

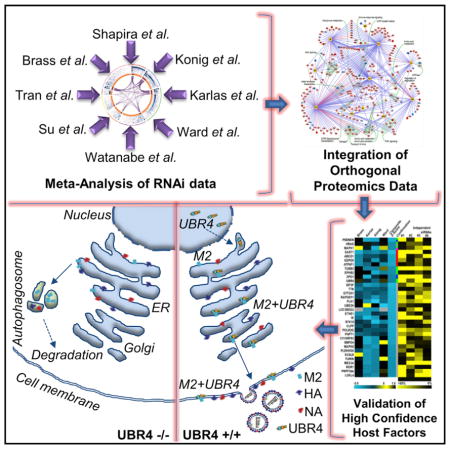
SUMMARY
Several systems-level datasets designed to dissect host-pathogen interactions during influenza A infection have been reported. However, apparent discordance among these data has hampered their full utility toward advancing mechanistic and therapeutic knowledge. To collectively reconcile these datasets, we performed a meta-analysis of data from eight published RNAi screens and integrated these data with three protein interaction datasets, including one generated within the context of this study. Further integration of these data with global virus-host interaction analyses revealed a functionally validated biochemical landscape of the influenza-host interface, which can be queried through a simplified and customizable web portal (http://www.metascape.org/IAV). Follow-up studies revealed that the putative ubiquitin ligase UBR4 associates with the viral M2 protein and promotes apical transport of viral proteins. Taken together, the integrative analysis of influenza OMICs datasets illuminates a viral-host network of high-confidence human proteins that are essential for influenza A virus replication.
Graphical Abstract
INTRODUCTION
Influenza A virus (IAV) continues to cause significant morbidity, mortality, and economical losses in epidemics and pandemics. The emergence of M2 and NA inhibitor-resistant viral mutants is cause for significant concern regarding the future efficacy of these antivirals for front-line therapy (Nicoll et al., 2008). In addition, limitations in vaccine efficacy in the older population, as well as the inadequate production of vaccines in response to global pandemics, further underscore the urgent need for novel antiviral therapies. Targeting host proteins for antiviral efficacy represent an attractive option for the development of antivirals. Host proteins constitute an expanded repertoire of therapeutically tractable antiviral targets and are immutable, which reduces the likelihood of developing drug resistance. However, understanding of the complex molecular interactions between the virus and the host is crucial to get critical insights toward their impact on viral pathogenesis.
Systems-level technologies, such as genome-scale RNAi screening and global affinity purification-mass spectrometry (APMS) approaches, have afforded unprecedented molecular insights into host-pathogen interactions (König and Stertz, 2015). Overlap of specific host proteins identified by various studies has been low (König and Stertz, 2015), and this lack of congruity has significantly hampered the leveraging of these important datasets to gain further mechanistic understanding of the role of specific host proteins in the viral replicative cycle and the development of host-directed therapeutic strategies. Reasons for the observed lack of direct correspondence in these global datasets likely include false positives and false negatives that are endemic to all systems-level approaches, including off-target activities and lack of silencing efficiency (RNAi) or non-specific binding and limit of detection issues (APMS).
Since it is difficult to assess the predictive value of each individual experimental system employed in these studies, we reasoned that host proteins that possess experimental support in multiple and/or orthogonal studies are more likely to act as bona fide regulators of in vivo replication. Therefore, we performed a meta-analysis of RNAi data from eight published studies. To mitigate the potential impact of individual analyses approaches biasing the interpretation of the data, we obtained the raw (previously unpublished) RNAi screening data from four of the published screens. Next, we integrated these data with three protein interaction datasets, two of which have been previously reported and one generated within the context of this study. The assimilation of these global genetic and proteomic studies resulted in the construction of a high-confidence network map that reflects the biochemical landscape of essential influenza-host interactions.
Among the host factors identified using this approach, we focused on the N-recognin E3 ligase family member protein UBR4 as a host factor required by IAV and interacting partner of M2. UBR4 belongs to the family of UBR-box containing N-recognin, which targets proteins for ubiquitination and proteasomal degradation, and has been shown to impact cell survival, membrane morphogenesis, and autophagy (Parsons et al., 2015). Among viral proteins, it interacts with human papillomavirus (HPV) E7 protein for cellular transformation and dengue virus (DENV) NS5 protein for STAT2 degradation (Morrison et al., 2013; White et al., 2012). Here we report that UBR4 is co-opted by IAV for efficient targeting of M2 to the cell membrane, a process that is essential for virus budding.
RESULTS AND DISCUSSION
Meta-analysis of RNAi Screens
To comprehensively survey the repertoire of host cellular factors affecting the replication of IAV, we analyzed the hit lists of eight independent RNAi datasets published previously (Brass et al., 2009; Karlas et al., 2010; König et al., 2010; Shapira et al., 2009; Su et al., 2013; Tran et al., 2013; Ward et al., 2012; Watanabe et al., 2014). A total of 1,257 genes was reported as confirmed host dependency factors and 192 genes as confirmed antiviral factors in at least one of the above-mentioned reports (Figures 1A or 1D, respectively, orange and red innermost circle segments). A pair-wise comparison to identify genes in common between screening sets resulted in only a modest overlap: 101 pro-viral and 2 anti-viral genes confirmed by multiple studies (red circle segments and purple inter-gene links in Figures 1A and 1D, respectively). To mitigate the influence of false-negative hit calls due to variances in data analysis methodologies and hit selection, we considered an expanded version of the respective hit lists based on the raw data of previously unpublished activity scores of four genome-wide RNAi screens (Brass et al., 2009; Karlas et al., 2010; König et al., 2010; Ward et al., 2012). We applied a statistical analysis to each screen set, termed the RSA algorithm, that utilizes an iterative accumulative hypergeometric distribution formula (König et al., 2007), to obtain a Z score for each gene in each individual screen (Figure 1, blue circles). The same principle was applied to obtain a consolidated Z score in order to prioritize genes based on the collective activities across all four genome-wide screening datasets (referred to as Z-RSA), both for pro-viral and anti-viral genes (Tables S1 and S2). The combined Z-RSA analysis outperforms the results of individual screens as exemplified by the recovery of 101 host factors confirmed in two or more screens (considered “gold standards”) in a receiver operating characteristic analysis (Figure S1A; Table S1, second tab). The addition of Z-RSA supported genes to the confirmed hit lists increased the observed overlap between reported screens (Figures 1B and 1D, respectively, black inter-gene links). For example, the activities of 52 host factors and 64 restriction factors, which were not confirmed in any report, are highly supported by at least two screens as significant Z-RSA hits (Figures 1B and 1D, respectively, green connectors; consolidated Z-RSA ≤ −2 and individual screen Z scores in at least 2 screens Z ≤ −2). Importantly, delineation of cellular factors that participate in similar cellular processes/pathways or represent members of a common biochemical complex further resolves the discrepancies in overlap between the datasets (Figure 1C, inter-gene connectors; Figures S1B–S1E). In fact, 613 host genes among the 1,257 confirmed pro-viral genes are encompassed in at least one of above-mentioned categories (Figure S1F), resulting in a significantly extended and cross-substantiated set of host cellular factors.
