

Abstract
With the advent of the -omics approaches our understanding of the chronic diseases like cancer and metabolic syndrome has improved. However, effective mining of the information in the large-scale datasets that are obtained from gene expression microarrays, deep sequencing experiments or metabolic profiling is essential to uncover and then effectively target the critical regulators of diseased cell phenotypes. Estrogen Receptor α (ERα) is one of the master transcription factors regulating the gene programs that are important for estrogen responsive breast cancers. In order to understand to role of ERα signaling in breast cancer metabolism we utilized transcriptomic, cistromic and metabolomic data from MCF-7 cells treated with estradiol. In this report we described generation of samples for RNA-Seq, ChIP-Seq and metabolomics experiments and the integrative computational analysis of the obtained data. This approach is useful in delineating novel molecular mechanisms and gene regulatory circuits that are regulated by a particular transcription factor which impacts metabolism of normal or diseased cells.
Keywords: Medicine, Issue 109, RNA-Seq, transcriptome, ChIP-Seq, cistrome, metabolomics, estrogen, breast cancer
Introduction
Estrogens are important regulators of many physiological processes in both females and males including reproductive tissues, metabolic tissues, brain and bone1. In addition to beneficial effects in these tissues, estrogens also drive cancers that arise from mammary and reproductive tissues. Estrogens mainly work through ERs to induce cell type specific effects. Deep sequencing of transcripts regulated by ERα using RNA-Seq and genome-wide ERα DNA binding sites analysis using ChIP-Seq proved to be useful to understand how ERα works in different tissues and cancers that arise from them. We and others have published gene expression profiles associated with different receptors (ERα v.s. ERβ)2,3, different ligands3-5 and different coregulators2,4,6,7.
RNA-Seq is the main method to examine the transcriptome, offering higher precision and efficiency compared to microarray based gene expression analysis8. RNA obtained from cell lines2-4,7, tissues or tumor samples are sequenced, mapped to available genomic assemblies and differentially regulated genes are identified. Chromatin Immunoprecipitation (ChIP) is employed to dissect the transcription factor and coregulator chromatin binding to known regulatory binding sites. ChIP-Seq (ChIP followed by high throughput sequencing) provides unbiased detection of global binding sites. Metabolomics is another increasingly used system biology approach, which quantitatively measures, dynamic multiparametric response of living systems to various stimuli including chemicals and genetic perturbations.
By performing global metabolic profiling, a functional readout can be obtained from cells, tissues, and blood. In addition, information from transcriptome experiments do not always reflect actual changes in the level of enzymes that contribute to biochemical pathways. Combined analysis of transcriptome and metabolome data enables us to identify and correlate changes in gene expression with actual metabolite changes. Harnessing the information from all these large scale datasets provide the mechanistic details to understand the role of transcription factors regulating complex biological systems, especially ones that pertain to human development and diseases like cancer and diabetes.
The complex nature of the mammalian genome makes it challenging to integrate and fully interpret the data obtained from the transcriptome, cistrome and metabolome experiments. Identifying functional binding events that would lead to changes in expression of target genes is important because once functional binding sites are identified; ensuing analysis, including transcription binding (TF) motif analysis, could be performed with higher accuracy. This leads to the identification of biologically meaningful TF cascades and mechanisms. Also direct comparison of RNA-Seq and ChIP-Seq experiments are not always possible since the data from each experiment have differing scales and noises and in some cases meaningful signals are obscured by population noise. We are not aware of any study that integrated information from these three independent but related approaches to understand the direct metabolic regulation by ERα in breast cancer. Therefore, our overall goal in this paper is to relate productive binding events to gene expression and metabolite changes. In order to achieve this goal, we integrated data from RNA-Seq, ChIP-Seq and metabolomics experiments and identified those estrogen induced ERα binding events that would lead to gene expression changes in metabolic pathways. For the first time, we provide a complete set of protocols (Figure 1) for generating ChIP-Seq, RNA-Seq and metabolomics profiling and performing integrative analysis of the data to uncover novel gene circuits regulating metabolism of breast cancer cell lines.
Protocol
1. Preparation of RNA-Seq Samples
- Cell Culture and Treatment
- Culture the MCF7 cells in Improved MEM (IMEM) plus phenol-red medium with 10% Heat inactive FBS, 1% penicillin/streptomycin at 37 °C in humidified 5% CO2 atmosphere. Note: Same cell line and cell culture condition is used throughout the study.
- Seed 100,000 MCF7 cells per well in 6-well plates. Have triplicates for each treatment. On day 3, remove the medium and add 2 ml fresh medium with or without 10-8 M E2 to each well. Incubate the plates for 24 hr in 37 °C in humidified 5% CO2 atmosphere.
- To harvest the cells, add 1 ml RNA isolation reagent (acid guanidinium thiocyanate-phenol-chloroform) into each well and incubate for 5 min at room temperature (RT). Then collect the cell lysate reagent by pipetting and deposit into 1.5 ml tubes
- RNA Isolation
- Add 200 µl chloroform to 1 ml cell lysate reagent (1.1.3) and vortex for 10 sec. Incubate at room temperature for 5 min. Centrifuge samples at 16,000 x g for 15 min at 4 °C. Following centrifugation, transfer the aqueous phase (top) to a new tube without disturbing the other phases.
- Add 500 µl 100% isopropanol to a new tube with the transferred-aqueous phase, and mix by inverting. Incubate at -20 °C overnight. Centrifuge the samples at 16,000 x g for 10 min at 4 °C. Observe a pellet at the bottom of the tube. Discard the supernatant and wash the pellet with 750 µl RNase-free 70% ethanol by brief vortex.
