

Abstract
Objective
Missense mutations in HIV-1 reverse transcriptase (RT) are frequently selected in response to therapy; we examined whether silent mutations were also selected for by HIV therapy.
Design
Retrospective, observational analysis. Biochemical assays.
Methods
A comparison of the RT gene from antiretroviral naïve (N=812) and experienced individuals (N=2212), reveals two silent mutations (K65K and K66K) that are strongly associated with treatment experience. To assess reverse transcription efficiency, steady-state kinetic assays were carried out using recombinant purified HIV-1 RT and a series of synthetic RNA/DNA template/primer substrates. The RNA templates spanned codons 60 to 77 in the RT and included different combinations of mutations at codons 65, 66, 67 and 70.
Results
Silent AAG mutations (or mixtures) at RT codons 65 and/or 66 were observed in 812 samples from 351 patients and 2129 samples from 829 patients, respectively. In clade B samples, there was a very strong relationship between the silent mutations and the thymidine analog mutations (TAMs), in particular D67N. Steady-state kinetic experiments demonstrated that HIV-1 RT exhibited a strong tendency to pause and/or dissociate at codons 65 and 66 on RNA templates that contained the D67N and K70R mutations. However, when the K66 or K66 AAA to AAG mutations were added to the background of the 67 and 70 mutational changes, these pausing and/or dissociation events were largely alleviated.
Conclusions
Silent mutations at codons 65 and/or 66 are strongly co-selected with TAMs. These data provide the first evidence for an RNA-level mechanism of direct relevance to drug resistance.
Keywords: HIV-1, reverse transcriptase, synonymous mutation, evolution, Highly Active Antiretroviral Therapy
INTRODUCTION
The HIV-1 reverse transcriptase (RT) facilitates the conversion of the viral single-stranded RNA genome into double stranded DNA. RT is a multifunctional enzyme and exhibits RNA-dependent DNA polymerase, DNA-dependent DNA polymerase, and ribonuclease H activities. Due to its essential role in HIV-1 replication, RT is a primary target for drug development [1]. Currently, two therapeutic classes of RT inhibitors (RTIs) – the nucleoside or nucleotide RTIs (NRTIs) and nonnucleoside RTIs (NNRTIs) [1] – are routinely used to treat HIV infection. However, the long-term efficacy of combination therapies that contain RTIs is limited by the selection of missense mutations which confer amino acid substitutions in the RT that lead to HIV drug resistance [2].
Amino acid substitutions in RT associated with NRTI and NNRTI resistance have been generally well-defined and well-characterized [3]. For example, NRTI-associated resistance mutations can be broadly categorized into two groups depending on their phenotypic mechanism of resistance. The K65R, K70E, L74V, Q151M and M184V mutations primarily increase the selectivity of RT for the incorporation of a natural dNTP substrate over a NRTI-triphosphate [4]. By contrast, the mutations M41L, D67N, K70R, L210W, T215F/Y and K219Q/E – typically referred to as thymidine analog mutations (TAMs) – augment the ability of HIV-1 RT to excise the chain-terminating NRTI-monophosphate from a prematurely terminated DNA chain [4].
In contrast to missense mutations, silent mutations do not result in a change to the primary amino acid sequence of the enzyme. As a result, they are typically treated as evolutionarily neutral and are largely overlooked in HIV genotypic analyses. However, in some cases, silent mutations have been shown to alter protein translational kinetics, which in turn may affect splicing, or transcriptional control, ultimately affecting subsequent protein expression and activity [5, 6]. In this study, we investigated whether HIV-1 co-selects for silent mutations in the HIV-1 RT gene under selective antiviral drug pressure.
METHODS
Prevalence of Mutations
The prevalence of all RT mutations with respect to a wild type reference sequence (HIV HXB2; GenBank Accession #K03455) was determined in 20,017 archived plasma samples. Using a list of key drug mutations modified from the International AIDS Society–USA (IAS) [3], we examined the prevalence of all mutations in samples stratified as either “wild-type” (WT) (i.e. not having IAS “key resistance mutations”) or “non-wild-type” (Figure 1A and B, respectively). The mutations we examined, by drug category were: lamivudine (184I/V); any other nucleoside reverse transcriptase inhibitors (41L, 62V, 65R, 67N, 69D or insertion, 70R, 74V, 75I, 151M, 210W, 215F/Y or 219E/Q); any non-nucleoside reverse transcriptase inhibitors (100I, 103N, 106A/M, 108I, 181C/I, 188C/H/L, 190A/S, P225H, M230L or 236L); and any protease-inhibitors (30N, 33F, 46I/L, 48V, 50L/V, 54V/L/M, 82A/F/S/T, 84V, or 90M). To show amino acid changes, we compared the difference in the prevalence of amino acid substitutions between “wild-type” and “non-wild-type” samples (B minus A) (Figure 1C).
