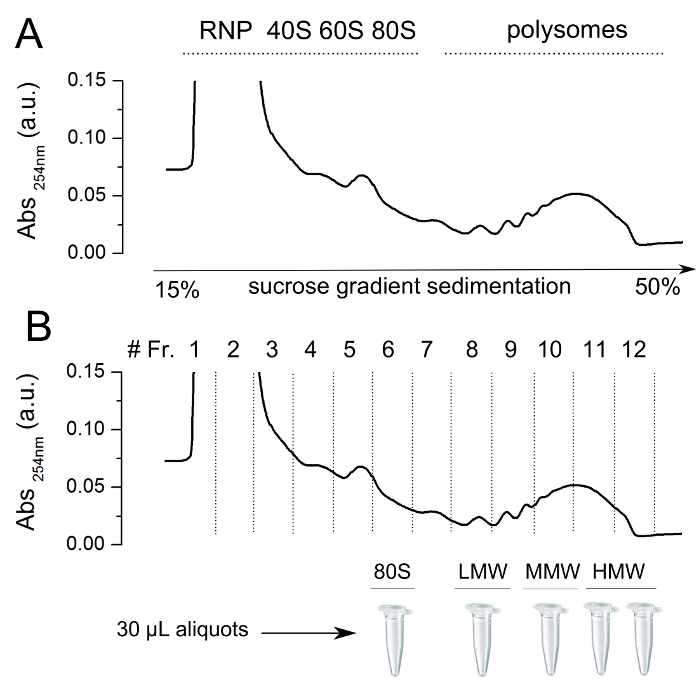 Figure 1. Representative absorbance profile for sucrose gradient sedimentation of whole mouse brain. (upper panel) A polysomal cytoplasmic lysate was obtained from P27 mouse brain and loaded onto a linear sucrose gradient (15-50% sucrose [w/v]). It is possible to observe clear peaks corresponding to absorbance at 254 nm of RiboNucleoParticles (RNP), ribosomal subunits (40S and 60S), ribosomes (80S) and polysomes. To avoid repeated freeze and thaw cycles of samples before AFM deposition on mica, it is convenient to split into aliquots both ribosome fraction and polysomal fractions (lower panel) with different number of ribosomes per transcript (i.e., low-, medium-, and high-molecular-weight polysomes (LMW, MMW, and HMW, respectively)). The aliquots can be stored at -80 °C for as long as two years.
Figure 1. Representative absorbance profile for sucrose gradient sedimentation of whole mouse brain. (upper panel) A polysomal cytoplasmic lysate was obtained from P27 mouse brain and loaded onto a linear sucrose gradient (15-50% sucrose [w/v]). It is possible to observe clear peaks corresponding to absorbance at 254 nm of RiboNucleoParticles (RNP), ribosomal subunits (40S and 60S), ribosomes (80S) and polysomes. To avoid repeated freeze and thaw cycles of samples before AFM deposition on mica, it is convenient to split into aliquots both ribosome fraction and polysomal fractions (lower panel) with different number of ribosomes per transcript (i.e., low-, medium-, and high-molecular-weight polysomes (LMW, MMW, and HMW, respectively)). The aliquots can be stored at -80 °C for as long as two years.
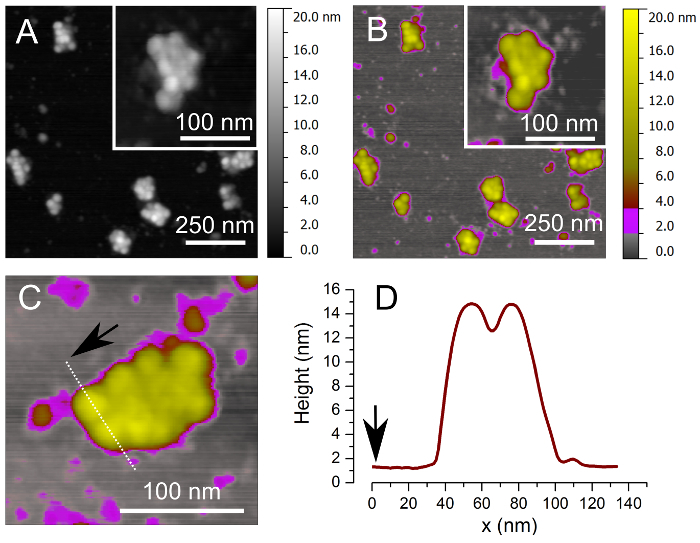 Figure 2. Images of brain polysomes by Tapping-Mode Atomic Force Microscopy. (A) Example of AFM image of MMW brain polysomes after absorption on mica using a linear grey color scale. (B) The same image shown in (A) is depicted using a different Z-ranges for the color scale: 0-0.5 nm gray for the background, 0.5-2 nm, and 2-10 nm yellow for ribosomes. In this case a better visual contrast is obtained for both low and high Z-range objects. (C) Magnification of a typical MMW polysome with cross-section profile (dotted white line). (D) Height profile of two ribosomes defined by cross-section profile in (C) shows ribosome height of 14 nm. This value is compatible with what previously observed in AFM for human ribosomes in polysomes27.
