

Abstract
Purpose
Sulfur mustard, nitrogen mustard (NM), and 2-chloroethyl ethyl sulfide all cause corneal injury with epithelial–stromal separation, differing only by degree. Injury can resolve in a few weeks or develop into chronic corneal problems. These vesicants induce microbullae at the epithelial–stromal junction, which is partially caused by cleavage of transmembranous hemidesmosomal collagen XVII, a component anchoring the epithelium to the stroma. ADAM17 is an enzyme involved in wound healing and is able to cleave collagen XVII. The activity of ADAM17 was inhibited in vesicant-exposed corneas by four different hydroxamates, to evaluate their therapeutic potential when applied 2 hours after exposure, thereby allowing ADAM17 to perform its early steps in wound healing.
Methods
Rabbit corneal organ cultures exposed to NM for 2 hours were washed, then incubated at 37°C for 22 hours, with or without one of the four hydroxamates (dose range, 0.3–100 nmol in 20 μL, applied four times). Corneas were analyzed by light and immunofluorescence microscopy, and ADAM17 activity assays.
Results
Nitrogen mustard–induced corneal injury showed significant activation of ADAM17 levels accompanying epithelial–stromal detachment. Corneas treated with hydroxamates starting 2 hours post exposure showed a dose-dependent ADAM17 activity inhibition up to concentrations of 3 nmol. Of the four hydroxamates, NDH4417 (N-octyl-N-hydroxy-2-[4-hydroxy-3-methoxyphenyl] acetamide) was most effective for inhibiting ADAM17 and retaining epithelial–stromal attachment.
Conclusions
Mustard exposure leads to corneal epithelial sloughing caused, in part, by the activation of ADAM17 at the epithelial–stromal junction. Select hydroxamate compounds applied 2 hours after NM exposure mitigated epithelial–stromal separation.
Keywords: ADAM17, TACE, nitrogen mustard, CEES, corneal injury, hydroxamate
Sulfur mustard (SM, bis-2-chloroethyl sulfide, HD, SM) was a warfare agent used by the Germans in World War I and by the Iraqis in the Iran–Iraq war of the 1980s. In August 2015, SM was used by ISIS in an attack on Kurdish forces in Iraq, as well as an attack in Syria. The victims developed blisters and had difficulty breathing, and US military tests in the area identified SM on a fragment of a bomb used in the attack. Mustard agents injure the eyes, the skin, and the lungs, with the eyes being the most sensitive. Because symptoms do not manifest until 2 to 4 hours after exposure, exposed persons do not immediately know they are exposed to mustard. This delay has contributed to confusion and panic when symptoms of exposure finally develop. For the eyes, these consist of blepharospasm, lacrimation, irritation, pain, and photophobia. Corneal ulceration occurs several hours later.1 A chronic injury phase, referred to as mustard gas keratopathy, is also found to recur in some previously healed corneas. For some patients this occurred a few weeks after exposure; in others it took years to manifest. This keratopathy is characterized by corneal conjuctivalization and limbal stem cell deficiency.2,3 Although much research has been done on SM exposure and injury, even after 100 years of study there are no US Food and Drug Administration (FDA)-approved therapies for mustard injury for any of the organs it affects. Identifying good therapies for ocular mustard exposure is desirable because these agents are considered by the US government to be chemical weapons that terrorists could use against US civilian or military populations. This is because the reagents used to synthesize SM are inexpensive and the synthesis is uncomplicated. It could be aerosolized and released in a shopping mall or train car, and the perpetrator could exit the area before any exposure effects are detected.