Fig.1.
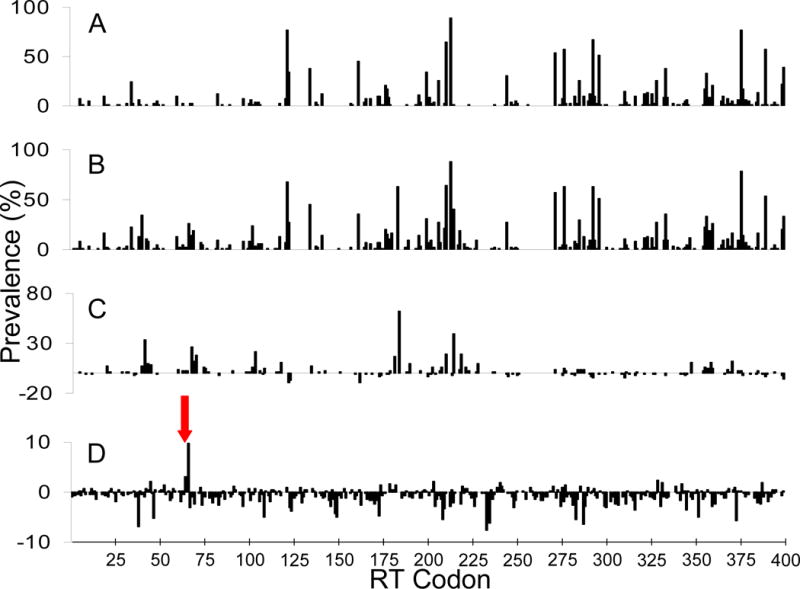
(A) Prevalence of all mutations at individual RT codons, with respect to HXB2, from all samples stratified as “wild-type” (i.e. having no IAS “key resistance mutations”). (B) Prevalence of all mutations at individual RT codons, with respect to HXB2, from all samples stratified as “non-wild-type” (i.e. having at least one IAS “key resistance mutation”). (C) Difference in prevalence between “wild-type” and “non-wild-type” sequences, at individual RT codons, for all mutations that conferred amino acid changes (B minus A). (D) Difference in prevalence between “wild-type” and “non-wild-type” sequences, at individual RT codons, for silent mutations only (B minus A). Codons 65 and 66 are highlighted by the red arrow.
Clinical Correlates of Silent Mutation Selection
In order to establish the clinical correlates of these silent mutations, data for silent AAG (or mixtures) at RT codons 65 and/or 66 was pooled from all individuals with available genotypes in members of the previously well-characterized HOMER (“HAART Observational Medical Evaluation and Research”) cohort [7, 8] infected with subtype B HIV-1 (N=2542). Patients were ≥ 18 years old and antiretroviral therapy naïve when they started HAART and initiated treatment between August 1, 1996 and November 30, 2005. All participants had a CD4 cell count and plasma viral load (pVL) measurement within six months of their first antiretroviral start date. Study data from eligible participants were extracted from the BC Centre for Excellence in HIV/AIDS’ monitoring and evaluation system. Associations between baseline characteristics (gender, AIDS diagnosis, history of injection drug use, year of first therapy, adherence strata, CD4 cell count, and pVL) as well as exposure to specific NRTI drugs during therapy (lamivudine, abacavir, zidovudine, stavudine, zalcitabine, didanosine, and tenofovir) were tested using the chi-square method, with Bonferroni corrections made for multiple comparisons. We also wished to assess the clinical impact of K65/66K silent mutations on pVL during therapy. Significance of the differences in pre and post K65/66K pVL was determined using the Wilcoxon signed-rank test.