Figure 1. Meta-analysis of Systems-Level Influenza A Datasets.

(A–D) Circos visualization (Krzywinski et al., 2009) of pro-viral (A, B, and C) or anti-viral (D) cellular factors supported by reported RNAi data. In addition to depicting the overlap of cellular genes confirmed by multiple studies (A), the visualization was extended to include host genes that were supported by raw data in primary screen sets (Z score of ≤ −2; B and C). Host factors that participate in common pathways or biochemical complex/networks are shown in (C): inter-screen inter-gene connectors display proteins from screens predicted to interact with each other based on protein-protein interaction databases (PPI) and genes sharing the same statistically enriched gene ontology (GO) functional groups. (D) Gene overlaps for antiviral (restriction) cellular proteins based on both reported confirmed genes and raw data (Z score ≤ −2). Each wedge of the Circos plots depicts data from one of eight screens (pro-viral, A) or six screens (anti-viral, D), respectively, denoted by the outermost colored line. The length of each circle segment corresponds to the number of confirmed or significant (Z-score ≤ −2) factors found in each screen. The innermost circle categorizes the cellular factors into the respective gene status: (1) gene was confirmed in the indicated screen and at least one additional screen (red); (2) gene reported confirmed in only the indicated screen (orange), (3) gene was not reported confirmed in any screen, but displays a high activity (Z score ≤ −2) in the raw datasets of the indicated screen (transparent white). The four blue circles display the calculated Z scores of each host factor (A, B, and C) or restriction factors (D) within four primary raw screen datasets, respectively (from outside to inside: Brass et al., 2009; Karlas et al., 2010; König et al., 2007; Ward et al., 2012). Intensity from white to blue indicates increasing significance of activity (lower Z score). Connecting lines denote the overlap of genes shared either by multiple screens (directly in A, B, and D or through networks/pathways in C). The color of the line indicates the category of the inter-screen gene links: (1) both genes are confirmed (purple), (2) one gene is confirmed and the other displays a high Z score of ≤ −2 (black), (3) both genes display high Z scores of ≤ −2 in their source screens (green).
See also Figure S1 and Tables S1 and S2.
Validation of Host Proteins
Thirty-four randomly selected putative IAV-relevant host factors with significant consolidated Z scores (Z-RSA) were experimentally validated in a lung epithelial cell line. Thirty of 34 predicted factors proved to reduce growth of A/WSN/33 by >75% upon knockdown by a minimum of two host factor-specific siRNAs (Figure 2A; Table S3, first tab), including 21 genes that were not previously called confirmed by any RNAi screen. Importantly, we were able to validate the activities of 14 host factors mutually called in at least two screens as RSA hits. Furthermore, we were able to corroborate 20 factors in primary lung fibroblast cells (WI38) (Figure 2B; Table S3, second tab), indicating that host genes identified by the meta-analysis are highly likely to play critical roles during the viral life cycle.
Figure 2. Validation of Host Protein Activities Called in Multiple Screens.

(A) Left: individual and composite Z scores derived from raw screening data of indicated screens. Middle: confirmation status of indicated genes across published screens. Right: 48 hr following siRNA-mediated gene knockdown A549 cells were infected with A/WSN/33 (MOI = 0.01) for 24 hr and supernatants were titered. Shown are virus titers (PFU) in percentage relative to siScr.
(B) At 48 hr following siRNA-mediated gene knockdown, WI38-GFP1–10 cells were infected with A/WSN/33:PB2-GFP11 (MOI = 0.1), and GFP counts were measured every 4 hr over a 44-hr period. Shown is the mean AUC of two experiments (triplicates).
(C) Raw data from published screens were analyzed for IAV restriction factors. Individual and composite Z scores from raw data of published screens are depicted (left). siRNAs targeting ISGs with significant composite Z scores were transfected into A549 cells. Cells (+/− INF) were challenged for 24 hr with A/Vietnam/1203/2004 HALo virus (MOI = 0.5). Mean fluorescence values from triplicate experiments are depicted as a fraction of replication in INF-treated versus INF-untreated cells.
Genetic and Chemical Perturbation of HRAS/MAPK Impedes IAV Entry
HRAS, a small GTPase of the RAS superfamily, and downstream-acting MAPK1 and MPK8 have been identified in multiple screens and were validated as host factors important to IAV replication (Figure 2A). HRAS was previously reported to mediate IAV VLP entry (Zona et al., 2013) and to be activated soon after infection with IAV (Fujioka et al., 2013). Knockdown of these factors significantly reduced IAV multi-cycle growth and was found to impact NP expression in the nuclei of infected cells (Figures 2A, S2A, and S2B). Consistent with this observation, we find that silencing of the candidates substantially reduced entry of IAV virus-like particles (Figure S2C). The requirement of HRAS, MAPK1, and MAPK8 expression for IAV entry was further substantiated by testing two inhibitors of this pathway: Lonafarnib and AS601245, which inhibit the activation of RAS proteins and c-Jun N-terminal kinases, respectively. Both compounds potently inhibited IAV multi-cycle growth and nuclear NP expression in a dose-dependent manner (Figures S2D and S2E). Thus, these data indicate that IAV entry is dependent on HRAS-mediated signaling cascades via MAP kinases and suggest the repurposing of Lonafarnib as a host-directed influenza antiviral.
Host Restriction of IAV Replication
To further explore host proteins that inhibit IAV replication, we re-analyzed raw data from genome-wide screens to enrich for genes, which, when inhibited, resulted in an increase in IAV replication (Tables S2 and S5). We focused on genes that are regulated by type I interferons (IFNs), since these IFN-stimulated genes (ISGs) are known to have potent antiviral activities. The effect of knockdown of eight ISGs on A/Vietnam/1203/2004 HALo reporter virus growth was assessed in the presence or absence of IFN. We found that targeting eight ISGs with each of two or more independent siRNAs significantly increased the number of infected cells compared with the control (siScr; p < 0.05; Figure 2C; Table S3, third tab).