- Centrifuge samples at 6,000 x g for 5 min at 4 °C. Remove the supernatant, and centrifuge the sample at 16,000 x g for 1 min at 4 °C to collect all the remaining ethanol. Remove the remaining ethanol by pipetting with a gel-loading tip and set the tubes up-side down to air-dry for 10 min. Dissolve the pellet in 20-50 µl DEPC treated water depending on the size of the pellets.
Purify the RNA using RNA clean-up kit following the protocol4. Measure the RNA concentration using a fluorometry based assay kit following the protocol9. NOTE: OD at 260 nm is taken to determine the RNA concentration and ratio of OD at 260 nm and 280 nm are used to determine the purity of sample. It is recommended checking the RNA quality using Bio-analyzer assay.
Take about 0.5 to 1 μg purified RNA for sequencing. NOTE: Biotech center prepares the sequencing libraries and performs paired-end read sequencing using methods detailed previously2.
2. Preparation of ChIP-Seq Sample
- Cell Culture and Treatment
- Seed 500,000 MCF7 cells per 10 cm plate in growth medium. NOTE: For each pull down 16 plates were used. On day 3, remove the medium and add 5 ml fresh medium with or without 10-8 M E2 to each plate. Treat the cells for 45 min (37 °C in humidified 5% CO2 atmosphere).
- Crosslink and Cell Harvest
- Add 250 µl 37% Formaldehyde into 5 ml medium to crosslink chromatin, incubate at room temperature for 15 min. Stop crosslinking by washing cells with 5 ml ice cold 1x MEM.
- Harvest the cells in 250 µl Cell Lysis buffer (10 mM EDTA, 50 mM Tri HCl, 1% SDS, 0.5% N,N-dimethyldodecan-1-amine oxide) per plate.
- Fragmentation of Chromatin
- Sonicate the cells for 30 sec x 8 at amp of 30 to reach required chromatin sizes. Cool each sample on ice after a sonication of 30 sec.
- Centrifuge at 16, 000 x g for 10 min at 4 °C, and carefully pipette the supernatant without disturbing the pellet at the bottom.
- Take a 5 µl aliquot of the supernatant, and check the DNA size by electrophoresis through 1.5% agarose gel as described previously10. The ideal size of DNA should be between 200-500 bp.
- Chromatin Immunoprecipitation
- Save 25 µl of cell lysate as input. In a 50 ml tube, mix 4 ml of cell lysate with 20 ml IP buffer (2 mM EDTA, 100 mM NaCl, 20 mM Tris-HCl, and 0.5% Triton X), ERα antibody (500 µl F10 and 500 µl HC-20), and 500 µl ProtA and ProtG protein coating magnetic beads. Incubate the mixture by rotation at 4 °C overnight to allow the formation of chromatin- antibody complex.
- Washes and Reverse-crosslinking
- Use a magnetic stand to collect the magnetic beads on the magnetized side to remove the wash buffer.
- Wash the magnetic beads in 25 ml Wash buffer I (2 mM EDTA, 20 mM Tris HCl, 01% SDS, 1% Triton X, and 150 mM NaCl). To thoroughly wash the magnetic beads without disturbing DNA-Protein complexes, add 25 ml wash buffer slowly to the tube along the side then invert the tubes until all the magnetic beads are mixed well with the wash buffer without appearing as a pellet. As an alternative, use the rotator to invert the tube. Do not vortex the samples. Wash the magnetic beads using the same method in the following steps.
- Wash the magnetic beads in 25 ml Wash-buffer II (2 mM EDTA, 20 mM Tris HCl, 01% SDS, 1% Triton X, and 500 mM NaCl).
- Wash the magnetic beads in 25 ml Wash-buffer III (1.6 mM EDTA, 10 mM Tris HCl, 1% NP-40, 1% Dexycholate and 250 mM LiCl).
- Wash the magnetic beads in 25 ml 1x TE buffer (1 mM EDTA, and 10 mM Tris HCl) twice.
- Elute DNA in 500 µl Elution buffer (1% SDS, and 10 mM NaHCO3) and then allocate the 500 µl DNA elution into 4 tubes. For the DNA from inputs, elute DNA in 125 µl Elution buffer.
- Incubate the tubes at 65 °C overnight on a heating block. Cover the tubes on the heating block with aluminum foil to keep the heat even over all the tubes. Place a weight on the tubes to keep them from opening.
- DNA Isolation
- Cool the tubes at RT for 5 min. Isolate DNA from the pull down samples using ChIP DNA isolation kit. Follow the directions from the manufacturer.
- Warm up the Elution buffer at 65 °C. Add 625 µl binding buffer to each tube. If the precipitate forms, add another 250 µl binding buffer until the solution is clear.
- Load the sample onto the column and centrifuge at 16,000 x g for 30 sec. Wash the sample with 250 µl wash buffer. Remember to add ethanol to the wash buffer when first time to use it. Repeat the wash.
- Elute the DNA in 15 µl Elution buffer. Combine the DNA of same pull down from 4 tubes to achieve around 55 µl DNA.
- Isolate the Input DNA Using a DNA Purification Kit.
- Warm up the Elution buffer at 65 °C. Add 625 µl binding buffer in each tube and incubate for 10 min. Load the sample onto the column in a 2 ml collection tube.
- Wash the sample with 750 µl wash buffer, centrifuge at 16,000 x g for 1 min. Repeat centrifuging to remove any remaining buffer. Elute the DNA into 30 µl of EB.
- Measure the DNA Concentration Using a Commercial Fluorochrome Assay with Modification11.
- Dilute the 20x TE buffer into 1x TE as the assay buffer for diluting the reagent and DNA samples. Take 5 µl isolated DNA and dilute it into 50 µl in 1x TE.