Steady-State Kinetic Assays
D67N/K70R/T215F/K219Q (TAM67) and TAM67/K65R HIV-1 RT were generated by site-directed mutagenesis of WT HIV-1LAI RT [9, 10]. Recombinant RTs were overexpressed and purified to homogeneity as described previously [11, 12]. RT concentration was determined spectrophotometrically at 280 nm, using an extinction co-efficient (ε280) of 260 450 M−1 cm−1 and the active site concentration was calculated from pre-steady-state burst experiments [13]. All experiments were performed using corrective active site concentrations.
DNA and RNA oligomers were synthesized by Integrated DNA technologies (Coralville, Iowa, USA). The RNA templates spanned codons 62 to 77 of the HIV-1 RT gene and included different combinations of mutations at codons 65 (AAA, AGA, AAG), 66 (AAA, AAG), 67 (GAC, AAC) and 70 (AAA, AGA) (Fig. S2). A complementary 20 nucleotide DNA primer (5′-TGAAATCTCTAATTTTCTCC-3′) was used to prime RT-mediated DNA synthesis reactions. The primer was 5′-radiolabeled with [γ-32P]-ATP (GE Healthcare, Piscataway, New Jersey, USA) and annealed to the respective RNA templates, as described previously [9, 10].
Steady-state DNA polymerization assays were carried out in 50 mM Tris-HCl pH 7.8 (37 °C), 50 mM KCl, 10 mM MgCl2 containing 200 nM of T/P and 5 μM dNTP. Reactions were initiated by the addition of 50 nM wild-type (WT) or mutant (TAM67 or TAM67/K65R) HIV-1 RT. Aliquots were removed at designated times (1; 2.5; 3.5; 5.5; 7.5; 10 and 20 min) and mixed with an equal volume of sample loading buffer (98 % deionized formamide, 10 mM EDTA, and 1 mg/mL each of bromophenol blue and xylene cyanol). Samples were denatured at 85 °C for 5 min, products were separated on a 7 M urea – 14 % polyacrylamide gel and detected using a Bio-Rad GS525 Molecular Imager (Bio-Rad Laboratories, Hercules, California, USA).
RNA Structural Determinations
Preliminary modeling of the minimum free energy secondary structure of isolated HIV-1 RT RNA sequences, spanning codons 55 to 75, were conducted using the Vienna RNA Secondary Structure Package (http://www.tbi.univie.ac.at/RNA/). Inputted sequences included wild type and several variants containing non-synonymous mutations at codons 65, 67, and 70, as well as silent mutations at codon 65 and 66.
RESULTS
Silent Mutations in HIV-1 RT are Associated with Treatment Experience
Figure 1A shows the prevalence of all mutations with respect to HXB2 at the individual RT codons, for samples that were classified as “wild-type”, while Figure 1B shows mutation prevalences for “non-wild-type” samples. The difference in prevalence between “wild-type” and “non-wild-type” samples for mutations that conferred amino acid changes is shown in Figure 1C. The prevalence of silent mutations, relative to HXB2 clade B consensus (Figure 1D), revealed that only two silent mutations at codons K65 and/or K66 in RT were strongly associated with treatment experience and both were highly significant after correction for multiple comparisons. The prevalence of silent mutations at codons K65 and/or K66 in treatment-experienced patients increased by 3.0 and 9.8 percent, respectively, relative to the naïve population. This degree of selection actually exceeds the degree of selection of most of the known IAS drug resistance mutations. Silent AAA to AAG (or mixtures) mutations at RT codons 65 and/or 66 were observed in 812 samples from 351 patients and 2129 samples from 829 patients, respectively. Similar selection of silent mutations at codons 65 and 66 was observed in the Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu; data not shown). We also investigated the prevalence of all mutations with respect to HXB2 at individual RT codons from first available pre-therapy genotypes, as well as the prevalence of mutations at the last available on-therapy genotypes, to ensure that only a single post-treatment sample per patient was examined. The results were similar, showing an increase in the prevalence of silent mutations at codons 65K and/or 66K between last available on therapy and first available pre-therapy sample (data not shown).
TAMs are Strongly Associated with Silent Mutations
Pooled data from available HIV-1 subtype B RT sequences in the British Columbia (BC) Centre for Excellence in HIV/AIDS database were used to identify the resistance mutations that are most commonly associated with the presence of 65K and/or 66K silent mutations (Table 1). In subtype B samples, there was a very strong correlation between the silent mutations and TAMs. In particular, at position 67 of the RT, 7% of wild-type D67 samples had the 65K and/or 66K silent mutations, compared with over 80% of samples with the D67N substitution (p<<0.001). These associations were confirmed in two independent resistance datasets (the Stanford HIV Drug Resistance Database and Virco; data not shown).
