

Abstract
Seasonal climatic shifts create peripheral habitats that alternate between habitable and uninhabitable for migratory species. Such dynamic peripheral habitats are potential sites where migratory species could evolve high genetic diversity resulting from convergence of immigrants from multiple regionally distant areas. Migrant populations of Helicoverpa zea (Boddie) captured during two different seasons were assessed for genetic structure using microsatellite markers and for host plant type using stable carbon isotope analysis. Individuals (N = 568) were genotyped and divided into 13 putative populations based on collection site and time. Fixation indices (F‐statistics), analysis of molecular variance (AMOVA), and discriminant analysis of principal components (DAPC) were used to examine within and among population genetic variation. Mean number of alleles per locus was 10.25 (± 3.2 SD), and allelic richness ranged from 2.38 to 5.13 (± 3.2 SD). The observed and expected heterozygosity ranged from 0.07 to 0.48 and 0.08 to 0.62, respectively. Low FST (0.01 to 0.02) and high FIS (0.08 to 0.33) values suggest captured migrants originated from breeding populations with different allele frequencies. We postulate that high genetic diversity within migrant populations and low genetic differentiation among migrant populations of H. zea are the result of asymmetrical immigration due to the high dispersal and reproductive behavior of H. zea, which may hinder the adaptation and establishment of H. zea to peripheral habitat. These findings highlight the importance of assessing peripheral population structure in relation to ecological and evolutionary dynamics of this and other highly reproductive and dispersive species.
Keywords: DAPC, Helicoverpa, heliothine, moth, population genetics, stable isotopes
Introduction
Migration is the primary mechanism species use to avoid competition, find resources and, in sexual systems, locate mates (Dingle and Drake 2007). Several species have evolved long distance, seasonal, migratory characteristics, which are generally prompted by environmental cues (Alerstam et al. 2003; Dingle and Drake 2007). Seasonal migratory species are ecologically important in providing ecosystem services (e.g., Kremen et al. 2007) and causing economic damage due to spreading disease and destruction of agricultural products. Assessing the population‐level process associated with seasonal migrant recolonization and invasion is necessary for understanding impacts on biodiversity (Hughes et al. 2008), developing conservation planning, and species management strategies.
Immigration is a fundamental process influencing seasonal population establishment and structure. Successful establishment of seasonal, peripheral populations at the extent of species’ geographic range is influenced by the genetic diversity of the local peripheral population (Slatkin 1987) and the gene flow (i.e., immigration) (Kolbe et al. 2004) and propagule pressure (Lockwood et al. 2005) of the colonizing population(s). If gene flow or propagule pressure of colonizing population(s) to a peripheral population is low, establishment may be hindered due to resulting low abundance (i.e., fewer propagules) and low genetic diversity resulting in lower adaptive potential or Allee effects (Lee 2002; Bock et al. 2015). Reduced adaptive potential can develop from low propagule pressure and gene flow resulting in reduced genetic diversity, which allows other evolutionary processes, such as drift, to reduce genetic diversity and for deleterious genes to be maintained longer. Alternatively, if immigration of colonizers is high enough to promote admixture and increase standing genetic variation, locally adaptive phenotypes are more likely to be selected and proliferate allowing establishment and expansion to occur (Lenormand 2002). However, if gene flow is too high, such that admixture is dominated by colonizing migrants compared to local genotypes, adaptation for the peripheral habitat may be hindered (due to influx of less fit genotypes; Verhoeven et al. 2010). Poor adaptation of immigrants for the peripheral habitat can in turn result in localized extinction, especially for extreme environments (Bridle and Vines 2007).
Migratory immigration and peripheral population structure can also be inferred from stable isotopic analysis of animal tissue, including insect wings (Jackson et al. 2006; Head et al. 2010). Carbon δ 13C, particularly, has been used to track the origin of several herbivorous insect species (Gould et al. 2002; Jackson et al. 2006; Flockhart et al. 2013). Carbon isotope values of plants depend on the photosynthetic pathway during uptake such that δ 13C ratios differ between C3, C4, and intermediary plants (Abelson and Hoering 1961; Hatch and Slack 1970; Monson et al. 1984). Herbivores’ carbon isotopic values, in turn, reflect the plants they feed on and thus may indicate separate source populations related to immigrant versus local populations (Gould et al. 2002; Head et al. 2010).
Helicoverpa zea (Bodie) (bollworm, corn earworm) is a polyphagous pest, feeding on a wide range of C3 (e.g., tomatoes and cotton) and C4 (e.g., corn and sorghum) plant species (Kennedy and Storer 2000). While estimates of economic damage due to H. zea are currently unknown, the estimated cost of control and yield loss due to damage in cotton and soybean in the southern United States in 2013–2014 exceeded $10 per acre (Musser et al. 2014; Williams 2014). Preference for C3 or C4 plants has been documented for H. zea and reflects larval feeding habits and may indicate the geographic sources of immigrating populations by relating isotopic patterns to crop type abundances (Gould et al. 2002). Helicoverpa zea are capable of high reproductive rates with females typically laying between 500 and 1000 eggs (Fye and McAda 1972), and up to 3000 eggs within a 8‐ to 10‐day period (Quaintance and Brues 1905). After pupal emergence, adult H. zea will facultatively migrate long distances, with suspected overnight migrations upwards of 400 km, aided by low‐level jet streams (Westbrook and López 2010). Helicoverpa zea primarily overwinters as diapausing pupae, which burrow into the soil as larva. However, H. zea is not currently known to overwinter north of the 40th parallel (Blanchard 1942; Hardwick 1965, 1996), due to lack of adaptation for cold temperatures (Blanchard 1942; Morey et al. 2012). Several generations of H. zea occur during late spring through early fall, depending on the geographic region, with 4 to 8 generations occurring in southern populations and 1 to 2 generations occurring in peripheral, northern populations due to cold intolerance (Quaintance and Brues 1905; Morey et al. 2012). Expansion of the winter range of H. zea could lead to significant changes (e.g., earlier, greater) in seasonal time of pest presence, population levels, and degree of crop damage it causes in northern latitudes, which typically do not observe infestation until late summer (Blanchard 1942; Morey et al. 2012). Despite the high impact on agriculture, little is known regarding the genetic diversity and the potential impacts on range expansion of this species.