- From 2 µg/ml DNA stock solution make a DNA standard ranging from blank to 1, 10, 100, 1000 µg/ml. In a 96 well plate, allocate 25 µl of each standard as well as DNA dilution in duplicate. (Triplicate for standard is recommended.)
- Prepare the working reagent by making a 200-fold dilution from dsDNA reagent in 1x TE buffer in plastic tube. Cover the working reagent solution with aluminum foil to avoid light. Add 200 µl working reagent into each sample. Incubate at room temperature for 5 min. Cover the plate with aluminum foil to avoid light.
- Measure the florescence of the samples using a fluorescence plate reader (excitation at 480 nm, emission 520 nm). Generate the DNA standard curve of florescence versus DNA concentration. Determine the concentrations of the samples based on the generated DNA standard curve.
- Verification of ChIP Experiment Using qPCR
- Take 2 µl isolated DNA and diluted into 20 µl in water. Set up the qPCR reaction in triplicate by mixing 2.5 µl DNA dilution, 5 µl Green I PCR master mix, 1 µl primer, and 1.5 µl nuclease free water.
- Run the qPCR with the Fast Real-Time PCR system using the following program: Step 1: 95 °C for 60 sec, Step 2: 95 °C for 10 sec, 60 °C for 60 sec, repeat Step 2 39 times.
Send 10 ng DNA sample to Biotech center for sequencing. NOTE: The biotech center prepares the ChIP DNA into libraries according to instruction, and performs single-read sequencing using methods detailed previously2.
3. Metabolomics Assay Sample Preparation
- Cell Culture and Treatment
- Seed 400,000 MCF7 cells per 10 cm plate in growth medium. For each treatment prepare three samples. Use two plates in each sample to receive enough cells for the detection. On day 3, remove the medium and add 5 ml fresh medium with or without 10-8 M E2 to the cells. Treat the cells for 24 hr (37 °C in humidified 5% CO2 atmosphere).
- Sample Collection
- Add 5 ml ice cold 1x PBS to the plate, aspirate the PBS after briefly tilting the plate several times. Repeat twice, and remove as much PBS as possible after the last wash.
- Add 750 µl of pre-cold acetone to each plate and scrape the cells. Combine the cells in acetone from two plates into a 2 ml tube on ice.
- Cell count for normalization
- For each treatment, use two extra plates for cell quantification. To de-attach the cells from plate, add 2 ml of HE buffer (1x HBSS, 10 mM HEPES, 0.075% NaHCO3, and 1 mM EDTA) into each plate and incubate for 5 min.
- Collect and mix well the cells by pipetting cells in the HE buffer. Use total of 20 µl cell solution to count the cell number using a hemocytometer.
Store the samples at -80 °C before sending to the Metabolomics center to identify and quantify the metabolites using Gas chromatography mass spectrometry (GC-MS) analysis.
4. Integrative Analysis
- RNA-Seq Data Analysis:
- Perform Quality check of FASTQ files using FastQC:Read QC (http://galaxy.illinois.edu)
- Select FastQC:Read under the tool of NGS:QC and manipulation. Upload the raw data file from current history. NOTE: It is recommended to give a title for the output file to remind you what the job was for since there might be multiple outputs.
- Execute and the output file will appear under the History. Repeat the same procedure for each sequence file. Trim adapters using Clip under NGS:FASTX Toolkit.
- Align reads to hg19 using Tophat2 with RefSeq genome reference annotation12 (default values).
- Select Tophat2 under tool of NGS:RNA analysis. Indicate the library paired type (single-end vs paired-end).
- Upload the input FASTQ file from the current history. Select the reference genome. Choose defaults settings. Or use the full parameter list to modify.
- Execute and it will produce two output files: one is UCSC BED track of junctions; another one is a list of read alignments in BAM format.
- Upload BAM files to sequencing data analysis tool. Calculate expression values using Quantification Tool.
- Identify differentially up-regulated genes using Expression Analysis Tool using default values and fold change >2 and p-value <0.05. Likewise, identify differentially down-regulated genes using Expression Analysis Tool using default values and fold change <0.5 and p-value <0.05.
- ChIP-Seq Data Analysis:
- Align FASTQ Reads to hg19 using Bowtie213 (Default Values) in Galaxy.
- Select Bowtie2 under the tool of NSC:Mapping. Verify if the library is mate-paired (single-end vs paired-end). Upload the FASTQ file from current history.
- Select the reference genome. Use the default parameter settings. Execute and it will produce output file with mapped sequence reads as BAM file. Repeat the procedure for every sequence file.
- Identify peaks using MACS14 a p-value cutoff of 6.0e-7 and FDR of 0.01, as previously described2.
- Metabolomics Data Analysis:
- Convert chemical names of metabolites to KEGG_IDs.
- Identify differentially regulated metabolites by comparing levels detected in Vehicle vs. E2 treated samples (a 2 fold cut-off was used in this study).
- Integrative Analysis:
- Identify genes that have ERα binding sites using sequencing data analysis tool. Translate Regions to genes function using 20 kb as distance cut-off.
- Identify differentially up- and down-regulated genes from RNA-Seq data analysis using fold change >2 and fold change <0.5, respectively with the p-value <0.05 as cut off. Compare to the list of ERα binding site containing genes using Venn diagram comparison.
- Use this list to identify E2 modulated metabolic pathways using Metscape15 module of Cytoscape.
- By selecting Metscape module from apps, choose Build network and then pathway-based option.
- Select the Organism (Human). Select data file of E2 unregulated compound list (KEGG ID) as well E2 up-regulated gene list.