Of the eight validated ISGs, two host proteins, IFITM3 and BST2, have been previously reported to block infection by IAV (García-Sastre, 2011). In addition, ANGPTL4, which has been implicated in IAV infection (Li et al., 2015), was also among the validated antiviral factors. Furthermore, we identified CCL3L3, a cytokine that binds to several chemokine receptors, including CCR5, and acts as a chemotactic for lymphocytes and monocytes. Interestingly, CCR5 deficiency is associated with severe influenza (Keynan et al., 2010). Another identified antiviral protein, NAT1, is predicted to associate with a densely connected predicted protein complex containing BST2 and RPL27 (Figure S3D). Further investigation of the mechanistic bases for the antiviral activities of these genes is likely to provide critical insight into host defense strategies and determinants of IAV pathogenesis.
Mapping Physical Host-Protein Interactions
To integrate biochemical evidence with functional data provided by the meta-analysis, we assimilated three different proteomics datasets. Two previously published datasets encompass cellular factors interacting with IAV proteins identified by mass spectrometry or yeast-two-hybrid approaches (Shapira et al., 2009; Watanabe et al., 2014). In addition, we performed our own systematic affinity-tag purification approach of each IAV protein followed by mass-spectrometry on proximal interacting host proteins. The subsequent mass-spectrometry analysis refined by CompPASS and MIST algorithm (Verschueren et al., 2015; Wenger et al., 2011) resulted in 849 co-precipitated host proteins forming 925 interaction pairs (Tables S4 and S5). APMS results were validated by co-immunoprecipitation analysis of a subset of interactions (Figure S4A). We combined the three independent host-viral protein interaction datasets and then incorporated databases of human protein-protein interactions. By overlaying high-confidence functionally validated host components derived from the meta-analysis of genetic screens (Z-RSA scores ≤ −2 and/or confirmed by at least one of the RNAi screens [pro-viral or anti-viral]), we were able to visualize the topography of the resulting functionally validated influenza-host biochemical landscape. A simplified version of this highly interconnected network is displayed in Figure 3 (see also Table S6). MCODE analysis revealed several subnetworks with high local network connectivity (Figure S3; Table S6). Importantly, cellular processes known to directly support IAV replication, such as vATPase activity and PI3K signaling, were identified as highly overrepresented in this analysis (Ehrhardt and Ludwig, 2009; König et al., 2010; Stertz and Shaw, 2011). Additional cellular processes are significantly enriched within these MCODE clusters (Table S7), including COPI vesicle transport (Figure S3A), splicing (Figure S3B) (Dubois et al., 2014; Sun et al., 2013), or the HRAS-MAPK-mediated signaling cascade (Figure S3C), further corroborating the importance of the latter pathway in IAV replication. Interestingly, the COP9 signalosome complex, which regulates cullin-RING-E3 ubiquitin ligase activity by deneddylation, was also identified as a critical component of the viral-host interface (Kato and Yoneda-Kato, 2009) (Figure 3). Indeed, knockdown of individual subunits of the COP9 complex by RNAi hampered IAV growth in A549 cells (Figure S4B). Unanticipated functional activities associated with IAV-encoded proteins include a possible function of M2 and NA in COPI-mediated vesicle transport, and a putative role of M1 and M2 in deneddylation processes (Table S8). Further studies are required to understand the role of these viral-host interactions in IAV replication and pathogenesis.
Figure 3. The Functionally Validated Landscape of IAV-Host Protein Interactions.

An interaction network (Cytoscape) between host and influenza proteins was generated. Three IAV interactomes were integrated: (1) Yeast-two-hybrid data from Shapira et al. (2009), (2) APMS data confirmed by RNAi from Watanabe et al. (2014), (3) APMS data generated in this study with a MIST score cutoff of ≥0.7 or a top 5% ComPASS score (see Experimental Procedures). Viral proteins are depicted as yellow nodes. Displayed host nodes constitute proteins that were reported confirmed as host dependency or restriction factors in one (light red, light blue) or two or more RNAi screens (dark red, dark blue) and interact with no more than three IAV proteins. Blue nodes reflect host proteins additionally identified through the analysis of raw datasets (Z-RSA ≤ −2). Protein-protein interactions that were reported by a single proteomics dataset or by both Watanabe et al. (2014) and this publication are highlighted as blue or red edges, respectively. Selected complexes and overrepresented biological processes are displayed as colored clouds, and the enriched functions are denoted. Human-human based interactions are only depicted inside the colored clouds. The resulting network contained 398 virus-host edges, connecting 264 confirmed host cellular factors and 11 IAV proteins.
See also Figures S3 and S4 and Tables S4, S5, S6, S7, and S8.
UBR4 Interacts with the M2 Ion Channel and Is Required for IAV Replication
M2 is an ion channel protein that plays crucial roles during IAV entry and exit (Edinger et al., 2014; Rossman and Lamb, 2011). UBR4 was identified as a M2 interactor in our APMS analysis, as well as by Kawaoka and colleagues (Watanabe et al., 2014). Additionally, it was also found to be required for IAV replication in multiple siRNA screens, including this study (Figure 2A). Therefore, we investigated the contribution of UBR4 in IAV replication. The interaction of UBR4 and M2 was confirmed in IAV-infected A549 cells by immunoprecipitation (Figure 4A). To map the domain required for interaction with UBR4, we made a series of N-terminal GST-tagged deletions in M2 and performed immunoprecipitations; only the ectodomain was dispensable for UBR4 binding (Figure 4B), indicating that the transmembrane domain and C-terminal tail contribute to UBR4 binding. Interestingly, these regions of M2 have been implicated in IAV assembly and budding (Rossman et al., 2010), suggesting a role for M2-UBR4 interaction in late events of IAV replication. We then studied cellular localization of M2 and UBR4 during IAV infection. We observed that during early stages of IAV infection M2 and UBR4 co-localized in the perinuclear ER region and that their localization coordinately progressed to the cell membrane during late stages of infection (Figure 4C). This suggests that M2-UBR4 interaction initiates in the endoplasmic reticulum (ER) and that UBR4 may play a role in translocation of M2 to the cell surface. Interestingly, UBR4 displayed primarily nuclear localization in uninfected cells, but upon IAV infection, it translocated out of the nucleus to the ER region. The trigger that governs UBR4 translocation out of the nucleus is not clear; however, transfection of M2 alone does not induce UBR4 movement (data not shown), suggesting it to be an IAV infection-associated event.
Figure 4. UBR4 Interacts with the M2 Ion Channel and Is Required for IAV Replication.