Here, we used microsatellite loci to assess genetic variation of 13 putative, migrant, peripheral populations of H. zea immigrating to seasonal habitat north of the known overwintering range (Morey et al. 2012). Putative populations are described below and are based on known H. zea migration behavior (Fitt 1989). We assessed the extent of genetic structure attributed to immigrating populations of H. zea to get insights into the underlying evolutionary, ecological, and demographic processes. Additionally, we assessed differences in colonizer sources using stable carbon isotope analysis. Specifically, we aimed to infer the extent of putative migratory genetic structure, and the potential implications for the geographic structure of H. zea. Alternatively, observation of random genetic variation would suggest asymmetrical gene flow or geographically admixed populations. Overall, genetic structure is characterized in relation to the dispersal dynamics of an important, but generally overlooked insect species, which has direct implications for land management and dispersal dynamics of other similarly high dispersal and reproductive species.
Methods
Sampling
Adult H. zea were sampled from July through September during 2002 and 2005 near the Russel E. Larson Research and Education Center at Rock Springs, PA (40°42′38.1″N 77°57′52.2″W) and from July through September 2005 at the Southeast Research Center at Landisville, PA (40°07′04.7″N 76°25′30.5″W; Fig. 1). Adults were trapped using Hartstack pheromone traps (Hartstack et al. 1979) positioned near corn fields, which is a preferred food source for H. zea. All trapped individuals (N = 576) were collected and stored at −20°C for additional laboratory analysis. Wings dissected from each individual were preserved in 70% ethanol, and genomic DNA from thorax and head of each individual extracted using the Qiagen DNeasy Kit (Valencia, CA) at Penn State were sent to the USDA‐ARS Southern Insect Management Research Unit (SIMRU) for stable carbon isotope analysis and for microsatellite genotyping, respectively.
Figure 1.
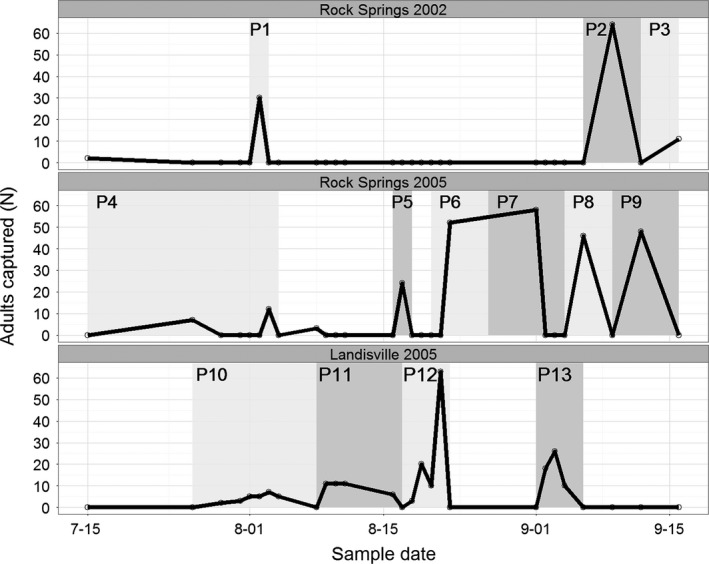
Adults captured (N) in pheromone traps from 26 July to 09 September (sample date), shown as month–day, for Helicoverpa zea sampled from Landisville, PA in 2005, and Rock Springs, PA in 2002 and 2005. The numbers depict putative populations (P1‐P13) of new migrants entering the respective sampling sites.
Stable carbon isotope analysis
The distal half of a forewing from each moth was cut into small pieces and placed into a 5 × 9 mm tin capsule that was tightly folded. Conversion of wing tissue in each tin capsule to CO2 was done at SMRT using micro‐Dumas combustion using a Costech ECS4010 Elemental Analyzer (Costech Analytical Technologies Inc., Valencia, CA) coupled to a Thermo Finnigan Delta plus Advantage Mass Spectrometer using a Conflo II Interface (Thermo Scientific, Waltham, MA). The isotope standard reference material was bovine muscle powder, from the National Institute of Standards and Technology (NIST‐RM 8414). Moths were then determined to either feed primarily on C3 (δ 13C ≤ −20) or C4 (δ 13C ≥−15) plants based on previous assessment of stable carbon isotope analysis of H. zea (Head et al. 2010).
Microsatellite analysis
Prior to microsatellite genotyping, DNA samples were quantified using the Quanti‐iT PicoGreen assay kit (Invitrogen, Carldbad, CA) for double‐stranded DNA and then adjusted to 20 ng/μL. A set of 12 polymorphic microsatellite markers previously developed for H. zea was used to analyze the DNA samples (Perera et al. 2007). Polymerase chain reactions (PCR) for each sample included 1 μL of DNA, 0.4 pM forward primer, 1.2 pM reverse primer, 1.2 pM 6‐carboxyfluorescein (6‐FAM)‐labeled universal primer, 1 μL of 10 mM dNTP mix, 0.5 μL of 10× titanium Taq polymerase buffer, 0.1 μL of titanium Taq polymerase (BD Bioscience, San Jose, CA), and 1.6 μL of DNase free water for a 5 μL reaction. Cycling conditions included an initial denaturation step for 2 min at 95°C, an initial annealing step of 1 min at 60°C, followed by 30 denaturing and annealing cycles (15 sec at 95°C, 15 sec at 60°C, 30 sec at 72°C). Reactions were then diluted 1/9 and amplicons separated on an ABI 3700xl genetic analyzer with ROX‐labeled markers. Peaks were scored as genotypes using the software GeneMapper (Applied Biosystems, Inc., Foster City, CA) and confirmed manually.
Genetic structure
All loci were checked for null alleles and allelic drop out using MICROCHECKER 2.2.3 (van Oosterhout et al. 2004). Individuals missing three or more loci were removed from the analysis. All additional genetic and statistical analyses were performed using the program R version 3.2.1 (R Development Core Team 2012). Markers were tested for linkage disequilibrium using the index of association (IA) and 999 permutations (Brown et al. 1980) implemented in the package poppr (Kamvar et al. 2014). Deviations from Hardy–Weinberg equilibrium (HWE) were assessed per loci across all populations using an exact test, based on Monte Carlo permutations, with 999 permutations, implemented in the package pegas (Paradis 2010). Significant levels for multiple comparisons of loci across samples were adjusted using a sequential Bonferroni correction (Rice 1989). The package hierfstat (Goudet 2005) was used to calculate expected (H E) and observed (H O) heterozygosity, within (F IS) and among (F ST) population fixation indices, pairwise F ST and allelic richness (Ar).