- Choose Compound -Reaction -Enzyme-Gene as the Network type. Choose compound/genes as the query.
- Build network. Repeat using the E2 down-regulated compound and genes.
Representative Results
Transcriptomics To analyze differentially expressed genes by E2 treatment, we chose to perform an RNA-Seq experiment. In addition to providing information about mRNA levels, RNA-Seq data can also be used to monitor changes in non-coding RNA (long non-coding RNAs, microRNAs) and alternative splicing events. We did not provide information on the analysis of non-coding RNAs or alternatively transcribed genes, since scope of our study is to identify protein coding genes that are important for metabolic regulation. However, RNA-Seq is an excellent way of obtaining information on differential gene expression events.
In order to obtain high quality reads, it is important to get high purity RNA. After isolating RNA we purified the RNA using a RNA clean up kit. Unfortunately nearly 50% of the RNA is lost during this process and amount of starting RNA should be taken into consideration to obtain required amount (100-500 ng) by the end of purification step.
We quantified the RNA yields using fluorometer RNA HS assay, which is very accurate and is preferred method for quantification of low-abundance RNA. Next, the integrity and overall quality of the RNA samples were examined using bioanlayzer, which is a viable alternative to gel electrophoresis using a minimal amount of RNA. The analysis shows all the samples have sharp and clean bands of 18S and 28S rRNAs indicating the integrity and purity of the samples (Figure 2B). RNA Integrity Number (RIN) was used as a standard for RNA quality control. RIN 10 indicates intact RNA, RIN 6 partially degraded and RIN3 strongly degraded16.
It is recommended that the RIN to be at least 7 for sequencing experiment. All of our RNA samples received RIN value between 9.6-9.9 (Figure 2A). Sequencing libraries were constructed from 500ng of high-quality RNA from each sample using ultra-high-throughput sequencing system such as Illumina 2500. On average 18,000,000 High-quality sequencing reads were obtained for each sample (Table 1), which was ready for downstream analysis. To check the overall quality of the high-throughput sequence raw data, FastQC was run for each sample's reads. A representative FastQC report for one vehicle RNA-Seq data was shown (Figure 3). The sequence length of the most reads was about 100 bp (Figure 3A). For each read, the quality score was 32-34 for the first 5 bp and 36-38 from 6-100bp (Figure 3B). Among 17,990,863 reads generated from this sample, most of them had quality score higher than 34 (Figure 3C). The GC content per read in the sequence also followed the normal distribution with an average of 48% GC. Overall the quality score of the reads was very high.
Cistromic analysis of ERα binding sites To achieve the efficient deep sequencing, obtaining DNA fragments at desirable size is one of the important factors. Current sequencing approaches may have different requirements on the DNA fragments, for example 300-600 bp for ultra-high-throughput sequencing system, 100-200 bp for AB/SOLiD17. In this experiment most of the DNA fragments are between 300 bp and 600 bp as shown on gel (Figure 4). As an alternative, readers might chose to prepare their chromatin using MNase digestion, however we feel our established protocol provides the required samples with high-quality and provides reproducible results. Ten ng of DNA from each treatment and its corresponding input sample was prepared in EB buffer. The ChIP DNA was single-read sequenced using ultra-high-throughput sequencing system. ChIP DNA samples yielded about 20,000,000 reads while the input DNA yielded more than 35.000,000 reads (Table 1).
Metabolomics To quantify the metabolites in mammalian cells using GCMS, at least 10 µg of cell mass are required. We started with 400,000 MCF7 cells with the experiments. By the time of harvest, there were about 500,000 cells in each plate. The overview of the metabolites represented by peaks were compared between the control and the Estradiol treated sample (Figure 5). About 100 metabolites were identified, which count for 60% of the total known in mammalians. Here we present a list of the top 10 metabolites, which show significant changes in E2 treatment (Table 2). Overall, E2 upregulated pathways including fructose and mannose metabolism, histidine metabolism, purine metabolism, cholesterol biosynthesis and vitamin B3 metabolism. The downregulated pathways were butanoate metabolism, de novo fatty acid biosynthesis, pentose phosphate pathway, urea cycle and metabolism of arginine (Table 3).
Data analysis and integration The data from RNA-Seq, ChIP-Seq and GC-MS were processed and analyzed through a series of steps (Figure 6). After the comparison between genes identified in RNA-Seq and ChIP-Seq, one set of up-regulated and one set of down-regulated genes by Estradiol were extracted. From the metabolic data, we obtained two sets of up- and down-regulated compounds by the Estradiol treatment. Next we used Metscape, an app for Cytoscape, to visualize and interpret the gene expression and metabolic data in the context of human metabolism15. First we chose to include elements of compound, reaction, enzyme, and gene in the network. We selected the compounds/genes as query and two networks of the E2 up-regulation (Figure 7A) and down-regulation (Figure 7B) were shown. The Metscape also provides an option to build networks on selective pathways. For example, using the same set of data as building the down-regulated overall network, a subnetwork of androgen-estrogen biosynthesis and metabolism pathway was generated by switching the query from the compound/gene to specific pathway. The genes or compounds with significant changes were highlighted in brighter color. Besides selecting a specific pathway from the drop-down window as described, one can also create a sub-network by applying "create subnetwork" function to the chosen area. As shown in Figure 6A and B, ERα activation and direct binding to gene regulatory regions impacted many metabolic pathways in breast cancer cells. Our integrative analysis indicated that, ERα directly binds to and regulates expression of FASN gene, which is important for de novo fatty acid biosynthesis pathway (Figure 7C). In addition E2 treatment downregulated androgen-estrogen biosynthesis pathways by repressing expression of CYP4Z1, CYP2E1, CYP4B1, CYP2C8, TPO, GSTA4 and GSTM2 in these pathways.