Table 1.
HIV RT resistance mutations most commonly associated with the presence of 65K and/or 66K silent mutations. Data is pooled from available clade B RT sequences in the BC Centre for Excellence database. Data is sorted by p-value for illustration.
| RT Codon | Number With Silent Mutation (%) | Number Without Silent Mutation (%) | p-Value |
|---|---|---|---|
| 67 | 1755 (69.6) | 767 (30.4) | 2.6 × 10−153 |
| 219 | 1182 (64.6) | 649 (35.5) | 5.5 × 10−86 |
| 70 | 921 (51.4) | 871 (48.6) | 9.4 × 10−48 |
| 215 | 1691 (42.5) | 2291 (57.5) | 2.0 × 10−45 |
| 41 | 1408 (43.2) | 1851 (56.8) | 3.0 × 10−35 |
| 65 | 33 (15.1) | 185 (84.9) | 1.8 × 10−29 |
| 184 | 1973 (33.2) | 3969 (66.8) | 1.8 × 10−27 |
| 210 | 1054 (45.5) | 1262 (54.5) | 5.3 × 10−26 |
Clinical Correlates of Silent Mutation Selection
Although the TAMs are considered to arise on exposure to the thymidine analogues (stavudine or, particularly, zidovudine), exposure to zidovudine was not associated with the selection of the silent mutations. However, exposure to the other NRTIs (abacavir, stavudine, didanosine, or tenofovir) during therapy were associated with the selection of silent mutations at position 65 and 66 (Table 2) in a dose-dependent manner (ANOVA F-test p = 0.01) (Figure 2). Note that the amino acid substitution K65R has been implicated in resistance to many of these agents, and is itself antagonistic to TAMs. The mean pVL for individuals before the first positive test for K65/66K silent mutations was 3.89 log10 and 4.01 log10 at the first positive test (Wilcoxon signed-rank test p=0.0064). While statistically significant, this is not likely to be a clinically significant difference. Male gender, no history of injection drug use, and low baseline CD4 count were each associated with the presence of the K65/66K mutations (Table 2). However, these associations may not be causal, but could be correlated with silent mutations due to the association of these variables with the development of extensive resistance in general.
Table 2.
Association between baseline characteristics and drug exposures with K65/66K for members of the HOMER cohort. Participants initiated treatment between August 1, 1996 and November 30, 2005.
| Variable | Subcategory | Presence of 65K and/or 66K silent Mutation (N [%])
|
||
|---|---|---|---|---|
| No | Yes | p-Value | ||
| Gender | Male | 1531 (80.1) | 548 (87.0) | <0.001 |
| Female | 381 (19.9) | 82 (13.0) | ||
| AIDS Diagnosis | Yes | 264 (13.8) | 103(16.4) | 0.12 |
| No | 1648 (86.2) | 527 (83.7) | ||
| History of Injection Drug Use | Yes | 693 (36.2) | 186 (29.5) | 0.0021 |
| No | 1219 (63.8) | 444 (70.5) | ||
| Year of First Therapy | 1996 – 1999 | 1104 (57.7) | 367 (58.3) | 0.83 |
| 1999 – 2003 | 616 (32.2) | 196 (31.1) | ||
| 2003 – 2005 | 192 (10.0) | 67 (10.6) | ||
| Adherence | 0% – <40% | 355 (18.7) | 104 (16.6) | 0.066 |
| 40% – <80% | 459 (24.2) | 127 (20.3) | ||
| 80% – <95% | 281 (14.8) | 104 (16.6) | ||
| ≥95% | 804 (42.3) | 292 (46.6) | ||
| CD4+ Cell Count (cells/mm3) | <200 | 766 (40.5) | 295 (47.12) | 0.015 |
| 200 – <350 | 528 (27.9) | 155 (24.76) | ||
| ≥350 | 597 (31.6) | 176 (28.12) | ||
| Plasma Viral Load (log10) | <4 | 131 (7.9) | 43 (8.3) | 0.95 |
| 4 – <5 | 627 (37.7) | 195 (37.7) | ||
| ≥5 | 904 (54.4) | 279 (54.0) | ||
| Antiretroviral Drug Exposure | Lamivudine | 1801 (94.2) | 599 (95.1) | 0.40 |
| Abacavir | 308 (16.1) | 157 (24.9) | <0.001 | |
| Zidovudine | 1091 (57.1) | 359 (57.0) | 0.97 | |
| Stavudine | 1210 (63.3) | 444 (70.5) | 0.001 | |
| Zalcitabine | 67 (3.5) | 31 (4.9) | 0.11 | |
| Didanosine | 742 (38.8) | 286 (45.4) | 0.0035 | |
| Tenofovir | 271 (14.2) | 130 (20.6) | <0.001 | |
Figure 2.