Figure 2. Images of brain polysomes by Tapping-Mode Atomic Force Microscopy. (A) Example of AFM image of MMW brain polysomes after absorption on mica using a linear grey color scale. (B) The same image shown in (A) is depicted using a different Z-ranges for the color scale: 0-0.5 nm gray for the background, 0.5-2 nm, and 2-10 nm yellow for ribosomes. In this case a better visual contrast is obtained for both low and high Z-range objects. (C) Magnification of a typical MMW polysome with cross-section profile (dotted white line). (D) Height profile of two ribosomes defined by cross-section profile in (C) shows ribosome height of 14 nm. This value is compatible with what previously observed in AFM for human ribosomes in polysomes27.
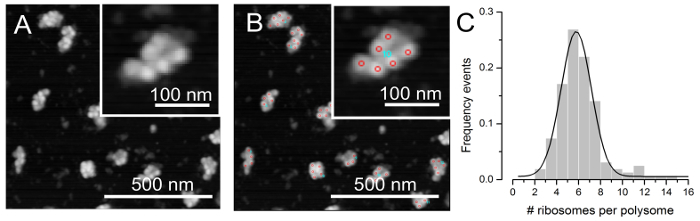 Figure 3. Example of counting the number of ribosomes per polysome in MMW brain polysomes. (A) AFM image that represents the input of the ImageJ macro, RiboPick. (B) After manually picking each ribosomes per polysome, RiboPick marks in the image the position of each ribosome (red circles). (C) The obtained number of ribosomes per polysomes can be plotted as a distribution that can be fitted with a Gaussian curve. The number of polysomes considered in this example is 164, obtained from 9 independent images. The fitting with the Gaussian curve returns the mean value of the population of polysomes in the MMW fraction (#ribosomes/polysome 5.8±1.3, R2= 0.99).
Figure 3. Example of counting the number of ribosomes per polysome in MMW brain polysomes. (A) AFM image that represents the input of the ImageJ macro, RiboPick. (B) After manually picking each ribosomes per polysome, RiboPick marks in the image the position of each ribosome (red circles). (C) The obtained number of ribosomes per polysomes can be plotted as a distribution that can be fitted with a Gaussian curve. The number of polysomes considered in this example is 164, obtained from 9 independent images. The fitting with the Gaussian curve returns the mean value of the population of polysomes in the MMW fraction (#ribosomes/polysome 5.8±1.3, R2= 0.99).
| Buffer | Composition | Application |
| Lysis Buffer | 10 mM Tris–HCl, pH 7.5 | Preparation of lysate (1.1) |
| 10 mM NaCl | ||
| 10 mM MgCl2 | ||
| 1% Triton-X100 | ||
| 1% Na-deoxycholate | ||
| 0.4 U/ml RNase Inhibitor | ||
| 1 mM DTT | ||
| 0.2 mg/ml cycloheximide | ||
| 5 U/ml Dnase I | ||
| 50% sucrose solution | 50% (w/v) sucrose in | Sucrose gradient preparation (1.2) |
| 100 mM NaCl | ||
| 10 mM MgCl2 | ||
| 10 mM Tris/HCl pH 7.5 | ||
| 15% sucrose solution | 15% (w/v) sucrose in | Sucrose gradient preparation (1.2) |
| 100 mM NaCl | ||
| 10 mM MgCl2 | ||
| 10 mM Tris/HCl pH 7.5 | ||
| Nickel Solution | 1 mM NiSO4 | Sample preparation for AFM (2) |
| Buffer-AFM | 100 mM NaCl | Sample preparation for AFM (2) |
| 10 mM MgCl2 | ||
| 100 µg/ml cycloheximide | ||
| 10 mM Hepes | ||
| 3% (w/v) sucrose, pH = 7.4 | ||
| Washing Solution | DEPC-Water | Sample preparation for AFM (2) |
| 100 µg/ml cycloheximide |
Table 1. Buffers.