Discriminant analysis of principal components (DAPC) (Jombart et al. 2010) was used as implemented in the R package adegenet (Jombart 2008) to determine population genetic structure of H. zea using (1) a set of prior putative H. zea populations, (2) assuming no prior population assignment, and (3) C3 versus C4 plant preference. Prior putative populations were determined as peak abundance periods during the collection periods, which signaled recent mass immigration of H. zea (Fig. 1) (Fitt 1989). Trapped moths, especially from earlier months when there are no local individuals present, are expected to be immigrant individuals. Later months may include local individuals, which may include progeny from earlier migrants as 1 to 2 generations do occur in Pennsylvania per year (Quaintance and Brues 1905; Morey et al. 2012). In total, 13 putative populations were assigned with populations sizes ranging from 11 to 95 per grouping, with an overall mean population size of 43. The nonprior population assignment DAPC analysis was used to evaluate number of clusters (K) between 2 and 20 for H. zea. Bayesian information criterion (BIC) was then used to evaluate the relevance of different K values to population structure. Assignment values for the selected number of clusters were then generated for each individual using DAPC (Jombart et al. 2010). DAPC first transforms the data using principal components analysis, which ensures that the variables are not correlated and that the number of variables is smaller than the number of individuals. Then, discriminant analysis partitions the variance into among‐ and within‐group components, maximizing separation between groups. DAPC does not assume a population genetics model and it is not constrained by Hardy–Weinberg or linkage equilibrium assumptions, making it a robust method to test for genetic differentiation. The DAPC findings were complemented by use of the program STRUCTURE to perform a Bayesian clustering analysis (Pritchard et al. 2000) to assess the possible occurrence of 1 to 13 distinct populations (K = 1 to K = 13; used 100,000 burn in and an additional 100,000 Markov chains). Analysis of molecular variance (AMOVA) was performed using Arlequin v3.5 (Excoffier and Lischer 2010) by partitioning populations into groups relative to the large peak trap catch that occurred in 2005 at Rock Springs and Landisville, PA (prepeak trap catch: P1, P4, P5, P10, P11; peak trap catch: P6, P7, P12; postpeak trap catch: P2, P3, P8, P9, P13), collection site and year, and host plant type (C3 or C4). The significance of the components of locus‐by‐locus AMOVA was tested with 10,000 permutations.
Results
Stable isotope analysis
For all 568 adults genotyped, irrespective of putative population assignment, 90.84% had δ 13C >−15 suggesting they developed on C4 host plants. Only 8.45% had δ 13C <−20, which suggested utilization of C3 host plants as larvae, regardless of sampling location. Host plant type could not be determined in only 4 individuals whose δ 13C was between −15 and −20. For adults across each putative population (Fig. 1), an average of 87% (± 10% SD) developed on C4 host plants compared to 12% (± 9% SD) that developed on C3 host plants (Fig. 2). Adults collected at Rock Springs prior to August 4 in 2002 and 2005 (Fig. 1, P1 and P4) had the highest proportion (>25%) of moths that developed on C3 host plants within a population (Fig. 2) suggesting higher proportion of founders to this area were feeding on different host plants prior to immigrating.
Figure 2.
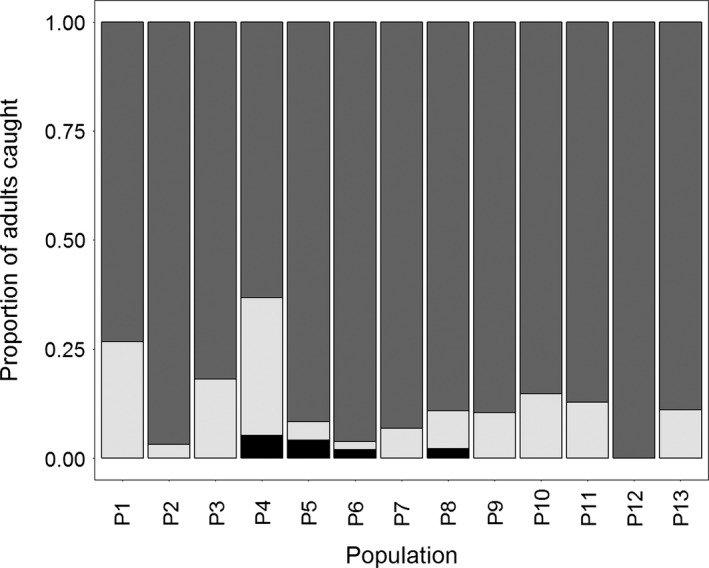
Proportion of Helicoverpa zea that developed on C4 (gray) C3 (light gray) or unknown (black) plants per putative population (see Fig. 1), based on stable carbon isotope analysis. Sample sizes for each population are given in Table 2.
Microsatellite analysis
Significant deviation from HWE occurred for 6 of the 12 loci used (Table 1). Deviation from HWE are nontrivial and need to be appropriately assessed with regard to the overall analysis as they imply genotyping error (Morin et al. 2009), potentially selected loci, or loci linked to selected loci, which do not reflect neutral evolutionary dynamics, including gene flow and drift (Holderegger and Wagner 2006), which are the focus of this study. Additionally, three loci (HzMS4_10, HzMS4_14, and HzMS4_16) had a significant deficit of heterozygotes across all or most populations, suggesting the presence of null alleles at these loci. Two of these loci were also suspected of genotyping error due to stuttering (HzMS4_14 and HzMS_16). These loci were also reported out of HWE and suspected of null alleles and excluded from analyses in previous studies (Perera et al. 2007; Perera and Blanco 2011). Individual removal of each locus, with significant deviation from HWE, from the analysis revealed that the four loci HzMS1_6, HzMS4_10, HzMS4_14, and HzMS4_16 greatly influenced the genetic structure of the H. zea populations, and they were not used in subsequent analyses (Table 1). The remaining two loci (HzMS3_41 and HzMS4_23) out of HWE, which were not suspect null alleles, did not influence genetic structure when removed from the analysis and were kept in further analyses (Table 1).
Table 1.