(A) A549 cells were infected with A/WSN/33 (MOI = 2) for 24 hr and lysed in IP buffer. Lysates were subjected to immunoprecipitation using antibodies against M2 and UBR4. Immunoprecipitated protein samples and 5% input were subjected to SDS-PAGE/western blotting using indicated antibodies.
(B) N-terminal GST-tagged deletion constructs of A/WSN/33 M2 (top) were transfected in HEK293T cells. At 48 hr later, cells were lysed and subjected to immunoprecipitation with anti-GST antibody. Immunoprecipitated samples and 10% input were subjected to SDS-PAGE/western blotting (bottom).
(C) A549 cells were infected with A/WSN/33 (MOI = 2), and cells were fixed at indicated time points for immunostaining. Nuclei are depicted in blue, M2 in green, and UBR4 in red. Arrows allocate M2-UBR4 co-localization. Images of three representative independent experiments are shown. Scale bar represents 10 μm.
(D) A549 cells were transfected with siNP, siUBR4, or siScr; 48 hr later, cell viability and UBR4 expression levels were determined. Alternatively, cells were infected with A/WSN/33 (MOI = 0.01) for 24 hr, and supernatants were titered. Shown is one of two independent duplicate experiments ± SD.
(E) UBR4 stable knockdown or control A549 cells were infected with IAV luciferase (MOI = 0.2), Dengue Luciferase, or Herpes 1 luciferase reporter virus (MOI = 0.1); 48 hr later cells were lysed, and luciferase activity was measured. The mean luminescence values ± SD of three independent experiments relative to control were plotted. * and # indicate p values compared with control and Scr shRNA, respectively.
See also Figures S4 and S5.
Next, we tested the impact of UBR4 depletion on IAV growth. Knockdown of UBR4 in A549 and WI38 cells reduced the amount of released infectious virus in the supernatant by 10- to 100-fold (Figures 4D and S4C) while cell viability was unaltered. Also, A549 cells stably expressing an UBR4-targeting shRNA were shown to display reduced UBR4 protein expression and IAV replication (Figures S5A and S5B). UBR4 is also known to be essential for dengue virus (DENV) replication (Morrison et al., 2013). To test the specificity of UBR4 requirement across different viruses, we compared the effect of UBR4 knockdown on replication of IAV, DENV, and HSV-1 luciferase reporter viruses: IAV and DENV were susceptible to UBR4 knockdown, while HSV-1 replication was unaffected (Figure 4E). This suggests that the inhibitory effect of UBR4 knockdown is virus specific and not a general defect. We next tested whether M2 ion channel activity is linked to the requirement of UBR4 for IAV replication. Treatment with amantadine, an M2 ion channel inhibitor, did not affect the susceptibility of A/Udorn/72 to UBR4 depletion (Figure S4D), indicating that the ion channel activity of M2 is not directly related to IAV dependence on UBR4 for efficient replication.
UBR4 Facilitates M2 Translocation to the Cell Membrane during Late Stages of IAV Infection
We then assessed the step of the viral life cycle in which UBR4 is required by IAV: UBR4 knockdown did not affect NP levels in the nuclei of infected cells after 3 hr of infection (Figure S4E). Even after 18 hr of infection, NP expression was similar in siUBR4 and siScr transfected cells, while virus titers in the cell supernatant from the same experiment were significantly reduced upon UBR4 knockdown (Figure S4F). These data suggest that UBR4 is not required during IAV entry, genome transcription, and early replication. However, virus release in UBR4 stable knockdown cells was reduced significantly after one cycle of replication (Figure 5A), confirming our postulate of its role in late events of IAV infection.
Figure 5. UBR4 Facilitates M2 Translocation to the Cell Membrane.

(A) UBR4 stable knockdown or control A549 cells were infected with A/WSN/33 (MOI = 2). Supernatants were titered at 12- and 18-hr post-infection.
(B) UBR4 stable knockdown or control A549 cells were infected with A/WSN/33 (MOI = 2) for 24 hr and subjected to immunostaining. Nuclei are depicted in blue and M2 in green. Arrows allocate M2 localization. Scale bar represents 10 μm. Shown are representative images of three independent experiments.
(C) UBR4 stable knockdown, scrambled, and control A549 cells were infected with A/WSN/33 (MOI = 2) for 24 hr. Cells were harvested, and M2 expressed on the non-permeabilized cell surface was measured by flow cytometry. Top shows the percentage of cells positive for M2 surface expression relative to control. Lower shows corresponding histograms.
(D) UBR4 WT or UBR4 KO HEK293T cells were infected with A/WSN/33 (MOI = 2) for 20 hr. Cells were harvested and M2 surface expression in non-permeabilized cells, and total M2 expression in permeabilized cells was measured by flow cytometry. Top shows the percentage of cells positive for M2 surface expression relative to control. Lower shows the corresponding M2 geometric mean intensity.
(E) UBR4 WT or UBR4 KO HEK293T cells were infected with A/WSN/33 (MOI = 2). At 20-hr post-infection, supernatants were titered (top). Corresponding M2, UBR4, and β actin levels were determined by western blot.
(F) UBR4 WT or UBR4 KO HEK293T cells were transfected with the autophagosome marker plasmid DIRAS3-N-RFP (red); 24 hr later, cells were infected with A/WSN/33 (MOI = 2). After 16 hr, cells were subjected to immunostaining. Nuclei are depicted in blue and M2 in green. M2-DIRAS3 co-localization is marked by arrows, and scale bar represents 10 μm. Images of three representative independent experiments are shown. Graphs represent mean ± SD of three independent experiments in (A) and (C); * and # indicate p values compared with control and Scr shRNA, respectively. In (D) and (E), # indicates p value compared to UBR4 WT samples. Immunofluorescence images are representative of three independent experiments, and scale bar represents 10 μm.
See also Figure S5.
Next, we tested the impact of UBR4 knockdown on M2 apical membrane targeting. In UBR4-depleted cells, M2 localization on cell surface was greatly reduced (Figure 5B). Consistently, quantification of M2 surface expression in non-permeabilized cells revealed a significant reduction of M2 cell membrane expression upon UBR4 knockdown (Figure 5C). To further confirm this phenotype, we generated HEK293T cells with CRISPR-Cas9-mediated UBR4 knockout (KO). These cells were characterized for absence of UBR4 expression and reduced IAV replication (Figure S5B). We also observed that in UBR4 KO cells, M2 cells surface expression was greatly reduced (Figure 5D). Interestingly, total M2 expression was also reduced in UBR4 KO cells (Figures 5D and 5E). Additionally, virus release was reduced by 10-fold after a single replication round in these cells (Figure 5E). To test whether UBR4 KO affects other viral and cellular proteins, we measured cell surface expression of the viral glycoproteins hemagglutinin (HA) and neuraminidase (NA) and the cellular proteins transferrin receptor (TFR) and LC3 in UBR4-depleted cells. Surface expression of HA and NA, but not TFR, was reduced in UBR4 KO cells (Figure S5C), suggesting that UBR4 requirement is specific for viral proteins. M2 is known to interact with LC3 and to induce its re-localization to the cell membrane (Beale et al., 2014). UBR4 knockdown did not affect virus-induced LC3 re-localization or overall LC3 expression (Figure S5D). Taken together, these data demonstrate that UBR4 is required for apical targeting of viral, but not cellular, proteins.