As vesicating agents, mustard compounds lead to a loss of epithelial–stromal attachment. In the skin, SM induces blisters that resemble those produced by UV exposure. In the cornea, large blisters are not formed. Instead, microbullae form, and once enough have accrued, the epithelium cannot hold fast to the basement membrane, causing the epithelial tissue to slough. A good postexposure therapy for SM could be one that enhances the ability of the epithelium to remain attached to the stroma. This might allow some basal epithelia the opportunity to recover in situ, maintaining their connections with their basement membrane and stroma. One of the key players in epithelial–stromal integrity is collagen XVII (i.e., BP180), a transmembranous component of the hemidesmosome.4 Cleavage of collagen XVII by ADAM9, ADAM10, and/or ADAM17 after injury releases epithelial cells from their basement membrane, and this cleavage allows them to migrate.5–7
ADAM17, also known as TNF-α converting enzyme or TACE, is a general response to injury8 as well as a “sheddase” for releasing collagen XVII. Since hydroxamate compounds are able to bind Zn+2, we reasoned they could potentially inhibit Zn+2-dependent ADAM17 and therefore be therapeutic for retaining epithelial–stromal integrity after mustard exposure, at least partially. This idea was based on the fact that ADAM17 would be activated very soon after injury to release wound-healing cytokines, but epithelial–stromal separation takes hours to manifest. By adding ADAM17 inhibitors at 2 hours post exposure, retention of epithelial–stromal integrity could be facilitated while not affecting the immediate postinjury release of cytokines that contribute to healing. Air-lifted rabbit corneal organ cultures were used to test this. The 2-hour delay before adding the hydroxamates mimics the time that transpires before victims realize they are exposed and begin to seek medical attention. Treatment thereafter consisted of three additional applications of hydroxamate compounds spaced out over a period of 22 hours. ADAM17 activity and epithelial–stromal detachment were then analyzed. The data presented here demonstrated that nitrogen mustard (NM) contributes to ADAM17 activation, and that corneal epithelial–stromal separation from mustard exposure can be appreciably decreased by topical application of select chemically synthesized hydroxamate compounds. At 24 hours post exposure, the corneas treated with the hydroxamates showed a range of injury that was less severe than that of exposed corneas receiving no inhibitor.
Materials and Methods
Rabbit ADAM17
Genbank and Ensembl9 databases were probed with human ADAM17 cDNA sequences to find corresponding rabbit (Oryctolagus cuniculus) cDNA sequences. No rabbit ADAM17 sequence was identified in the Genbank search, but the blast search of the open access Ensembl database yielded two highly conserved matches, ENSOCUT00000010473 and ENSOCUT00000031438. Aligning the translation products of the human ADAM17 coding sequence with those of the rabbit sequences indicated a 93% amino acid identity. When conservative amino acid substitutions were considered, the human and rabbit ADAM17 showed 95.5% similarity. In the rabbit and human comparison, the signal peptide exhibited the least identity (64.6%), while the transmembrane domain was 100% conserved. The next most conserved region (98.8% identity) was the peptidase M12B domain located in the ADAM17 catalytic region (amino acids [aa] 223–474 in human enzyme). This conservation suggested that anti-human ADAM17 monoclonal antibodies would cross-react with rabbit ADAM17.
Preparation of Hydroxamates
The synthetic steps for the hydroxamates termed NDH4385 (i.e., Retro-OH-7, or N-heptyl-N-hydroxy-2-[4-hydroxy-3-methoxyphenyl] acetamide), NDH4409 (i.e., olvanil hydroxamate, or (9Z)-N-hydroxy-N-[(4-hydroxy-3-methoxyphenyl)methyl]-9-octadecenamide), NDH4417 (i.e., Retro-OH-8, or N-octyl-N-hydroxy-2-[4-hydroxy-3-methoxyphenyl] acetamide), and NDH4450 (i.e., Abbott ABT-518 [Abbott Park, IL, USA] or N-[(1S)-1-((4S)-2,2-dimethyl-[1,3]dioxolan-4-yl)-2-(4-(4-(trifluoromethoxyphenoxy)phenylsulfonyl)ethyl]-N-hydroxyformamide) are described in the Supplementary Material. The cLogP values predicting lipophilicity were derived by using Perkin Elmer Corporation's (Waltham, MA, USA) ChemBioDraw Ultra 14.0, part of the ChemBio Office 2014 package. Each of the four hydroxamates was prepared as 15, 50, 150, 500, 1500, and 5000 μM solutions. To make these solutions, the appropriate amount of solid hydroxamates was dissolved in 0.1% dimethyl sulfoxide at 1/10 the desired final volume. After several vortexings separated by 1-minute 10,000g centrifugations, Major's LiquiTears (Medline, Mundeleine, IL, USA) was added as the remaining 9/10 volume, achieving the desired molarity. The dissolved hydroxamates were then placed in a 50°C water bath overnight. A 20-μL volume of each was used for application to corneas.