Percentage of patients with a 65K and/or 66K silent mutation as a function of the number of NRTIs exposed to during treatment (abacavir, stavudine, didanosine, or tenofovir). Total number of patients for each exposure, including those without K65/66K mutations, are shown below each grouping. ANOVA F-test p = 0.01
RNA Structural Determinations
Preliminary investigations modeling the secondary structure of isolated wildtype HIV-1 RNA templates and several templates with various combinations of codon changes at positions 65, 66, 67, and 70 showed most of the silent changes occurred in a loop of the secondary RNA structure, and therefore would not be expected to result in significant structural changes between secondary structures.
Silent Mutations Compensate for Replication Block of D67N/K70R
To address a possible mechanism by which the silent mutations at codons 65 and 66 in the HIV-1 RT gene might affect virus replication, we assessed the ability of recombinant HIV-1 RT that was WT or contained the TAMs D67N, K70R, T215F and K219Q (TAM67 RT) to copy RNA templates that spanned codons 62 to 77 in RT and included different combinations of mutations at codons 65 (AAA, AGA, AAG), 66 (AAA, AAG), 67 (GAC, AAC) and 70 (AAA, AGA). Under steady-state kinetic conditions, both the WT and TAM67 RTs exhibited a stronger tendency to pause/dissociate at codons 65 and 66 on RNA templates containing the D67N/K70R mutations (template RH1) in comparison to the wild-type sequence (Figure 3A, 3C). However, when the K65 or K66 AAA to AAG mutations were added to the background of the D67N and K70R mutational changes (templates RH3 and RH2, respectively), these pausing and/or dissociation events were largely alleviated (Figures 3A, 3C). Interestingly, for both RT enzymes, the introduction of the K65R mutation (AAA to AGA) into the RNA template (template RH4) also alleviated the replication block resulting from the 67 and 70 mutational changes (Figure 3B, 3C). Together, these results suggest that mutations at codons 65 and 66 in the RNA template compensate for a replication block triggered by the D67N/K70R mutations.
Fig. 3.
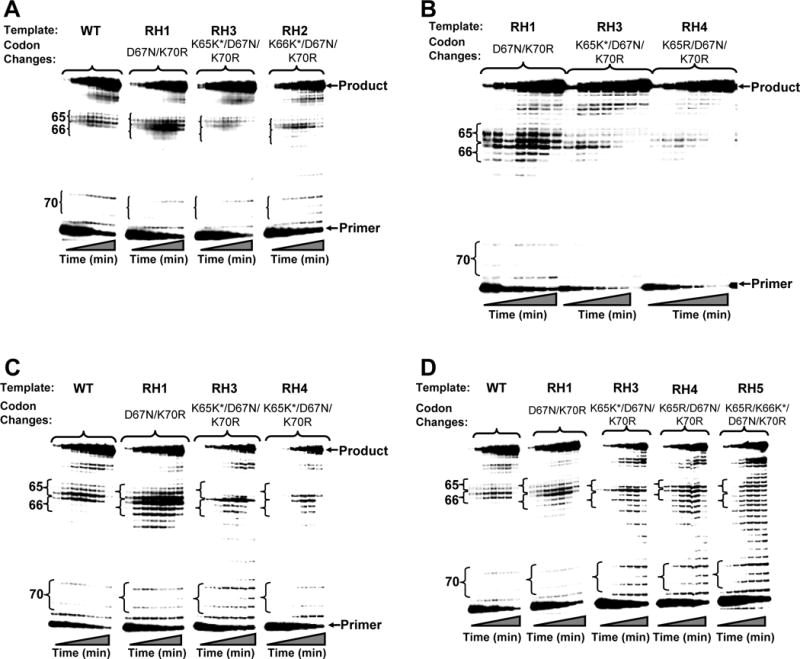
DNA synthesis reactions carried out by wild-type, TAM67, and TAM67/K65R HIV-1 RT on RNA templates with substitutions at codons 70 (AAA to AGA), 67 (GAC to AAC), 66 (AAA to AAG) and/or 65 (AAA to AAG or AGA).