IAV M2 Is Targeted to Autophagosomes for Degradation in Absence of UBR4
Both M2 and UBR4 are known regulators of autophagy: M2 is known to interact with LC3 and inhibit autophagosome fusion with lysosomes (Beale et al., 2014; Gannagé et al., 2009), whereas UBR4 is required for efficient execution of autophagy (Tasaki et al., 2013). Although overall autophagy was not changed in the absence of UBR4, co-localization of M2 with the autophagosomal marker DIRAS3 was enhanced in KO cells compared with WT cells (Figure 5F). As overall M2 levels are decreased in UBR4 KO cells (Figures 5D and 5E), we hypothesized that in absence of UBR4 IAV M2 is diverted from ER to autophagosomes for degradation. The cellular endoplasmic reticulum-associated degradation (ERAD) machinery is known to target membrane proteins from ER for degradation and is frequently exploited by different viruses (Morito and Nagata, 2015). Interestingly, DBeQ, an inhibitor of the ERAD regulatory protein p97 (Dugre et al., 1990), significantly rescued both M2 total and cell surface expression (Figure S5E). In addition, siRNA-mediated p97 knockdown partially rescued M2 surface expression, although not as effectively as DBeQ, which may be due to incomplete inhibition of p97 and siRNA toxicity (Figure S5F). In contrast, the autophagy inhibitor Bafilomycin A (Baf A) could partially rescue M2 expression, but had no impact on M2 surface levels (Figure S5E). Because of cytotoxic effects, we could not assess the impact of DBeQ on viral replication (data not shown). Thus, these data suggest that, in the absence of UBR4, M2 is likely diverted to autophagosomes for degradation, potentially through involvement of the ERAD machinery. As a result, M2 surface expression and overall virus release are reduced.
UBR4 Knockdown Mitigates IAV Replication and Pathogenesis In Vivo
As UBR4 KO is lethal in mice (Tasaki et al., 2013), we used peptide-conjugated phosphorodiamidate morpholino oligomers (PPMOs) to understand the impact of UBR4 depletion in mouse lungs on IAV replication in vivo. We first evaluated the efficacy of two PPMOs designed against UBR4 (Figure S5G) and chose PPMO-2 for further studies. Scr or UBR4-targeting PPMO were administered intranasally to mice for 2 days (Figure 6A). On the following day, mice were infected with 250 PFU of A/Puerto Rico/8/34. Lung tissue was harvested on day 0 (before infection), day 3, and day 6 post-infection, and UBR4 expression, virus titers, and lung histopathology were assessed. Transient knockdown of UBR4 expression in mouse lungs was observed after 2 days of PPMO-2 treatment (Figure S5H). Following infection, the UBR4 PPMO-treated group of mice lost less weight (Figure 6B) and showed prolonged survival compared with the PBS or Scr PPMO-treated group (Figure 6C). Of note, UBR4 PPMO treatment by itself induced some body weight loss; however, uninfected mice recovered within 3 days following PPMO treatment (Figure S5I). Lung virus titers declined sharply upon UBR4 knockdown on day 3 and recovered partially on day 6 post-infection (Figure 6D). These data confirm the requirement of UBR4 for successful IAV replication in vivo. Furthermore, PPMO treatment itself did not induce any marked damage to the lung epithelium prior to infection (Figure 6E). UBR4 PPMO-treated mice showed less inflammation in lung tissue compared with control (Figure 6E). Notably, we found the M2 cell surface expression in lung tissue to be reduced upon UBR4 knockdown on day 3 post-infection (Figures S5J and S5K). This effect was more prominent in CD45-negative epithelial cells, as compared with CD45-positive immune effector cells (Figures S5J and S5K) and likely results from greater exposure of CD45-negative cells to UBR4 targeting PPMOs compared with CD45-positive cells, which generally infiltrate the lung tissue later during infection. Overall, these results indicate an essential role for UBR4 in efficient IAV replication in vivo, and depletion of UBR4 protects mice from IAV-induced pathogenesis.
Figure 6. UBR4 Knockdown Mitigates IAV Replication and Pathogenesis In Vivo.

Six-week-old female BALB/c mice (20 per group) were administered PBS or PPMOs (100 μg in 40 μl PBS, the equivalent of approximately 5 mg/kg) intranasally for 2 consecutive days. Five mice from each group were used to study PPMO toxicity without IAV infection (Figure S7C). On day 0, 15 mice per group were infected with A/Puerto Rico/8/34 (250 PFU) intranasally. Five mice per group were euthanized on days 3 and 6 post-infection. Lungs were harvested to determine virus titer, UBR4 expression, and histopathology. In five mice per group, survival was studied until day 14.
(A) Upper shows experimental setup.
(B) Graph shows mouse body weight ± SEM up to day 7 post-infection, for at least five mice per group.
(C) Graph shows mice survival (five per group).
(D) Graph shows mean lung virus titer ± SD on days 3 and 6 post-infection. * and # represent p value compared with PBS and Scr PPMO group, respectively.
(E) Mouse lungs were isolated on day 0 (before infection), day 3, and day 6 post-infection, and H&E staining was performed on lung sections. Representative images are shown. Areas showing extensive inflammation are marked by arrows. Scale bars represent 300 μm.
See also Figure S6.