Discussion
As the structure of DNA was proven to be of paramount importance to describe the process of transcription and as the organization of chromatin advanced our understanding of transcriptional control of gene expression, it is essential to analyze the organization and structure of polysomes to improve the truthful comprehension of translation and its regulation.
With the described protocol, an optimal density of polysomes gently immobilized on a flat surface, suitable for AFM imaging is obtained. Using these conditions with conveniently prepared samples, nearly 50 polysomes per hour can be acquired, ready for further analyses and characterization. Moreover, dried samples stored at RT have proven to be suitable for imaging after more than one year.
To successfully obtain polysome images with our protocol, there are some crucial steps: to use tissues that has been properly flash-frozen immediately after dissection and stored at -80 °C to minimize RNA degradation; to use the lysis buffer containing cycloheximide and RNase inhibitors to avoid ribosome dissociation from mRNA; to perform all incubations on ice during the sample deposition on mica; and when preparing the sample for AFM imaging on air, to extensively wash the sample on the mica with buffers and RNase free water in the presence of cycloheximide. The last point is particularly important. If the image appears to contain smooth surfaces at approximately the right height but 100-1,000 nm wide, this is a clear indication of a poorly washed sample (often ribosomes/polysomes can be still observed as faint, embedded objects). A method to resolve this issue is to repeat the washing procedure, but this does not usually mitigate the issue on an already dried sample. In this case it is worth starting again with a new aliquot.
Given the resolution of AFM, this method for studying polysomes cannot resolve the structural details of ribosomal interfaces in polysomes. In fact, AFM is a complementary and simpler approach to cryo-EM, representing an effective and robust technique, to acquire and analyze thousands of polysomes, and better understand the overall ribosome organization in polysomes. Importantly, this protocol has the unique advantage of identifying filaments compatible with RNA27. This very same procedure can be used for any polysomal sample obtained from any biological tissue or cell line. More importantly it can also be successfully employed to obtain transcript-specific information using in vitro translation systems, as recently demonstrated by Lauria and co-workers35. This methodology will pave the way for several experiments, to gain understanding of the kinetics of polysome formation or changes in polysome organization under different cellular or tissue conditions.
Disclosures
The authors have no competing financial interests in this paper and nothing to disclose.
Acknowledgments
This research was supported by the AXonomIX research project financed by the Provincia Autonoma di Trento, Italy.
References
- Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J. 1995;312:163–167. doi: 10.1042/bj3120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JB, Cook GM. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol Rev. 1995;59(1):48–62. doi: 10.1128/mr.59.1.48-62.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, et al. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol. 2010;6:400. doi: 10.1038/msb.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Tebaldi T, et al. Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells. BMC Genomics. 2012;13:220. doi: 10.1186/1471-2164-13-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov N, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145(3):383–397. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Svitkin YV, Sonenberg N, Shatkin AJ. Cap and cap-binding proteins in the control of gene expression. Wiley Interdiscip Rev RNA. 2011;2:277–298. doi: 10.1002/wrna.52. [DOI] [PubMed] [Google Scholar]
- Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher A, et al. An mRNA-derived noncoding RNA targets and regulates the ribosome. Mol Cell. 2014;54(1):147–155. doi: 10.1016/j.molcel.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, et al. An active role for the ribosome in determining the fate of oxidized mRNA. Cell Rep. 2014;9(4):1256–1264. doi: 10.1016/j.celrep.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushar ML, et al. Temporally defined neocortical translation and polysome assembly are determined by the RNA-binding protein Hu antigen R. Proc Natl Acad Sci U S A. 2014;111(36):E3815–E3824. doi: 10.1073/pnas.1408305111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heesch S, et al. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014;15(1):R6. doi: 10.1186/gb-2014-15-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, et al. Not4-dependent translational repression is important for cellular protein homeostasis in yeast. EMBO J. 2015;34(14):1905–1924. doi: 10.15252/embj.201490194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade GE. A small particulate component of the cytoplasm. J Biophys Biochem Cytol. 1955;1(1):59–68. doi: 10.1083/jcb.1.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillen K, Roberts RB, Britten RJ. Synthesis of nascent protein by ribosomes in Escherichia coli. Proc Natl Acad Sci U S A. 1959;45(9):1437–1447. doi: 10.1073/pnas.45.9.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein FO, Staehelin T, Noll H. Ribosomal aggregate engaged in protein synthesis: characterization of the ergosome. Nature. 1963;2(197):430–435. doi: 10.1038/197430a0. [DOI] [PubMed] [Google Scholar]