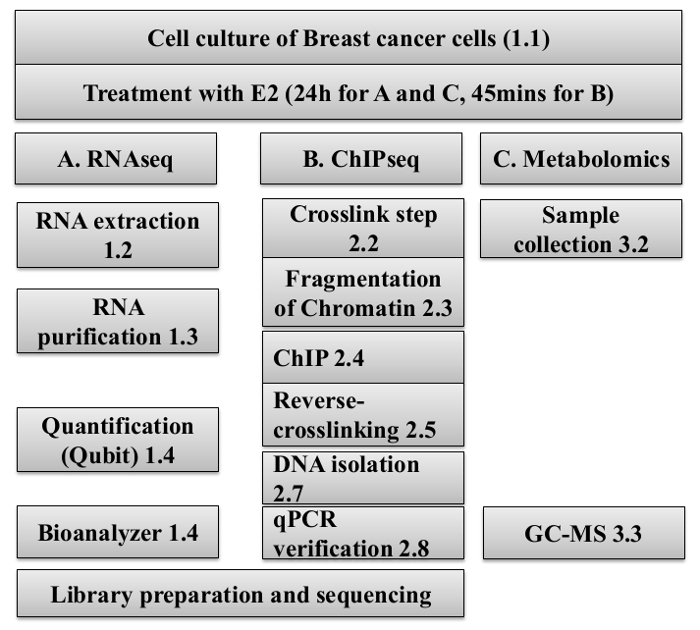 Figure 1: Workflow of the preparation of samples of RNA, DNA and cell lysates for the RNA-Seq, ChIP-Seq, and Metabolomic analysis.
Figure 1: Workflow of the preparation of samples of RNA, DNA and cell lysates for the RNA-Seq, ChIP-Seq, and Metabolomic analysis.
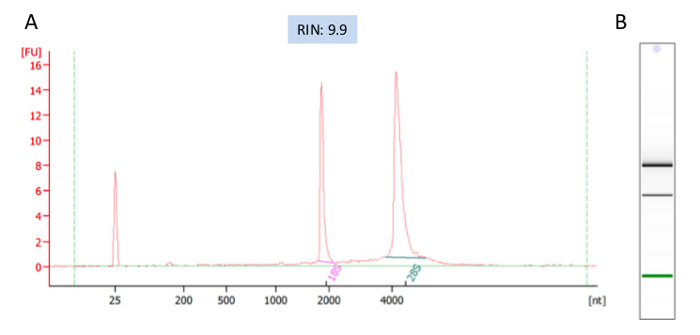 Figure 2: A representative electropherogram of the RNAs analyzed with Agilent 2100 Bioanalyzer. RNA Integrity Number (RIN), indicator of RNA quality, was given by the analysis as 9.9. (A) On a virtual gel picture of the RNA sample both bands of 26S and 18S are sharp.(B) Fluorescent signals of individual trace in the RNA shows that the intensity of 28S peak is greater than 18S peak and no contamination is seen. Please click here to view a larger version of this figure.
Figure 2: A representative electropherogram of the RNAs analyzed with Agilent 2100 Bioanalyzer. RNA Integrity Number (RIN), indicator of RNA quality, was given by the analysis as 9.9. (A) On a virtual gel picture of the RNA sample both bands of 26S and 18S are sharp.(B) Fluorescent signals of individual trace in the RNA shows that the intensity of 28S peak is greater than 18S peak and no contamination is seen. Please click here to view a larger version of this figure.
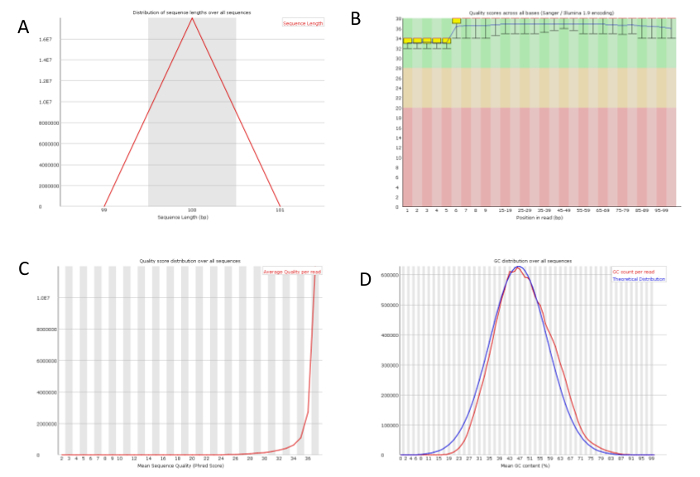 Figure 3: A representative quality check on the RNA-Seq raw data. One of the vehicle sample's sequence data was analyzed using FastQC. (A) The sequence length distribution of the library which is 100 bp. (B) The quality score along the position in read. (C) The quality score per sequence showed the universal high quality of all sequences. (D) The GC content per read matched the theoretical distribution. Please click here to view a larger version of this figure.
Figure 3: A representative quality check on the RNA-Seq raw data. One of the vehicle sample's sequence data was analyzed using FastQC. (A) The sequence length distribution of the library which is 100 bp. (B) The quality score along the position in read. (C) The quality score per sequence showed the universal high quality of all sequences. (D) The GC content per read matched the theoretical distribution. Please click here to view a larger version of this figure.