Mammalian UBR4 Is Dispensable for Replication of Avian IAV Strains In Vitro
We next investigated the range of IAV strains that are dependent upon UBR4 for replication. We used A/WSN/33 (H1N1), A/Hong Kong/68 (H3N2), and A/Udorn/72 (H3N2) as representative human IAV strains and the avian strains A/duck/Ukraine/1/1963 (H3N8), A/duck/England/1/1956 (H11N6), and A/duck/Alberta/35/1976 (H1N1). All human IAV strains tested were sensitive to UBR4 knockdown (Figure S6A). The avian strains, however, were less dependent on the presence of UBR4. Only A/duck/Ukraine/1/1963 exhibited reduced growth upon knockdown of UBR4, but this effect was less pronounced than for the human isolates tested (Figure S6A). This could suggest that adaptations that enable the appropriation of mammalian UBR4 may be critical to zoonotic transmission and/or pathogenesis. To test this hypothesis, we generated a recombinant influenza virus in the backbone of A/WSN/33 in which we replaced M2 of WSN with M2 of A/duck/England/1/1956 and tested replication upon UBR4 depletion. The recombinant virus was still dependent on UBR4 expression even though the reduction in replication was not as pronounced as for the human viruses (Figure S6B). These results suggest that, while there is a difference between avian and human viruses in UBR4 dependence, M2 is not the sole determinant of this variance. We next tested the UBR4 dependency of an avian virus, A/duck/England/1/1956, in vivo using the PPMO model described above (Figure 6). Even though UBR4 PPMO-treated mice lost less weight compared with scrambled PPMO-treated mice (Figure S6C), this effect was statistically not significant (Figure S6D). Lung virus titers at day 3 post-infection showed a reduction in virus growth in response to UBR4 knockdown (Figure S6E). However, when comparing the relative weight loss and the relative reduction in virus growth at day 3 post-infection between the human (Figure 6) and the avian virus isolate, A/duck/England/1/1956 appeared to be less dependent on the presence of UBR4 (Figures S6D and S6F).
Summary and Conclusion
The meta-analysis presented in this study provides a comprehensive assimilation of IAV RNAi screens, including, when available, previously unpublished raw data from these assays. In addition, we have integrated data from three proteomics-based datasets, including a proteome-wide IAV interactome conducted within the context of this study. This compilation of influenza OMICs data creates a valuable resource for the community on multiple distinct levels. First, it provides an opportunity to reconcile seemingly disparate results from individual RNAi screens through the interrogation of raw data, as well as pathway- and biochemical complex-level analyses. Second, the unification of proteomic and genetic datasets provides critical spatial context to RNAi studies (Figure 3). Finally, the underlying data for these analyses provide an unprecedented resource for “wet-bench” scientists to access and action data from this comprehensive compendium of IAV genome-level datasets. Simplified analyses of these datasets enable the elucidation of host proteins found in multiple independent and/or orthogonal datasets, and thus are more likely to play bona fide roles in IAV replication and pathogenesis since this approach can help circumvent experimental variables that may account for false negatives within a single dataset. Specifically, we provide a consolidated table of host protein activities that can be differentially analyzed to elucidate likely critical regulators of IAV replication and pathogenesis based on selected levels of orthogonal support and to identify potential therapeutic targets (Tables 1, S4, and S5). We have also made the underlying data accessible through a user-friendly web portal (http://www.metascape.org/IAV) that enables the user to customize thresholds and criteria. We anticipate that these will both improve confidence in IAV systems-level studies and facilitate simplified access to IAV “big data” to a critical segment of the research community.
Table 1.
Selected Druggable, Transmembrane, and/or Secreted Host Factors with Support from Multiple and/or Orthogonal OMICs Datasets
| Gene | Gene ID | Known Function | RNAi Support
|
Proteomics Support
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brass et al. (2009) | Watanabe et al. (2014) | König et al. (2007) | Karlas et al. (2010) | Shapira et al. (2009) | Ward et al. (2012) | Su et al. (2013) | Tran et al. (2013) | Watanabe et al. (2014) | This Report | |||
| ATP6AP1 | 537 | ATPase; organelle acidification | ✔ | ✔ | ✔ | ✔ | ||||||
|
|
|
|||||||||||
| ATP6V0B | 533 | ✔ | ✔ | ✔ | ||||||||
|
|
|
|||||||||||
| ATP6V0C | 527 | ✔ | ✔ | ✔ | ||||||||
|
|
|
|||||||||||
| ATP6V1B2a | 526 | ✔ | ✔ | ✔ | ||||||||
|
| ||||||||||||
| CAD | 790 | de novo pyrimidine biosynthesis | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| FASNa | 2194 | fatty acid synthase | ✔ | ✔ | ||||||||
|
| ||||||||||||
| DNMT1a | 1786 | DNA-methyltransferase | ✔ | ✔ | ||||||||
|
| ||||||||||||
| HLA-B | 3106 | major histocompatibility complex | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| ITGA3 | 3675 | integrin; cell surface adhesion | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| LGALS3BP | 3959 | galectin; cell-cell interaction | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| FUS | 2521 | RNA binding/processing | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| EIF4A3 | 9775 | translation initiation factor | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| RPL3a | 6122 | ribosome; translation | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| PSMB2a | 5690 | proteasome | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| PSMD1a | 5707 | proteasome | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| PSMD2a | 5708 | proteasome | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| SLC16A1 | 6566 | solute carrier; monocarboxylate transporter | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| SLC1A3 | 6507 | solute carrier; glutamate transporter | ✔ | ✔ | ||||||||
|
| ||||||||||||
| SLC25A5a | 292 | solute carrier; solute carrier; ADP transporter | ✔ | ✔ | ||||||||
|
| ||||||||||||
| TTN | 7273 | cytoskeletal protein | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| TUBA4A | 7277 | cytoskeletal protein | ✔ | ✔ | ||||||||
|
| ||||||||||||
| TUBBa | 203068 | cytoskeletal protein | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| CDK1 | 983 | cyclin-dependent kinase; cell cycle | ✔ | ✔ | ||||||||
|
| ||||||||||||
| PHB | 5245 | regulation of DNA synthesis and proliferation | ✔ | ✔ | ✔ | |||||||
|
| ||||||||||||
| CD81 | 975 | tetraspanin receptor; signaling | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| PRKACA | 5566 | cAMP-dependent protein kinase; signaling | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| JAK1a | 3716 | Janus tyrosine kinase; immune response/signaling | ✔ | ✔ | ||||||||
|
| ||||||||||||
| OSMR | 9180 | type I cytokine receptor; signaling | ✔ | ✔ | ✔ | ✔ | ||||||
|
| ||||||||||||
| HRAS | 3265 | GTPase; signaling | ✔ | ✔ | ||||||||
|
| ||||||||||||
| MTORa | 2475 | S/T Kinase; target of rapamycin | ✔ | ✔ | ||||||||
Druggable, transmembrane, and secreted gene annotations derived from Hopkins and Groom (2002) and Uhlén et al. (2015).
Reported targets of Food and Drug Administration-approved molecules (Law et al., 2014).