Summary of locus‐level genetic parameters, number of unique alleles (A), observed heterozygosity (Hobs), expected heterozygosity (Hexp), F ST across populations, and whether the loci were included in the final analysis (Included). F ST values that significantly differed from HWE expectations based on Monte Carlo permutations are indicated with an asterisk (*)
| Locus | GenBank Accession | A | Hobs | Hexp | F ST | Included |
|---|---|---|---|---|---|---|
| HzMS1_4 | EF152205 | 9 | 0.326 | 0.321 | 0.001 | Yes |
| HzMS3_1 | EF152207 | 6 | 0.354 | 0.377 | 0.003 | Yes |
| HzMS3_11 | EF152209 | 11 | 0.481 | 0.514 | 0.000 | Yes |
| HzMS3_41 | EF152210 | 14 | 0.390 | 0.496 | 0.034* | Yes |
| HzMS3_48 | EF152211 | 7 | 0.446 | 0.518 | 0.017 | Yes |
| HzMS3_86 | EF152212 | 14 | 0.068 | 0.075 | 0.004 | Yes |
| HzMS4_3 | EF152213 | 8 | 0.269 | 0.282 | 0.006 | Yes |
| HzMS4_23 | EF152217 | 13 | 0.486 | 0.622 | 0.000* | Yes |
| HzMS1_6 | EF152206 | 21 | 0.378 | 0.536 | 0.122* | No |
| HzMS4_10 | EF152214 | 14 | 0.057 | 0.321 | 0.013* | No |
| HzMS4_14 | EF152215 | 14 | 0.214 | 0.422 | 0.001* | No |
| HzMS4_16 | EF152216 | 9 | 0.231 | 0.759 | 0.016* | No |
The eight loci used in the final genetic analyses had no linkage disequilibrium, allelic dropout, or potential null alleles. Mean number of unique alleles per locus was 10.25 (±3.2 SD). Observed heterozygosity ranged from 0.07 to 0.48, while expected heterozygosity ranged from 0.08 to 0.62 (Table 1). Among putative populations, allelic richness ranged from 2.38 to 5.13 (±3.2 SD). The F IS ranged from 0.06 to 0.33, with two populations (P1 and P3) not significantly deviating from zero (Table 2), suggesting inbreeding and nonrandom mating across the putative populations. Additionally, the observed F IS may have resulted from pheromone traps capturing a mixture of immigrants originated from populations with different allele frequencies. Estimates of F ST (Table 2) for all populations ranged between 0.01 and 0.02 and remained unaffected by the removal of individual loci from the analysis. Global F ST was 0.01 with an F IS of 0.16 and total allelic richness of 82 unique alleles genotyped. Pairwise F ST ranged from 0.002 to 0.038 (Table S1).
Table 2.
Summary of population‐level genetic parameters, number of individuals genotyped (N), mean number of alleles per locus (A), observed heterozygosity (Hobs), expected heterozygosity (Hexp), mean pairwise F ST, and F IS with its 95% confidence interval (F ISCI)
| Population | N | A | Hobs | Hexp | F ST | F IS | F ISCI |
|---|---|---|---|---|---|---|---|
| P1 | 30 | 2.75 | 0.346 | 0.386 | 0.011 | 0.082 | −0.02 to 0.22 |
| P2 | 64 | 5.13 | 0.357 | 0.421 | 0.008 | 0.125 | 0.01 to 0.28 |
| P3 | 11 | 2.38 | 0.261 | 0.334 | 0.016 | 0.244 | −0.13 to 0.52 |
| P4 | 19 | 3.38 | 0.362 | 0.452 | 0.014 | 0.172 | 0.04 to 0.35 |
| P5 | 24 | 3.63 | 0.302 | 0.390 | 0.012 | 0.207 | 0.15 to 0.31 |
| P6 | 52 | 4.75 | 0.394 | 0.422 | 0.009 | 0.059 | 0.00 to 0.14 |
| P7 | 57 | 4.25 | 0.345 | 0.496 | 0.008 | 0.295 | 0.19 to 0.37 |
| P8 | 45 | 4.63 | 0.342 | 0.503 | 0.014 | 0.329 | 0.17 to 0.39 |
| P9 | 48 | 4.13 | 0.326 | 0.422 | 0.009 | 0.240 | 0.12 to 0.31 |
| P10 | 27 | 3.13 | 0.347 | 0.418 | 0.013 | 0.167 | 0.06 to 0.28 |
| P11 | 38 | 3.00 | 0.327 | 0.403 | 0.009 | 0.156 | 0.05 to 0.21 |
| P12 | 95 | 4.25 | 0.294 | 0.427 | 0.005 | 0.331 | 0.16 to 0.39 |
| P13 | 54 | 4.63 | 0.370 | 0.477 | 0.008 | 0.229 | 0.10 to 0.32 |
Genetic structure
Results of the DAPC analysis using prior assignment of putative populations (Fig. 3) revealed very little differentiation among the putative populations. In the nonprior DAPC analysis (Table S2; Fig. S1), K = 7 was selected as the most parsimonious clustering based on differences between successive values of BIC summary statistics and successive cluster assignment from repeated DAPC runs (Jombart 2008; Jombart et al. 2010). This additional clustering of H. zea individuals predicted using nonprior DAPC, despite showing some variation among individuals, did not correspond to any putative population structure, location, or utilization of C3 or C4 host plant, suggesting that these H. zea populations are largely panmictic (Fig S2). Specifically, individuals from each putative population were generally found across all DAPC clusters with a mean of 2 to 14 (SD = 1.89 to 4.22) individuals from each putative population grouping in each of the DAPC cluster. Analysis with STRUCTURE showed even less support for genetic differentiation among populations compared to the DAPC analysis (Fig. S2). STRUCTURE clusters had mean coefficient of ancestry ranging from 0.17 (SD = 0.01) to 0.29 (SD = 0.08) with 28 to 121 individuals assigned to one of seven clusters. Molecular variance analysis (Excoffier and Lischer 2010) of genotype data partitioned by host plant type (C4 or C4), or by collection site/year, indicated that the greatest molecular variance (>87%) occurred in individuals within the total population. Genetic variation among populations was minimal and not significant (Table S3).
Figure 3.