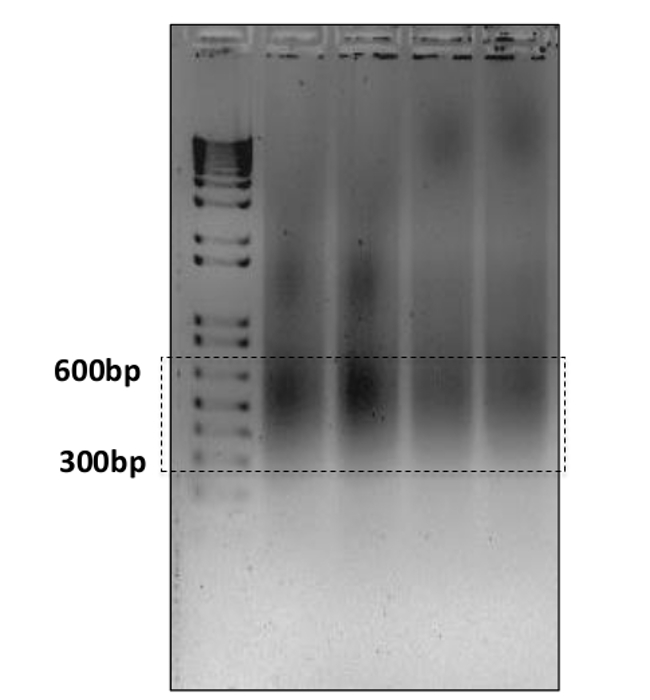 Figure 4: Gel electrophoresis of the sonicated chromatin fragments. 5 µl of the cell lysate from each sample was run on a 1.5% agarose gel with a 1,000 bp plus ladder on the first lane. The majority of the fragments are between 300 and 600 bp. Please click here to view a larger version of this figure.
Figure 4: Gel electrophoresis of the sonicated chromatin fragments. 5 µl of the cell lysate from each sample was run on a 1.5% agarose gel with a 1,000 bp plus ladder on the first lane. The majority of the fragments are between 300 and 600 bp. Please click here to view a larger version of this figure.
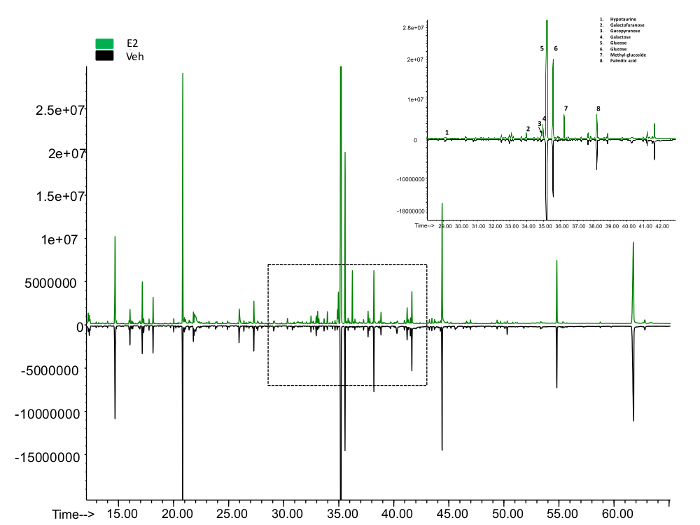 Figure 5: The overview of peaks representing metabolites from the GC-MS. The upper panel shows the peaks identified in the E2 treated samples while the lower panel from the control. The area enclosed was roomed in to show the exact metabolite represented by each peak. Please click here to view a larger version of this figure.
Figure 5: The overview of peaks representing metabolites from the GC-MS. The upper panel shows the peaks identified in the E2 treated samples while the lower panel from the control. The area enclosed was roomed in to show the exact metabolite represented by each peak. Please click here to view a larger version of this figure.
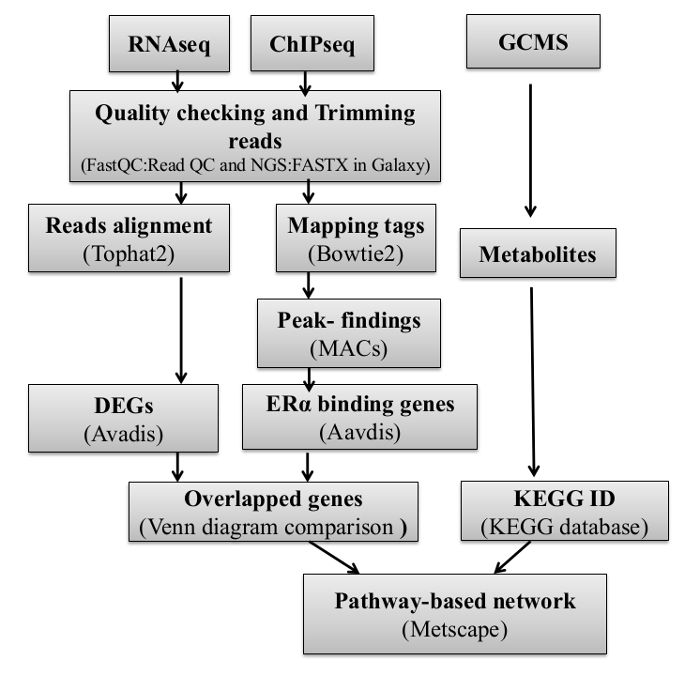 Figure 6: Flowchart of the Integrative clustering method.
Figure 6: Flowchart of the Integrative clustering method.