The utility of this approach is underscored both by the extension of known IAV-host interaction biology, as well as previously unknown host mechanisms, highlighted in this report. For example, this analysis led to the identification of 20 previously unrecognized host proteins required for IAV replication in primary cells, as well as 6 previously unreported ISGs that block replication. Importantly, UBR4 was defined by this meta-analysis as a proviral factor and interacting partner of IAV M2. Our data suggest that in the absence of UBR4 IAV M2 is not targeted efficiently to the cell membrane; rather, it is likely directed to autophagosomes for degradation. We hypothesize that IAV M2 may be co-opting UBR4 to counteract a yet unidentified ER resident host restriction factor, which interferes with transport of viral proteins across the ER-Golgi network. Thus the recruitment of UBR4 by IAV M2 enables safe passage of viral glycoproteins to the cell membrane and facilitates efficient budding and replication of IAV.
Taken together, the results from this study offer important molecular understanding of a key host component that is co-opted by IAV to coordinate late stage viral replication and, critically, also provides a consolidated and cross-validated compendium of influenza OMICs data. The latter is likely to focus strategies toward the development of next-generation host-targeted antiviral therapies.
EXPERIMENTAL PROCEDURES
Affinity Purification and Mass Spectrometry
Pull-down assays with HEK293 cells individually expressing the 11 FLAG-tagged viral proteins were performed followed by mass spectrometry. APMS samples were scored using ComPASS and MiST (Jäger et al., 2012; Smoot et al., 2011; Sowa et al., 2009; Verschueren et al., 2015), as described in the Supplemental Experimental Procedures.
Bioinformatic Analysis of Screening Data
Eight independent RNAi datasets and two previously published interactome datasets and the systematic affinity-tag mass spectroscopy interactions produced by this study were used to perform the meta-analysis as described in the Supplemental Experimental Procedures.
Immunofluorescence Microscopy
Cells were fixed in 4% paraformaldehyde in PBS, permeabilized with 0.5% Tween-20 or Triton X-100 in PBS, and blocked with 2% bovine albumin in PBS. Primary and secondary antibody incubation was performed in 0.5% BSA in PBS. For mounting, Slowfade Gold antifade mounting medium with DAPI (Life Technologies) was used. Antibodies used are listed in the Supplemental Experimental Procedures.
Immunoprecipitation
For UBR4-M2 coimmunoprecipitation experiments, cells were lysed in 1% NP-40 IP Lysis Buffer (Pierce) supplemented with protease inhibitor cocktail (Pierce). Cell lysates were incubated with primary antibody overnight followed by incubation with protein A Dynabeads (Life Technologies) for 2 hr. Beads were washed three times with cold PBS. Immunoprecipitates were eluted by boiling the beads in SDS-PAGE sample buffer (BioRad). Samples were resolved on 4%–12% gradient Bis-Tris gels (Life Technologies) and transferred to polyvinylidene fluoride (PVDF) membrane (Life Technologies) by standard methods. Membranes were blocked with 3% BSA in TBS-Tween (20 mM Tris-HCl [pH 7.4], 150 mM NaCl; 1% Tween) and then incubated with antibodies and subjected to western blot. Antibodies are listed in the Supplemental Experimental Procedures.
Flow Cytometry
For in vitro fluorescence-activated cell sorting (FACS), cells were harvested by trypsinization and washed with FACS buffer (3% BSA, PBS). Where indicated, cells were stained using the LIVE/DEAD Fixable Blue Dead dye (Thermo). Where indicated, cells were permeabilised by incubating with 1% Tween-20 in PBS for 10 min. Cells were blocked with NRS (normal rabbit serum) (Abcam), followed by incubation with primary antibody (see Supplemental Experimental Procedures) and Alexa Flour tagged secondary antibody (Life Technologies). For in vivo FACS, mouse lungs were minced, treated with collagenase, and forced through a 70 μM filter to produce single-cell suspensions. Erythrocytes were removed by lysis in NH4Cl red blood cell lysis buffer. For blocking, cells were incubated with anti-mouse CD16/CD32 antibody (FcBlock, BD) and NRS. Cells samples were run on the BD LSRII Flow cytometer (BD Biosciences), and FACS data were analyzed using FLOWJO software (FLOWJO LLC).
Animal Experiments
Six- to 8-week-old female BALB/c mice purchased from Jackson Laboratories were used. Mice were anesthetized by intraperitoneal injection of a mixture of Ketamine and Xylazine (100 μg and 5 μg per gram of body weight). Mouse were inoculated intranasally with the indicated doses of PPMOs or viruses in 40 μl of PBS. Mice were monitored daily for weight loss and clinical signs. Lung homogenates were prepared using a FastPrep24 system (MP Biomedicals). After addition of 800 μl of PBS containing 0.3% BSA, lungs were subjected to two rounds of mechanical treatment for 10 s each at 6.5 m/s. Tissue debris was removed by low-speed centrifugation, and virus titers in supernatants were determined by plaque assay.
Ethics Statement
All research studies involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committees of the Icahn School of Medicine at Mount Sinai and were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals.
Supplementary Material
Highlights.
Meta-analysis of influenza OMICs datasets reveals high-confidence virus-host interactions
Integration of orthogonal data exposes unique host and restriction factor activities
Experimental validation of virus-host circuits supports robustness of approach
The host E3 ligase UBR4 is identified as essential for virus budding and pathogenesis
Acknowledgments
We thank Andrea Gamarnik and David Leib for providing luciferase reporter viruses. Microscopy and FACS experiments were performed at the Microscopy Center of Research Excellence (CORE) and Flow Cytometry Core of the Icahn School of Medicine at Mount Sinai (ISMMS). We thank Rumana Huq and Lauren O’Rourke for their help in microscopy. We thank Tom Moran for providing anti-M2 E10 and anti-HA PY102 antibodies and Patricia Nigg and Nina Hein-Fuchs for help with figure graphics. These studies were partially supported by NIAID research grant U19 AI106754 to A.G.-S., R.A.A., D.S., H.M., P.D., L.P., S.K.C., N.K., and M.S. This work was also supported by a grant from the Swiss National Science Foundation (31003A_135278) to S. Stertz. M.O.P. is the beneficiary of a doctoral grant from the AXA Research Fund. Additionally, this work was supported by the NIH P50 GM085764 (S.K.C.). A.B. is supported by a grant (1R01AI091786) from the National Institute of Allergy and Infectious Diseases of the NIH, the Burroughs Wellcome Fund, and the Bill and Melinda Gates Foundation. S.E. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and eight tables and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2015.11.002.