- Dimitriadis EK, et al. Tetrameric organization of vertebrate centromeric nucleosomes. Proc Natl Acad Sci U S A. 2010;107(47):20317–20322. doi: 10.1073/pnas.1009563107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MJ, et al. Atomic force microscope measurements of nucleosome cores assembled along defined DNA sequences. Biochemistry. 1993;32(33):8390–8396. doi: 10.1021/bi00084a002. [DOI] [PubMed] [Google Scholar]
- Ban N, Nissen P, Hansen J, Moore P, Steitz T. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289(5481):905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- Schluenzen F, et al. Structure of functionally activated small ribosomal subunit at 3.3 Å resolution. Cell. 2000;102(5):615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ben-Shem A, et al. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334(6062):1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- Anger AM, et al. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497(7447):80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- Brandt F, et al. The native 3D organization of bacterial polysomes. Cell. 2009;136:261–271. doi: 10.1016/j.cell.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Myasnikov AG, et al. The molecular structure of the left-handed supra-molecular helix of eukaryotic polyribosomes. Nat. Commun. 2014;5:5294. doi: 10.1038/ncomms6294. [DOI] [PubMed] [Google Scholar]
- Afonina ZA, Myasnikov AG, Shirokov VA, Klaholz BP, Spirin AS. Formation of circular polyribosomes on eukaryotic mRNA without cap-structure and poly(A)-tail: a cryo electron tomography study. Nucleic Acids Res. 2014;42(14):9461–9469. doi: 10.1093/nar/gku599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonina ZA, Myasnikov AG, Shirokov VA, Klaholz BP, Spirin AS. Conformation transitions of eukaryotic polyribosomes during multi-round translation. Nucleic Acids Res. 2015;43(1):618–628. doi: 10.1093/nar/gku1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viero G, et al. Three distinct ribosome assemblies modulated by translation are the building blocks of polysomes. J Cell Biol. 2015;208(5):581–596. doi: 10.1083/jcb.201406040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt F, Carlson L-A, Hartl FU, Baumeister W, Grünewald K. The three-dimensional organization of polyribosomes in intact human cells. Mol. Cell. 2010;39:560–569. doi: 10.1016/j.molcel.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Wu X, Liu WY, Xu L, Li M. Topography of ribosomes and initiation complexes from rat liver as revealed by atomic force microscopy. Biol Chem. 1997;378(5):363–372. doi: 10.1515/bchm.1997.378.5.363. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Wakiyama M, Yazaki K, Miura K. Transmission electron and atomic force microscopic observation of polysomes on carbon-coated grids prepared by surface spreading. J. Electron Microsc (Tokyo) 1997;46(6):503–506. doi: 10.1093/oxfordjournals.jmicro.a023550. [DOI] [PubMed] [Google Scholar]
- Mikamo EC, et al. Native polysomes of Saccharomyces cerevisiae in liquid solution observed by atomic force microscopy. J. Struct. Biol. 2005;151:106–110. doi: 10.1016/j.jsb.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Mikamo-Satoh EA, et al. Profiling of gene-dependent translational progress in cellfree protein synthesis by real-space imaging. Anal. Biochem. 2009;394:275–280. doi: 10.1016/j.ab.2009.07.033. [DOI] [PubMed] [Google Scholar]
- Bernabò P, et al. Studying translational control in non-model stressed organisms by polysomal profiling. J. Insect Physiol. 2015;76:30–35. doi: 10.1016/j.jinsphys.2015.03.011. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. IH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria F, et al. RiboAbacus: a model trained on polyribosome images predicts ribosome density and translational efficiency from mammalian transcriptomes. Nucleic Acids Res. 2015. [DOI] [PMC free article] [PubMed]
- Al Omran AJ, et al. Live Imaging of the Ependymal Cilia in the Lateral Ventricles of the Mouse Brain. J Vis Exp. 2015. p. e52853. [DOI] [PMC free article] [PubMed]
- Warner JR, Rich A, Hall CE. Electron microscope studies of ribosomal clusters synthesizing hemoglobin. Science. 1962;138:1399–1403. doi: 10.1126/science.138.3548.1399. [DOI] [PubMed] [Google Scholar]
- Yazaki K, Yoshida T, Wakiyama M, Miura K. Polysomes of eukaryotic cells observed by electron microscopy. J. Electron Microsc (Tokyo) 2000;49:663–668. doi: 10.1093/oxfordjournals.jmicro.a023856. [DOI] [PubMed] [Google Scholar]