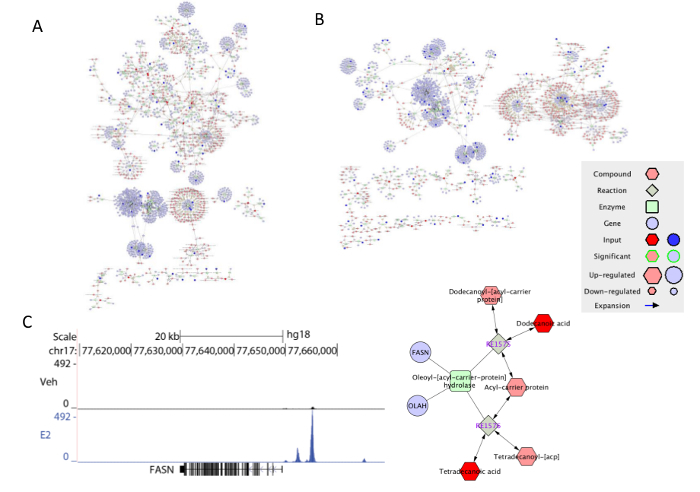 Figure 7: The visualization of the integrated gene-metabolite data in Metscape. (A) The network of pathways up-regulated by E2. (B) The network of pathways down-regulated by E2. (C) E2 stimulated ERα recruitment to the nearby region of FASN, which is found to be regulated in the de novo fatty acid biosynthesis network generated by applying subnetwork function. Please click here to view a larger version of this figure.
Figure 7: The visualization of the integrated gene-metabolite data in Metscape. (A) The network of pathways up-regulated by E2. (B) The network of pathways down-regulated by E2. (C) E2 stimulated ERα recruitment to the nearby region of FASN, which is found to be regulated in the de novo fatty acid biosynthesis network generated by applying subnetwork function. Please click here to view a larger version of this figure.
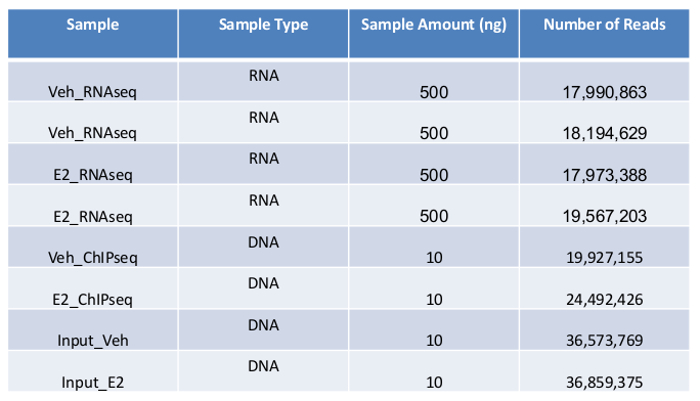 Table 1:
Summary of ultra-high-throughput sequencing of RNA-Seq and ChIP-Seq.
Table 1:
Summary of ultra-high-throughput sequencing of RNA-Seq and ChIP-Seq.
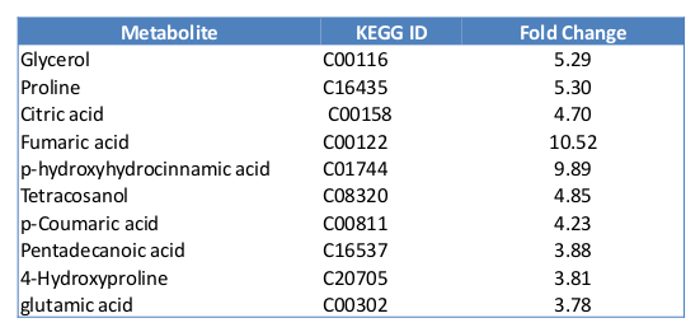 Table 2:
List of top 10 metabolites with significant changes with E2 treatment.
Table 2:
List of top 10 metabolites with significant changes with E2 treatment.
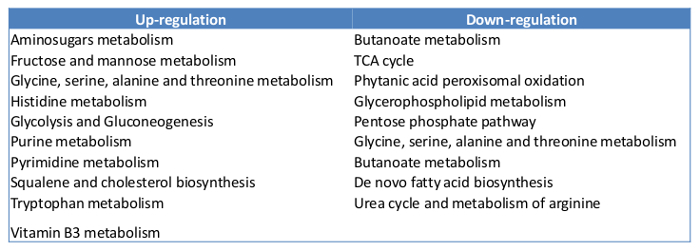 Table 3: Pathways up- and down-regulated by E2 treatment.
Table 3: Pathways up- and down-regulated by E2 treatment.
Discussion
In this paper, we described generation and integrative analysis of RNA-Seq, ChIP-Seq and metabolomics data from MCF-7 cells that are treated with E2. We developed a set of protocols strategizing to utilize the most efficient methods and user friendly soft wares which produce biologically relevant discovery. To our knowledge, ours is the first study to integrate -omics data from three different analysis and identified new metabolic pathways that ERα directly regulated in breast cancer cells.
Transcriptomics It is critical to start with high quality RNAs for a successful transcriptomic analysis. Good practice will eliminate the chance of RNA degradation, including creating RNase-free bench, changing gloves often, storing in -80 °C and keeping on ice in use. Addition of overnight incubation at -20 ºC improves RNA recovery. To avoid freezing and thawing RNA repeatedly, one can allocate the RNAs for downstream procedures upon the isolation and purification. For the existence of sequencing errors, qRT-PCR using the cDNA from the samples is often performed to verify the differentially expressed genes identified in RNA-Seq. Therefore, enough RNA should be reserved for this purpose. We perform paired-end-sequencing that enables us to analyze splice variants, non-coding RNAs and miRNAs. To avoid high-sequencing costs, single-end sequencing can be performed as well. Compared to microarrays, RNA-Seq is more sensitive and is not limited to the probes provided on the chip. We obtain information not only on coding sequences, but also on non-coding genes and differentially spliced transcripts. We are currently using this technique to analyze transcriptomes of various cell lines after ligand treatments as well as tissues obtained from mouse experiments.