Organ Culture of Corneas
A rabbit corneal organ culture model system was used to evaluate healing after exposure to NM or 2-chloroethyl ethyl sulfide (CEES) as previously reported.10 Briefly, rabbit eyes (8–12 weeks old) were purchased from Pel-Freez Biologicals (Rogers, AR, USA). Corneas with 2-mm scleral rims were dissected from the eyes, placed epithelial-side down into a spot plate, and the concavities were filled with 55°C molten agar (0.75%) in Dulbecco's modified Eagle's medium (DMEM). Once the solution gelled, the corneas were inverted so that the epithelial layer was accessible. Cultures were placed in 60-mm-diameter pyrex tissue culture dishes. High glucose DMEM was prepared containing 1× MEM-NEAA (minimal essential medium non-essential amino acids; Invitrogen), 1× RMPI 1640 Vitamin Solution (Sigma-Aldrich), 1× antibiotic/antimycotic (Invitrogen), ascorbic acid (0.45mM; Sigma-Aldrich), and ciprofloxacin (10μg/ml; Sigma-Aldrich). High glucose DMEM was added up to the scleral rims, leaving the corneas exposed to air. The dishes were placed in a 37°C humidified incubator with 5% CO2. The epithelium of each culture was moistened with 500 μL medium, added dropwise onto the central cornea every 7 to 9 hours. All other agents (CEES, NM, and/or hydroxamates) were also added dropwise onto the central cornea. Cornea samples (peeled off their agar support) were either put epithelial side down in cryomolds containing Optimal Cutting Temperature (OCT, Tissue-Tek; Sakura, Torrance, CA, USA) compound and flash frozen for histology and immunofluorescence, or directly snap frozen for further protein analyses including Western blot and ADAM17 activity assays (InnoZyme TACE activity assay kit; Calbiochem, Billerica, MA, USA). For DiI staining, 10-μm-thick frozen sections were fixed in cold 2% paraformaldehyde in phosphate-buffered saline (PBS) for 15 minutes, then slides were incubated with 5 μM Perchlorate (Dil Stain, 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine, Molecular Probes TM; Thermo Fisher Scientific, Inc., Eugene, OR, USA) for 20 minutes at room temperature before three 10-minute washes in PBS. Prolong Gold Antifade Mountant and 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes TM) were added before coverslipping. An Olympus Epi-fluorescent microscope (Center Valley, PA, USA) was used to collect fluorescent images.
Exposure of Cultured Corneas to Vesicants and Application of Hydroxamates
CEES or NM was used to induce mild injury. A 2M solution was made by adding 24 μL full-strength CEES (half mustard) liquid (catalog No. 242640; Sigma-Aldrich Corp., St. Louis, MO, USA) to 76 μL absolute ethanol. One microliter of the 2M CEES was then added to 1999 μL high-glucose DMEM medium, diluting the CEES to 1 mM. Each cornea received 20 μL of this solution (i.e., 20 nmol). For NM, the powdered solid (catalog No. 122564; Sigma-Aldrich) was first dissolved in PBS to 100 mM, and then diluted with medium to 10 mM. Ten microliters were applied to deliver 100 nmol vesicant to the cornea.
After applying CEES or NM onto the central corneas, the cultures were returned to the 37°C incubator for 2 hours without removing the vesicant. After this incubation, contaminated medium was removed, and fresh medium was added to the central cornea until the level in the dish reached the top of the scleral rim. Control unexposed and exposed corneas were then returned to 37°C for a 22-hour incubation, being removed for only three short periods to add 20 μL medium to the exposed samples not receiving hydroxamate therapy, or to add 20 μL of a particular hydroxamate as therapy to the central corneas. The first hydroxamate application was left on for 8 hours, the second for 9 hours, and the third for 5 hours. Thus, the length of the 2-hour exposure and the subsequent treatment was 24 hours in total.
For experiments analyzing how fast NM exposure induced ADAM17, cultures were set up as described. For the shortest exposure time, the NM solution was applied to a cornea, then immediately washed off and the sample was put in protein isolation extraction buffer. This was repeated with two other corneas to collect three 0-minute exposures. For the 5- and 10-minute exposures, NM was added to the sets of three corneas accordingly, insuring none were accidentally under- or overexposed to NM. All corneas were extracted and processed for ADAM17 activity assays.
Hydroxamate Lipophilicity
A computational approach was used to derive the lipophilicity value, cLogP. The most widely used software package is Perkin-Elmer Corporation's ChemBioDraw Ultra, which is part of the ChemBioOffice software suite. Since the partition function is a sum of the individual partition functions for component moieties of the structure, inputting the compounds structures via a drawing interface allowed the interactive data base (with built-in proprietary computational algorithms) to directly calculate the cLogP.