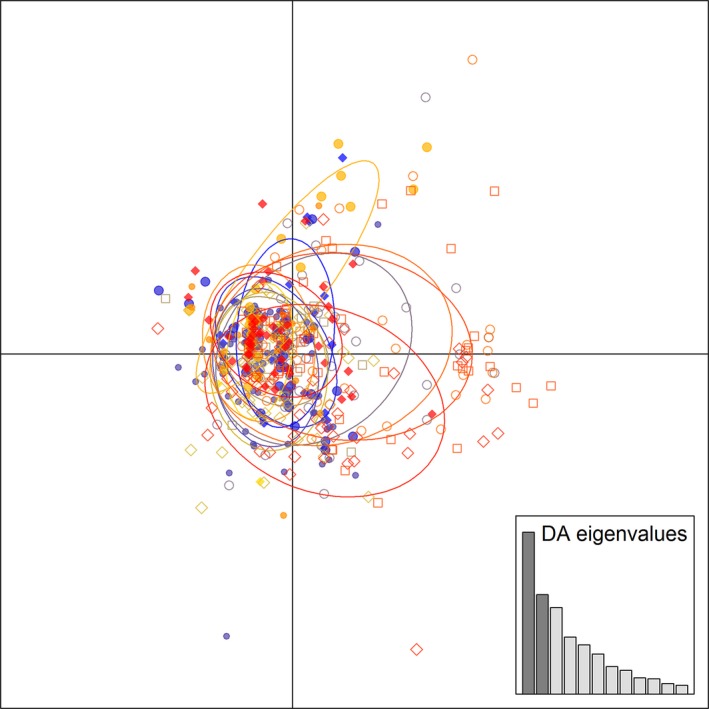
DAPC results using prior assignment of putative population showing the first two axes of the analysis (depicted in the insert plot). Each color and symbol represents a unique population cluster with the corresponding circles showing the prior unique groupings of the 13 putative populations.
Discussion
The high genetic diversity and low differentiation observed among the putative populations of H. zea sampled suggest that peripheral, northern populations are panmictic with asymmetrical immigration likely dominated by immigrants from southern populations occurring throughout the growing season. That there was no temporal or spatial relationships associated with the putative population clusters suggest immigrants are highly admixed possibly originating from multiple source populations. This tendency is potentially reflected in high F IS estimates due to capturing of multiple immigrating populations with differing allelic frequencies per trapping event. The continuous mass pooling and asymmetrical dispersal of H. zea is likely influenced by variation in emigration from different southern population generation cycles, which are known to be locally influenced by abiotic (e.g., weather fronts) and biotic (e.g., crop phenology) factors, and occur more frequently in southern (5 to 7 generations/year) than northern (1 to 2 generations/year) regions (Quaintance and Brues 1905; Fitt 1989). Additionally, asymmetrical dispersal may limit selection of H. zea to adapt to cooler peripheral habitats, thereby limiting the extent of the over wintering range by counteracting selection of local peripheral populations.
Studies have found generally low genetic variability of H. zea across North and South America using allozymes (Sell et al. 1974; Sluss et al. 1978; Mallet et al. 1993; Han and Caprio 2002), mitochondrial DNA (Behere et al. 2007), and microsatellites (Perera and Blanco 2011). However, the population genetic structure of H. zea is still largely unknown. Studies of the very closely related Helicoverpa armigera (Hübner) suggest that temporal and spatial genetic differentiation may exist in Australia (Scott et al. 2003, 2005), but also showed low genetic differentiation across other larger geographic areas (Nibouche et al. 1998; Zhou et al. 2000; Behere et al. 2007; Endersby et al. 2007) indicating high gene flow among populations. Migration (i.e., long range dispersal) of H. zea and other heliothine species is well documented (Hartstack et al. 1982; Farrow and Daly 1987; Gregg et al. 1995), with simulations suggesting that overnight dispersal of a few hundred kilometers is possible (Westbrook 2008; Westbrook and López 2010), and repeated dispersal events enabling semicontinental scale immigration for related noctuid species (Westbrook et al.2016).
Temporal structure used in this study is based on previously studied aspects of H. zea ecology including lack of adaptation (e.g., cold tolerance) to winter peripheral environments, low number (1 to 2) of generations occurring at peripheral sites, high reproductive rate, and rapid mass dispersal behavior (Quaintance and Brues 1905; Blanchard 1942; Fitt 1989; Morey et al. 2012). Like our findings, a smaller study across southern H. zea populations using the same genetic markers (Perera and Blanco 2011) found greater genetic diversity and similarly lower genetic differentiation and expected heterozygosity. Both findings fit well with the highly dispersive ecology of H. zea and suggest that peripheral seasonal populations harbor more genetic variation compared to southern populations where continuous selective pressures, such as insecticides, disease, and predation, may reduce genetic variation compared to peripheral populations, which are likely admixtures of immigrants from a much larger geographic area. Previous expectations are that peripheral populations go extinct annually, due to lower temperatures reducing survivability (Fitt 1989). However, recent studies suggest return southward migration of H. zea is possible (Pair et al. 1987; Gould et al. 2002), but yet to be confirmed by genetic or mark recapture approaches. Agrotis ipsilon (Hufnagel), another long distance migratory lepidopteran species, does utilize southerly airflows to return migrate to the Gulf Coast of the USA from the northern USA each autumn (Showers et al. 1993). Peripheral populations of H. zea, resulting from progeny of earlier immigrating populations, may therefore provide sources of genetic diversity and increase admixture with southern populations through return migration from northern populations, thus maintaining migrant (e.g., seasonally invasive) phenotypes (Bock et al. 2015).
Helicoverpa armigera is morphologically and phylogenetically closely related to H. zea. Recently, H. armigera became established in Brazil and is presumed to have originated from Europe (Tay et al. 2013; Murúa et al. 2014). There are severe economic concerns regarding the expansion of H. armigera as it is known to develop resistance to insecticides, which are currently being used to manage H. zea (Behere et al. 2007; Yang et al. 2013). Similarity in physiology (e.g., diapause) and population genetics between H. zea and H. armigera indicate that our findings could have direct implications regarding a hypothesis of delayed invasion of H. armigera from South America to North America, whereby low genetic diversity of H. zea may be limiting successful local adaptation (Kriticos et al. 2015). However, successful establishment is likely to occur, given the favorable climate, agricultural crop flora and landscape present across much of the USA (Kriticos et al. 2015). Overall, understanding the patterns and underlying mechanisms of gene flow in migrant populations is important for developing control strategies for invasive species, such as H. armigera, which are expected to be substantially cheaper compared to projected management cost (Kriticos et al. 2015).