A) DNA synthesis reactions carried out on the wild-type template as well as RH1, RH3 and RH2.
B) DNA synthesis reactions carried out on the RH1, RH3 and RH4 templates.
C) DNA synthesis reactions carried out by TAM67 HIV-1 RT on the wild-type, RH1, RH3 and RH4 templates.
D) DNA synthesis reactions carried out by TAM67/K65R HIV-1 RT on the wild-type, RH1, RH3, RH4 and RH5 templates.
Reactions were carried out and analyzed as described in Methods. K65K* and K66K* signify the AAA to AAG silent mutations at codons 65 and 66. Times each aliquot ran for, from left to right, are: 1, 2.5, 3.5, 5.5, 7.5, 10, and 20 minutes, respectively.
As described above, silent mutations at codons 65 and 66 in the RT gene are highly associated with TAMs, and in particular D67N. However, antagonism between missense mutations at K65 (e.g. K65R) and TAMs has previously been reported at the genomic, viral and enzyme level in subtype B RT, indicating that the virus cannot readily accommodate both TAMs and K65R in RT [10, 14, 15]. Accordingly, we were also interested in the ability of RT enzymes containing TAMs and K65R (i.e. TAM67/K65R RT) to copy RNA templates that contained codon changes at positions 65, 66, 67 and 70 (Figure 3D). In comparison with WT and TAM67 HIV-1 RT, the TAM67/K65R enzyme exhibited a decreased tendency to pause at codons 66 and 67 on the RH1 template. Furthermore, the silent AAA to AAG change at codon 65 in combination with the D67N and K70R (template RH3) had minimal effect of the overall polymerization pattern of the enzyme. However, on templates that included the K65R mutation (RH4), TAM67/K65R RT exhibited a very strong tendency to pause at codons 65, 66 and 67. Interestingly, the introduction of silent mutation into templates with 67N/70R/K65R mutations does not alleviate the block experienced by RT with K65R+TAMs (Fig. 3D; RH5). Furthermore, the addition of the K66 AAA to AAG silent mutation to this template (RH5) did not compensate for this increased pausing. In fact the enzyme exhibited a very distributive mode of polymerization; by contrast, both the WT and TAM67 RT could replicate very efficiently on this template (data not shown). Taken together, these data show that antagonism between RT codon 65 and TAMs may also extend to the RNA level.
DISCUSSION
Our results reveal the surprising finding that silent mutations at K65 and/or K66 of the HIV-1 RT are strongly selected for during antiretroviral therapy. These silent mutations are particularly observed in association with mutations at HIV-1 RT codon 67. Selection was associated with exposure to the NRTIs abacavir, didanosine, stavudine and/or tenofovir. Enzymatic data suggest that the silent mutations may play a role in the efficiency of viral DNA synthesis and also highlight a previously unobserved aspect of antagonism between K65 mutations and TAMs at the level of reverse transcription. Subsequently, there have been two independent follow-up studies which showed similar results [16, 17].
Other studies have focused on the predispostion of virus with silent mutations in the evolution of HIV drug resistance, particularly with respect to non-B subtypes, but none have tied the actual evolution of silent mutations to drug exposure. Loemba et al. identified several silent mutations at codons 62, 65, 106, 138 and 161 associated with antiretroviral resistance in subtype C [18]. Additionally, Turner et al. described a G48G silent mutation within the protease region of subtype C subtypes, as well as a T215T silent mutation present in subtype A and A/E subtypes [19]. However, to our knowledge no silent mutations have been demonstrated to be selected for by antiretroviral drug pressure, and indeed, this may represent the first observation of the selection of a synonymous mutation in response to a known selective pressure occurring in a natural setting.