AUTHOR CONTRIBUTIONS
Co-first authors S.T. and M.O.P. designed and performed experiments, analyzed the data, and wrote the paper. A.R.-F., D.A.S., H.M.M., P.D., L.M., E.Y., D.A., B.M., Q.N., R.A.A., N.K., and M.S. designed and/or performed experiments. Y.Z., R.K., A.R.-F., J.C., L.P., and A.G. analyzed the data. A.B., S.E., M.W., S. Shapira, N.H., and T.M. provided unpublished data and provided conceptual framework for the study. Y.Z., R.K., S.S., A.G.-S., and S.K.C. designed experiments and wrote the paper.
References
- Beale R, Wise H, Stuart A, Ravenhill BJ, Digard P, Randow F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014;15:239–247. doi: 10.1016/j.chom.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois J, Terrier O, Rosa-Calatrava M. Influenza viruses and mRNA splicing: doing more with less. MBio. 2014;5:e00070–e14. doi: 10.1128/mBio.00070-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugre V, Levillain P, Lemonnier A. Hyperphosphatemia in case of abnormal proteinemia. Presse Med. 1990;19:338. [PubMed] [Google Scholar]
- Edinger TO, Pohl MO, Stertz S. Entry of influenza A virus: host factors and antiviral targets. J Gen Virol. 2014;95:263–277. doi: 10.1099/vir.0.059477-0. [DOI] [PubMed] [Google Scholar]
- Ehrhardt C, Ludwig S. A new player in a deadly game: influenza viruses and the PI3K/Akt signalling pathway. Cell Microbiol. 2009;11:863–871. doi: 10.1111/j.1462-5822.2009.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka Y, Tsuda M, Nanbo A, Hattori T, Sasaki J, Sasaki T, Miyazaki T, Ohba Y. A Ca(2+)-dependent signalling circuit regulates influenza A virus internalization and infection. Nat Commun. 2013;4:2763. doi: 10.1038/ncomms3763. [DOI] [PubMed] [Google Scholar]
- Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–380. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sastre A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011;162:12–18. doi: 10.1016/j.virusres.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. Global landscape of HIV-human protein complexes. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14:1209–1225. doi: 10.1111/j.1365-2443.2009.01349.x. [DOI] [PubMed] [Google Scholar]
- Keynan Y, Juno J, Meyers A, Ball TB, Kumar A, Rubinstein E, Fowke KR. Chemokine receptor 5 ρ32 allele in patients with severe pandemic (H1N1) 2009. Emerg Infect Dis. 2010;16:1621–1622. doi: 10.3201/eid1610.100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König R, Stertz S. Recent strategies and progress in identifying host factors involved in virus replication. Curr Opin Microbiol. 2015;26:79–88. doi: 10.1016/j.mib.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, Chanda SK. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods. 2007;4:847–849. doi: 10.1038/nmeth1089. [DOI] [PubMed] [Google Scholar]
- König R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, et al. Human host factors required for influenza virus replication. Nature. 2010;463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chong HC, Ng SY, Kwok KW, Teo Z, Tan EH, Choo CC, Seet JE, Choi HW, Buist ML, et al. Angiopoietin-like 4 increases pulmonary tissue leakiness and damage during influenza pneumonia. Cell Rep. 2015;10:654–663. doi: 10.1016/j.celrep.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morito D, Nagata K. Pathogenic Hijacking of ER-Associated Degradation: Is ERAD Flexible? Mol. Cell. 2015;59:335–344. doi: 10.1016/j.molcel.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, Mulder LC, Fernandez-Sesma A, García-Sastre A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013;9:e1003265. doi: 10.1371/journal.ppat.1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll A, Ciancio B, Kramarz P Influenza Project Team. Observed oseltamivir resistance in seasonal influenza viruses in Europe interpretation and potential implications. Euro Surveill. 2008;13:pii. doi: 10.2807/ese.13.05.08025-en. [DOI] [PubMed] [Google Scholar]
- Parsons K, Nakatani Y, Nguyen MD. p600/UBR4 in the central nervous system. Cell Mol Life Sci. 2015;72:1149–1160. doi: 10.1007/s00018-014-1788-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA. Influenza virus m2 ion channel protein is necessary for filamentous virion formation. J Virol. 2010;84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stertz S, Shaw ML. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect Pasteur. 2011;13:516–525. doi: 10.1016/j.micinf.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su WC, Chen YC, Tseng CH, Hsu PW, Tung KF, Jeng KS, Lai MM. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc Natl Acad Sci USA. 2013;110:17516–17521. doi: 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun E, He J, Zhuang X. Dissecting the role of COPI complexes in influenza virus infection. J Virol. 2013;87:2673–2685. doi: 10.1128/JVI.02277-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Kim ST, Zakrzewska A, Lee BE, Kang MJ, Yoo YD, Cha-Molstad HJ, Hwang J, Soung NK, Sung KS, et al. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc Natl Acad Sci USA. 2013;110:3800–3805. doi: 10.1073/pnas.1217358110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AT, Rahim MN, Ranadheera C, Kroeker A, Cortens JP, Opanubi KJ, Wilkins JA, Coombs KM. Knockdown of specific host factors protects against influenza virus-induced cell death. Cell Death Dis. 2013;4:e769. doi: 10.1038/cddis.2013.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Verschueren E, Von Dollen J, Cimermancic P, Gulbahce N, Sali A, Krogan NJ. Scoring large-scale affinity purification mass spectrometry datasets with MiST. Curr Protoc Bioinformatics. 2015;49:8.19.1–18.19.16. doi: 10.1002/0471250953.bi0819s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SE, Kim HS, Komurov K, Mendiratta S, Tsai PL, Schmolke M, Satterly N, Manicassamy B, Forst CV, Roth MG, et al. Host modulators of H1N1 cytopathogenicity. PLoS ONE. 2012;7:e39284. doi: 10.1371/journal.pone.0039284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Kawakami E, Shoemaker JE, Lopes TJ, Matsuoka Y, Tomita Y, Kozuka-Hata H, Gorai T, Kuwahara T, Takeda E, et al. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe. 2014;16:795–805. doi: 10.1016/j.chom.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11:1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EA, Sowa ME, Tan MJ, Jeudy S, Hayes SD, Santha S, Münger K, Harper JW, Howley PM. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc Natl Acad Sci USA. 2012;109:E260–E267. doi: 10.1073/pnas.1116776109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zona L, Lupberger J, Sidahmed-Adrar N, Thumann C, Harris HJ, Barnes A, Florentin J, Tawar RG, Xiao F, Turek M, et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe. 2013;13:302–313. doi: 10.1016/j.chom.2013.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.