In summary, this study revealed that peripheral northern populations of H. zea possess high genetic diversity but low genetic differentiation among immigrant populations being invaded from several highly admixed source populations. Additionally, our findings support previous work suggesting that migratory H. zea are capable of dispersal over a large geographic range, likely resulting in genetic homogenization and panmixia (Behere et al. 2007; Perera and Blanco 2011). We highlight the general importance of peripheral populations to accumulate genetic diversity and suggest peripheral populations as potential sources for assessing genetic variability for the larger geographic area. In conclusion, it is likely that the previously documented high dispersal ability of H. zea and asymmetrical immigration is influencing the high genetic diversity (compared to previous studies) and low genetic differentiation observed. This process could have direct implications for management practices and future assessment of H. zea ecology at range limits likely to be affected as climate change is expected to extend species ranges northward due to increased mean annual temperatures.
Data Accessibility
Microsatellite genotype data and pertinent metadata used in this manuscript have been included in the supplementary Data file S01. Accession numbers of previously published microsatellite loci used in this analysis are listed in Table 1.
Conflict of Interest
None declared.
Supporting information
Figure S1. Simulation saturation for the allelic richness.
Figure S2. DAPC results using no prior assignment of putative population showing the first two axes of the analysis (depicted in the insert plot).
Figure S3. Outcome from STRUCTURE analysis assuming K = 7. Each vertical bar represents a unique individual (x‐axis) with their corresponding assignment score (y‐axis) for inclusion in a particular potential population (color).
Table S1. Pairwise F ST estimates (upper triangle) for putative population pairs.
Table S2. Summary of population level genetic parameters for the non‐prior DAPC analysis; the non‐prior populations cluster (Pop), number of individuals genotyped (N), mean number of alleles/locus (A), observed heterozygosity (Hobs), expected heterozygosity (Hexp), F ST and F IS with its 95% confidence interval (F ISCI).
Table S3. The results of AMOVA of Helicoverpa zea populations grouped by collection time to calculate ΦST (between populations when there was only one group), ΦCT (among groups), or ΦSC (among populations within groups).
Data S1. Microsatellite genotypes and collection data for Helicoverpa zea adults collected from Landisville and Rock Springs, PA. Size of each microsatellite allele (in Bp) in an individual is given in two consecutive sets of 3‐digits in the 6‐digit genotype.
Acknowledgments
We thank Prof. Jukka Jokela for his insightful discussion and technical assistance from Vanessa Russo (at Penn State), Les Price (at SIMRU), and Sharon Simpson (at the USDA‐ARS Genomics and Bioinformatics Research Unit, Stoneville, MS). Additionally, we thank Drs. Brad Coates and Maribel Portilla and Marie Leys for comments on earlier versions of the manuscript. Funding for work performed by HWF and SJF was from USDA‐ARS Specific Cooperative Agreement 58‐6402‐5‐066. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
References
- Abelson, P. H. , and Hoering T. C.. 1961. Carbon isotope fractionation in formation of amino acids by photosynthetic organisms. Proc. Natl Acad. Sci. USA 47:623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alerstam, T. , Hedenström A., and Åkesson S.. 2003. Long‐distance migration: evolution and determinants. Oikos 103:247–260. [Google Scholar]
- Behere, G. T. , Tay W. T., Russell D. A., Heckel D. G., Appleton B. R., and K. R. Kranthi. 2007. Mitochondrial DNA analysis of field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) and of its relationship to H. zea . BMC Evol. Biol. 7:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard, R. A. 1942. Hibernation of the corn earworm in the central and northeastern parts of the United States. United States Department of Agriculture, Technical Bulletin No. 838 [Google Scholar]
- Bock, D. G. , Caseys C., Cousens R. D., Hahn M. A., Heredia S. M., and Hübner S.. 2015. What we still don't know about invasion genetics. Mol. Ecol. 24:2277–2297. [DOI] [PubMed] [Google Scholar]
- Bridle, J. R. , and Vines T. H.. 2007. Limits to evolution at range margins: when and why does adaptation fail? Trends Ecol. Evol. 22:140–147. [DOI] [PubMed] [Google Scholar]
- Brown, A. H. , Feldman M. W., and Nevo E.. 1980. Multilocus structure of natural populations of Hordeum spontaneum . Genetics 96:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle, H. , and Drake V. A.. 2007. What is migration? Bioscience 57:113–121. [Google Scholar]
- Endersby, N. M. , Hoffmann A. A., McKechnie S. W., and Weeks A. R.. 2007. Is there genetic structure in populations of Helicoverpa armigera from Australia? Entomol. Exp. Appl. 122:253–263. [Google Scholar]
- Excoffier, L. , and Lischer H. E. L.. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10:564–567. [DOI] [PubMed] [Google Scholar]
- Farrow, R. A. , and Daly J. C.. 1987. Long‐range movements as an adaptive strategy in the genus Heliothis (Lepidoptera, Noctuidae)‐a review of its occurrence and detection in 4 pest species. Aust. J. Zool. 35:1–24. [Google Scholar]
- Fitt, G. P. 1989. The ecology of Heliothis species in relation to agroecosystems. Annu. Rev. Entomol. 34:17–52. [Google Scholar]
- Flockhart, D. T. T. , Wassenaar L. I., Martin T. G., Hobson K. A., Wunder M. B., and Norris D. R. 2013. Tracking multi‐generational colonization of the breeding grounds by monarch butterflies in eastern North America. Proc. Royal Soc. London B: Biol. Sci. 280: 20131087. doi: 10.1098/rspb.2013.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fye, R. E. , and McAda W. C.. 1972. Laboratory studies on the development, longevity, and fecundity of six lepidopterous pests of cotton in Arizona. United States Department of Agricultural, Technical Bulletin No 1454. [Google Scholar]