Cistromics The ultra-high-throughput sequencing method requires minimal 5 ng ChIP DNA for library construction. In standard ChIP-Seq protocol18, it is recommended to have at least 10 ng. Quantitation with fluorometry is critical. In this protocol we used double strand DNA detection assay to determine the DNA concentration. One can also use fluorometer assay for precise DNA quantification. For the differences in equipment and materials, it is very important for each lab to establish their own sonication protocol, which generates desirable DNA fragments sizes. Over-shearing the ChIP DNA might cause loss of enrichment of target DNAs while ChIP DNA of large sizes create problem for efficient sequencing. Before submitting the DNA to sequencing, it is always important to verify enrichment of some known regions by qPCR. The technique is still limited by the efficiency and specificity of the antibody used for immunoprecipitations. To obtain binding site information that could be reliably used in comparative analyses, we validate our reagents following ENCODE ChIP-Seq standards,19 and use standardized protocols that provide us with consistent data sets. Coupled with the post transcription factor binding site enrichment analysis, ChIP-Seq is still one of the best methods to determine direct binding of transcription factors to chromatin. We are currently using this protocol to study recruitment kinases, coregulators and other transcription factors to chromatin in other cell lines as well as estrogen responsive mouse tissues.
Metabolomics For a successful metabolic profiling in human breast cancer cells, a careful sample preparation is critical. The amount of cells in use and cell culture condition in this protocol is optimal for MCF7 cells. Other cell lines might require more or less cells for detection of a significant number of metabolites. We obtain around 100 metabolites in each experiment. This method is limited for detection of short lived metabolites or metabolites that are present at low concentrations. This method offers the ease of using a single sample prep to identify multiple metabolites. We are currently using this technique to identify estrogen regulated metabolites in serum and various tissues from mouse.
Data Integration Numerous transcriptomics and cistromics studies were done to investigate estrogen signaling and gene regulation in breast cancer cells, including MCF7 cells2,5-7,20. In this study, for the first time, we added metabolomics data and tried to infer metabolic networks that are directly regulated by ERα upon ligand addition. This novel approach enabled us to pin point molecular circuitries regulated by estrogens that impact cellular metabolism. Overall, our findings support the role of ERα as a master regulator of breast cancer biology.
In summary, we provided a detailed compendium of protocols for generation of RNA-Seq, ChIP-Seq and metabolomics samples from breast cancer cells after estrogen treatments and integrative analysis of the data obtained from these -omics approaches. These protocols will enable researchers to do similar analysis for other transcription factors, nuclear receptor ligands and nutrient conditions.
Disclosures
University of Illinois received an Investigator Initiated grant from Pfizer Inc. ZME and YCZ received salary support from this grant.
Acknowledgments
This work was supported by grants from the National Institute of Food and Agriculture, U.S. Department of Agriculture, award ILLU-698-909 (ZME) and Pfizer, Inc (ZME). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the U.S. Department of Agriculture.
References
- Hamilton KJ, Arao Y, Korach KS. Estrogen hormone physiology: reproductive findings from estrogen receptor mutant mice. Reprod Biol. 2014;14(1):3–8. doi: 10.1016/j.repbio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak-Erdogan Z, et al. Integrative genomics of gene and metabolic regulation by estrogen receptors alpha and beta, and their coregulators. Mol Syst Biol. 2013;9:676. doi: 10.1038/msb.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, et al. Transcriptomic analysis identifies gene networks regulated by estrogen receptor alpha (ERalpha) and ERbeta that control distinct effects of different botanical estrogens. Nucl Recept Signal. 2014;12:e001. doi: 10.1621/nrs.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi A, et al. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014;16(5):436. doi: 10.1186/s13058-014-0436-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak-Erdogan Z, et al. Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors. Mol Endocrinol. 2008;22(9):2116–2127. doi: 10.1210/me.2008-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Lupien M, Stossi F, Brown M, Katzenellenbogen BS. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol Cell Biol. 2011;31:226–236. doi: 10.1128/MCB.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak-Erdogan Z, Ventrella R, Petry L, Katzenellenbogen BS. Novel roles for ERK5 and cofilin as critical mediators linking ERalpha-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol Cancer Res. 2014;12(5):714–727. doi: 10.1158/1541-7786.MCR-13-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Invitrogen. Quant-iT™ PicoGreen ® dsDNA Reagent and Kits. 2008. Available from: http://www.nature.com/protocolexchange/system/uploads/3551/original/Quant-iT_Picogreen_dsDNA.pdf?1423512049.
- Lee PY, Costumbrado J, Hsu CY, Kim YH. Agarose gel electrophoresis for the separation of DNA fragments. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- Quant-iT™ Assays for high-throughput quantitation of DNA, RNA, and oligos. 2006. Available from: https://tools.thermofisher.com/content/sfs/brochures/F-065432%20quantit%20htp_FLR.pdf.
- Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon WB. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 2015;8 doi: 10.1186/s13040-014-0034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based analysis of ChIP-Seq(MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, et al. Metscape: a Cytoscape plug-in for visualizing and interpreting metabolomic data in the context of human metabolic networks. Bioinformatics. 2010;26:971–973. doi: 10.1093/bioinformatics/btq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder A, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7(3) doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokry M, et al. Efficient double fragmentation ChIP-seq provides nucleotide resolution protein-DNA binding profiles. PLoS One. 2010;5(11):e15092. doi: 10.1371/journal.pone.0015092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Kraus WL. Hormone-regulated transcriptomes: lessons learned from estrogen signaling pathways in breast cancer cells. Mol Cell Endocrinol. 2014;382:652–664. doi: 10.1016/j.mce.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]