Using data from the Los Alamos HIV sequence database, we noted that consensus codons 64, 65 and 66 in wild-type subtype C HIV-1 differ from other subtypes in this region. Whereas subtype C has AAA, AAG, and AAG at codons 64, 65, and 66 respectively, subtypes B–F have AAG, AAA, and AAA at codons 64, 65 and 66 respectively. There have been recent suggestions that silent mutations in the subtype C RT gene were responsible for the rapid selection of the K65R mutation [20, 21]. Another unique feature of subtype C virus is that the K65R mutation which develops in subtype C is AGG, whereas in subtype B it is almost exclusively AGA (data not shown). While our results are analogous, they are likely not directly relevant to subtype C HIV-1.
An important limitation of our preliminary investigations of secondary RNA structure was that they were conducted on isolated RNA sequences and we cannot make any inferences about the effects on a RNA sequence that is undergoing reverse transcription. In addition, we have only examined the effect of the silent mutations on RNA-dependent DNA polymerization. Since HIV-1 RT is a multi-functional enzyme, is it is also possible that silent changes in the RNA template affect the efficiency of the DNA-dependent DNA polymerization and/or ribonuclease H activity of HIV-1 RT, but these were not investigated.
The data show that mutations at codons 65 and/or 66 are co-selected with TAMs, and therefore provide an interesting and potentially clinically relevant example of situations where silent amino acid changes may not be evolutionarily neutral. These data also provide the first evidence for an RNA-level mechanism of direct relevance to drug resistance. Further investigations should be conducted to more accurately identify the mechanism by which these silent mutations are effecting viral evolution, along with other studies to try and identify any further examples of silent mutation selection in response to therapy across the HIV genome.
Acknowledgments
Role of Authors: This study was conceived and designed by P.R.H. and N.S.C. Data acquisition was performed by V.S.G., E.H., B.W. (genotypic data), as well as C.W.S. (steady-state kinetic assays). Data analysis was conducted by V.D.L., B.W., P.L., A.F.P., and R.A. Initial drafting of the manuscript was conducted by V.S.G., E.H., and N.S.C. All authors contributed to the interpretations of data and critical revision of the manuscript.
Funding/Support: This work was supported by the Canadian Institutes of Health Research (CIHR) through a fellowship for Dr. Lima, by GlaxoSmithKline/CIHR through a Chair in Clinical Virology for Dr. Harrigan, and by grants from the University of Pittsburgh Clinical and Translational Science Institute (CTSI RR024153) to Dr. Slui-Cremer.
Footnotes
Previous presentation: A portion of this work was presented at the XVI International HIV Drug Resistance Workshop, June 12–16, 2007, Needham’s Point, Barbados.
References
- 1.Warnke D, Barreto J, Temesgen Z. Antiretroviral drugs. J Clin Pharmacol. 2007;47:1570–1579. doi: 10.1177/0091270007308034. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch MS, Brun-Vezinet F, D’Aquila RT, Hammer SM, Johnson VA, Kuritzkes DR, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society-USA Panel. JAMA. 2000;283:2417–2426. doi: 10.1001/jama.283.18.2417. [DOI] [PubMed] [Google Scholar]
- 3.Johnson VA, Brun-Vezinet F, Clotet B, Gunthard HF, Kuritzkes DR, Pillay D, et al. Update of the drug resistance mutations in HIV-1: 2007. Top HIV Med. 2007;15:119–125. [PubMed] [Google Scholar]
- 4.Menendez-Arias L. Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 2008;134:124–146. doi: 10.1016/j.virusres.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 5.Brookes AJ. The essence of SNPs. Gene. 1999;234:177–186. doi: 10.1016/s0378-1119(99)00219-x. [DOI] [PubMed] [Google Scholar]
- 6.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 7.Hogg RS, Yip B, Chan KJ, Wood E, Craib KJ, O’Shaughnessy MV, et al. Rates of disease progression by baseline CD4 cell count and viral load after initiating triple-drug therapy. JAMA. 2001;286:2568–2577. doi: 10.1001/jama.286.20.2568. [DOI] [PubMed] [Google Scholar]