- Goudet, J. 2005. HIERFSTAT, a package for R to compute and test hierarchical F‐statistics. Mol. Ecol. Notes 5:184–186. [Google Scholar]
- Gould, F. , Blair N., Reid M., et al. 2002. Bacillus thuringiensis‐toxin resistance management: stable isotope assessment of alternate host use by Helicoverpa zea . Proc. Natl Acad. Sci. USA 99:16581–16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg, P. C. , Fitt G. P., Zalucki M. P., and Murray D. A. H. (1995) Insect migration in an arid continent. II. Helicoverpa spp. in eastern Australia Pp. 151–72 in Drake V. A., Gatehouse A. G., eds. Insect Migration: Tracking Resources Through Time and Space. Cambridge University Press, Cambridge. 478 pp. [Google Scholar]
- Han, Q. , and Caprio M. A.. 2002. Temporal and spatial patterns of allelic frequencies in cotton bollworm (Lepidoptera: Noctuidae). Environ. Entomol. 31:462–468. [Google Scholar]
- Hardwick, D. F. 1965. The corn earworm complex. Memoirs Entomol. Soc. Canada 97:5–247. [Google Scholar]
- Hardwick, D. F. 1996. A monograph to the North American Heliothentinae (Lepidoptera: Noctuidae). Agriculture Canada, Centre for Land and Biological Resources Research. [Google Scholar]
- Hartstack, A. W. , Witz J. A., and Buck D. R.. 1979. Moth traps for the tobacco budworm. J. Econ. Entomol. 72:519–522. [Google Scholar]
- Hartstack, A. W. , Lopez J. D., Muller R. A., Sterling W. L., and King E. G.. 1982. Evidence of long range migration of Heliothis zea (Boddie) into Texas and Arkansas. Southwestern Entomol. 7:188–201. [Google Scholar]
- Hatch, M. D. , and Slack C. R.. 1970. Photosynthetic CO2‐fixation pathways. Annu. Rev. Plant Physiol. 21:141–162. [Google Scholar]
- Head, G. , Jackson R. E., Adamczyk J., Bradley J. R., Van Duyn J., and Gore J. 2010. Spatial and temporal variability in host use by Helicoverpa zea as measured by analyses of stable carbon isotope ratios and gossypol residues. J. Appl. Ecol. 47:583–592. [Google Scholar]
- Holderegger, R. , and Wagner H.. 2006. A brief guide to landscape genetics. Landscape Ecol. 21:793–796. [Google Scholar]
- Hughes, A. R. , Inouye B. D., Johnson M. T. J., Underwood N., and Vellend M.. 2008. Ecological consequences of genetic diversity. Ecol. Lett. 11:609–623. [DOI] [PubMed] [Google Scholar]
- Jackson, R. E. , Gould F., and Bradley J. R.. 2006. Genetic variation for resistance to Bacillus thuringiensis toxins in Helicoverpa zea (Lepidoptera: Noctuidae) in eastern North Carolina. J. Econ. Entomol. 99:1790–1797. [DOI] [PubMed] [Google Scholar]
- Jombart, T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. [DOI] [PubMed] [Google Scholar]
- Jombart, T. , Devillard S., and Balloux F.. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamvar, Z. N. , Tabima J. F., and Grünwald N. J.. 2014. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. 2:e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy, G. G. , and Storer N. P.. 2000. Life systems of polyphagous arthropod pests in temporally unstable cropping systems. Annu. Rev. Entomol. 45:467–493. [DOI] [PubMed] [Google Scholar]
- Kolbe, J. J. , Glor R. E., Rodriguez Schettino L., Lara A. C., Larson A., and Losos J. B.. 2004. Genetic variation increases during biological invasion by a Cuban lizard. Nature 431:177–181. [DOI] [PubMed] [Google Scholar]
- Kremen, C. , Williams N. M., Aizen M. A., Gemmill‐Herren B., LeBuhn G., and Minckley R.. 2007. Pollination and other ecosystem services produced by mobile organisms: a conceptual framework for the effects of land‐use change. Ecol. Lett. 10:299–314. [DOI] [PubMed] [Google Scholar]
- Kriticos, D. J. , Ota N., Hutchison W. D., Beddow J., Walsh T., and Tay W. T.. 2015. The potential distribution of invading Helicoverpa armigera in North America: is it just a matter of time? PLoS ONE 10:e0119618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C. E. 2002. Evolutionary genetics of invasive species. Trends Ecol. Evol. 17:386–391. [Google Scholar]
- Lenormand, T. 2002. Gene flow and the limits to natural selection. Trends Ecol. Evol. 17:183–189. [Google Scholar]
- Lockwood, J. L. , Cassey P., and Blackburn T.. 2005. The role of propagule pressure in explaining species invasions. Trends Ecol. Evol. 20:223–228. [DOI] [PubMed] [Google Scholar]
- Mallet, J. , Korman A., Heckel D. G., and King P.. 1993. Biochemical genetics of Heliothis and Helicoverpa (Lepidoptera: Noctuidae) and evidence for a founder event in Helicoverpa zea . Ann. Entomol. Soc. Am. 86:189–197. [Google Scholar]
- Monson, R. K. , Edwards G. E., and Ku M. S. B.. 1984. C3‐C4 intermediate photosynthesis in plants. Bioscience 34:563–574. [Google Scholar]
- Morey, A. C. , Hutchison W. D., and Venette R. C.. 2012. Cold hardiness of Helicoverpa zea (Lepidoptera : Noctuidae) Pupae. Environmental Entomolog 41:172–179. [DOI] [PubMed] [Google Scholar]
- Morin, P. A. , Leduc R. G., Archer F. I., Martien K. K., Huebinger R., Bickham J. W., and Taylor B. L.. 2009. Significant deviations from Hardy‐Weinberg equilibrium caused by low levels of microsatellite genotyping errors. Mol. Ecol. Resour. 9:498–504. [DOI] [PubMed] [Google Scholar]
- Murúa, M. G. , Scalora F. S., Navarro F. R., Cazado L. E., Casmuz A., Villagrán M. E., et al. 2014. First record of Helicoverpa armigera (Lepidoptera: Noctuidae) in Argentina. Florida Entomologist 97:854–856. [Google Scholar]
- Musser, F. R. , Catchot A. L., Davis J. A., Herbert D. A., Lorenz G. M., and Reed T.. 2014. 2013 Soybean Insect Losses in the Southern US. Midsouth Entomologist 7:15–28. [Google Scholar]
- Nibouche, S. , Buès R., Toubon J.‐F., and Poitout S.. 1998. Allozyme polymorphism in the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae): comparison of African and European populations. Heredity 80:438–445. [Google Scholar]
- van Oosterhout, C. , Hutchinson W. F., Wills D. P. M., and Shipley P.. 2004. MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4:535–538. [Google Scholar]