- 8.Brumme ZL, Brumme CJ, Heckerman D, Korber BT, Daniels M, Carlson J, et al. Evidence of Differential HLA Class I-Mediated Viral Evolution in Functional and Accessory/Regulatory Genes of HIV-1. PLoS Pathog. 2007;3:e94. doi: 10.1371/journal.ppat.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sluis-Cremer N, Arion D, Parikh U, Koontz D, Schinazi RF, Mellors JW, et al. The 3′-azido group is not the primary determinant of 3′-azido-3′-deoxythymidine (AZT) responsible for the excision phenotype of AZT-resistant HIV-1. J Biol Chem. 2005;280:29047–29052. doi: 10.1074/jbc.M503166200. [DOI] [PubMed] [Google Scholar]
- 10.Parikh UM, Zelina S, Sluis-Cremer N, Mellors JW. Molecular mechanisms of bidirectional antagonism between K65R and thymidine analog mutations in HIV-1 reverse transcriptase. AIDS. 2007;21:1405–1414. doi: 10.1097/QAD.0b013e3281ac229b. [DOI] [PubMed] [Google Scholar]
- 11.Le Grice SF, Gruninger-Leitch F. Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Eur J Biochem. 1990;187:307–314. doi: 10.1111/j.1432-1033.1990.tb15306.x. [DOI] [PubMed] [Google Scholar]
- 12.Le Grice SF, Cameron CE, Benkovic SJ. Purification and characterization of human immunodeficiency virus type 1 reverse transcriptase. Methods Enzymol. 1995;262:130–144. doi: 10.1016/0076-6879(95)62015-x. [DOI] [PubMed] [Google Scholar]
- 13.Kati WM, Johnson KA, Jerva LF, Anderson KS. Mechanism and fidelity of HIV reverse transcriptase. J Biol Chem. 1992;267:25988–25997. [PubMed] [Google Scholar]
- 14.Parikh UM, Bacheler L, Koontz D, Mellors JW. The K65R mutation in human immunodeficiency virus type 1 reverse transcriptase exhibits bidirectional phenotypic antagonism with thymidine analog mutations. J Virol. 2006;80:4971–4977. doi: 10.1128/JVI.80.10.4971-4977.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parikh UM, Barnas DC, Faruki H, Mellors JW. Antagonism between the HIV-1 reverse-transcriptase mutation K65R and thymidine-analogue mutations at the genomic level. J Infect Dis. 2006;194:651–660. doi: 10.1086/505711. [DOI] [PubMed] [Google Scholar]
- 16.Svicher V, Alteri C, Lagana A, Pigola G, Dimonte S, Mussini C, et al. Synonymous mutations in the highly ordered regulation of nucleoside reverse transcriptase inhibitor resistance [Abstract] Antivir Ther. 2008;13(Suppl 3):A62. [Google Scholar]
- 17.Gonzalez LMF, Pinto AFN, Martins AN, Ahuiar RS, Afonso AO, Brindeiro RM, et al. Silent mutations at reverse transcriptase codons 65 and 66 in B and C strains found in Brazil are very strongly associated with treatment and these signatures can impact on zidovudine mutation acquisition in vitro [Abstract] Antivir Ther. 2008;13(Suppl 3):A63. [Google Scholar]
- 18.Loemba H, Brenner B, Parniak MA, Ma’ayan S, Spira B, Moisi D, et al. Genetic divergence of human immunodeficiency virus type 1 Ethiopian clade C reverse transcriptase (RT) and rapid development of resistance against nonnucleoside inhibitors of RT. Antimicrob Agents Chemother. 2002;46:2087–2094. doi: 10.1128/AAC.46.7.2087-2094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner D, Brenner B, Moisi D, Detorio M, Cesaire R, Kurimura T, et al. Nucleotide and amino acid polymorphisms at drug resistance sites in non-B-subtype variants of human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2004;48:2993–2998. doi: 10.1128/AAC.48.8.2993-2998.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brenner BG, Oliveira M, Doualla-Bell F, Moisi DD, Ntemgwa M, Frankel F, et al. HIV-1 subtype C viruses rapidly develop K65R resistance to tenofovir in cell culture. AIDS. 2006;20:F9–13. doi: 10.1097/01.aids.0000232228.88511.0b. [DOI] [PubMed] [Google Scholar]
- 21.Invernizzi CF, Coustinos D, Moisis D, et al. Introduction of the 64/65 Nucleotide Polymorhpisms of Subtype C into Subtype B HIV-1 Selects for the K65R Mutational Pathway in Cell Culture [Abstract]. 15th Conference on Retroviruses and Opportunistic Infections; 2008; Boston. [Google Scholar]