- Pair, S. D. , Raulston J. R., Rummel D. R., Westbrook J. K., Wolf W. W., Sparks A. N., and Schuster M.F.. 1987. Development and production of corn earworm and fall armyworm in the Texas high plains: evidence for reverse fall migration. Southwestern Entomol. 12:89–99. [Google Scholar]
- Paradis, E. 2010. pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26:419–420. [DOI] [PubMed] [Google Scholar]
- Perera, O. P. , and Blanco C. A.. 2011. Microsatellite variation in Helicoverpa zea (Boddie) populations in the Southern United States. Southwestern Entomol. 36:271–286. [Google Scholar]
- Perera, O. P. , Blanco C. A., Scheffler B. E., and Abel C. A.. 2007. Characteristics of 13 polymorphic microsatellite markers in the corn earworm, Helicoverpa zea (Lepidoptera: Noctuidae). Mol. Ecol. Notes 7:1132–1134. [Google Scholar]
- Pritchard, J. K. , Stephens M., and Donnelly P.. 2000. Inference of population structure using multilocus genotype data. Genetics 155:945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaintance, A. L. , and Brues C. T.. 1905. The cotton bollworm. US Department of Agriculture, Bureau of Entomology, Bulletin No 50. [Google Scholar]
- R Development Core Team 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: URL: https://www.r-project.org. [Google Scholar]
- Rice, W. R. 1989. Analyzing tables of statistical tests. Evolution 43:223–225. [DOI] [PubMed] [Google Scholar]
- Scott, K. D. , Wilkinson K. S., Merritt M. A., Scott L. J., Lange C. L., and Schutze M. K.. 2003. Genetic shifts in Helicoverpa armigera Hübner (Lepidoptera: Noctuidae) over a year in the Dawson/Callide Valleys. Crop Pasture Sci. 54:739–744. [Google Scholar]
- Scott, K. D. , Lawrence N., Lange C. L., Scott L. J., Wilkinson K. S., and Merritt M. A.. 2005. Assessing moth migration and population structuring in Helicoverpa armigera (Lepidoptera: Noctuidae) at the regional scale: example from the Darling Downs, Australia. J. Econ. Entomol. 98:2210–2219. [DOI] [PubMed] [Google Scholar]
- Sell, D. K. , Whitt G. S., Metcalf R. L., and Lee L. K.. 1974. Enzyme polymorphism in the corn earworm, Heliothis zea (Lepidoptera: Noctuidae). Hemolymph esterase polymorphism. Can. Entomol. 106:701–709. [Google Scholar]
- Showers, W. B. , Keaster A. J., Raulston J. R., Hendrix W. H., Derrick M. E., McCorcle M. D., et al. 1993. Mechanism of southward migration of a noctuid moth [Agrotis ipsilon (Hufnagel)]: a complete migrant. Ecology 74:2303–2314. [Google Scholar]
- Slatkin, M. 1987. Gene flow and the geographic structure of natural populations. Science 236:787–792. [DOI] [PubMed] [Google Scholar]
- Sluss, T. P. , Sluss E. S., Graham H. M., and Dubois M.. 1978. Allozyme differences between Heliothis virescens and H. zea . Ann. Entomol. Soc. Am. 71:191–195. [Google Scholar]
- Tay, W. T. , Soria M. F., Walsh T., Thomazoni D., Silvie P., Behere G. T., et al. 2013. A brave new world for an old world pest: Helicoverpa armigera (Lepidoptera: Noctuidae) in Brazil. PLoS ONE 8:e80134. doi: 10.1371/journal.pone.0080134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven, K. J. F. , Macel M., Wolfe L. M., and Biere A.. 2010. Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc. Biol. Sci. 278:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook, J. K. 2008. Noctuid migration in Texas within the nocturnal aeroecological boundary layer. Integr. Comp. Biol. 48:99–106. [DOI] [PubMed] [Google Scholar]
- Westbrook, J. K. , and López J. D.. 2010. Long‐distance migration in Helicoverpa zea: what we know and need to know. Southwestern Entomol. 35:355–360. [Google Scholar]
- Westbrook, J. K. , Nagoshi R. N., Meagher R. L., Fleischer S. J., and Jairam S.. 2016. Modeling seasonal migration of fall armyworm moths. Int. J. Biometeorol. 60:255–267. [DOI] [PubMed] [Google Scholar]
- Williams, M (2014) Cotton Insect Losses‐2013 In: 2014 Beltwide Cotton Conferences (January 6–8, 2014). NCC. [Google Scholar]
- Yang, Y. , Li Y., and Wu Y.. 2013. Current status of insecticide resistance in Helicoverpa armigera after 15 years of Bt cotton planting in China. J. Econ. Entomol. 106:375–381. [DOI] [PubMed] [Google Scholar]
- Zhou, X. , Faktor O., Applebaum S. W., and Coll M.. 2000. Population structure of the pestiferous moth Helicoverpa armigera in the Eastern Mediterranean using RAPD analysis. Heredity 85:251–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Simulation saturation for the allelic richness.
Figure S2. DAPC results using no prior assignment of putative population showing the first two axes of the analysis (depicted in the insert plot).
Figure S3. Outcome from STRUCTURE analysis assuming K = 7. Each vertical bar represents a unique individual (x‐axis) with their corresponding assignment score (y‐axis) for inclusion in a particular potential population (color).
Table S1. Pairwise F ST estimates (upper triangle) for putative population pairs.
Table S2. Summary of population level genetic parameters for the non‐prior DAPC analysis; the non‐prior populations cluster (Pop), number of individuals genotyped (N), mean number of alleles/locus (A), observed heterozygosity (Hobs), expected heterozygosity (Hexp), F ST and F IS with its 95% confidence interval (F ISCI).
Table S3. The results of AMOVA of Helicoverpa zea populations grouped by collection time to calculate ΦST (between populations when there was only one group), ΦCT (among groups), or ΦSC (among populations within groups).
Data S1. Microsatellite genotypes and collection data for Helicoverpa zea adults collected from Landisville and Rock Springs, PA. Size of each microsatellite allele (in Bp) in an individual is given in two consecutive sets of 3‐digits in the 6‐digit genotype.
Data Availability Statement
Microsatellite genotype data and pertinent metadata used in this manuscript have been included in the supplementary Data file S01. Accession numbers of previously published microsatellite loci used in this analysis are listed in Table 1.