ADAM17 Activity Assays
The InnoZyme ADAM17/TACE Activity Kit (Calbiochem) was used to quantify the enzyme's activity from corneal extracts according to the vendor's provided protocol. Briefly, 400 μL wash buffer (from the InnoZyme kit) was applied to 96-well plates precoated with anti-human ADAM17 antibody, followed by two washes. Triplicate samples of corneal extracts and InnoZyme kit standards (100 μL) were each added to three sets of wells. Plates were sealed and incubated 1 hour with gentle shaking at room temperature. Then, plates were washed with 400 μL wash buffer five times. ADAM17 substrate supplied in the kit (100 μL) was added to each well and incubated for 5 hours at 37°C. Fluorescence was measured at an excitation wavelength of 324 nm and an emission wavelength of 405 nm, and was reported as relative fluorescence units on graphs.
Hydroxamate Dose-Response Experiments
The experiment to determine the dose response of each hydroxamate was set up by using the kit directions. First, proteins were extracted from NM-exposed corneas (n = 3) receiving no hydroxamate (n = 3), then were assayed by following the InnoZyme kit instructions to yield the maximal ADAM17 activity value induced by the 2-hour NM exposure used in all experiments. This was found to be 118 ng/mL of active ADAM17. Next, after a 2-hour NM exposure, sets of corneas were treated three times over the course of 22 hours with 20 μL solutions to deliver 0.3, 1, 3, 10, 30, or 100 nmol doses to three corneas for each concentration of each of the four hydroxamates. The first of the three treatments was at 2 hours post NM exposure, the second 8 hours later, and the third 9 hours later. Five hours after this third hydroxamate application, proteins were extracted from each of the corneas and assessed for ADAM17 activity with the kit. The values determined for each of the sets of three corneas were averaged and plotted as a semilog graph with ADAM17 activity (y-axis) versus the log dose of each concentration of hydroxamate (x-axis). This showed the decrease in enzyme activity as a function of decreased levels of hydroxamate. The half-maximal ADAM17 concentration was read off the graph at the point where the y-axis was 59 ng/mL (i.e., half the maximal ADAM17 activity with no treatment). The 3-nmol dose was found to be most effective for each hydroxamate, and was used for all subsequent analyses using the same treatment schedule.
Hematoxylin and Eosin (H&E) Histology
Frozen sections (10 μm) were fixed in Pen-Fix (Thermos Fisher Scientific, Bellefonte, PA, USA) and stained with a modified H&E staining procedure as previously reported.10 The H&E-stained corneas were viewed with a Leica microscope using ProgRes software (Jenoptik, Jena, Germany). Digital images were captured by using an ×10 and an ×40 objective. The measurement scale was taken from the stage micrometer.
Assessment of the Percentage of Epithelial–Stromal Attachment
To view a whole central cornea across its diameter, composites images of the H&E-stained histologic sections across the entire width were made by using the ×10 micrographs, overlapping images of the sections with Adobe Photoshop CS3 (Adobe Systems, Inc., San Jose, CA, USA). The attached and detached areas of these Photoshop-derived full-width central corneas were measured in millimeters by using the program's rulers. As verification, composite images of the corneas across the entire diameter were enlarged for printing on 8.5 × 11–inch paper to ensure proper image alignment. The percentage detachment and remaining epithelial–stromal attachment preserved after NM exposure, plus and minus countermeasures, was calculated as follows:
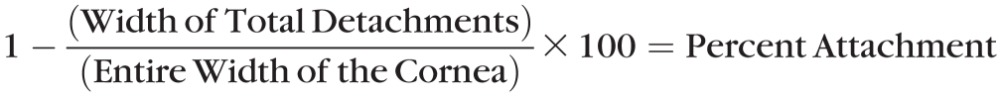 |
Next, to evaluate a correlation between the ADAM17 activity and epithelial–stromal separation, histograms of the percentage epithelial–stromal attachment (y-axis) were plotted for each of three NM-exposed corneas after the four applications of 3 nmol hydroxamate and were compared with the determined ADAM17 activity.
Immunofluorescence
For immunodetection of ADAM17, OCT-embedded sections on slides were first fixed in −20°C methanol for 10 minutes. Nonspecific binding was blocked for 1 hour with 5% normal goat serum (NGS) in PBS with 0.05% Tween-20 (PBST). A mouse monoclonal antibody against the ectodomain (amino acids 18–671) of human ADAM 17 (5 μg/mL in 1.5% NGS, MAB9304; R&D Systems, Minneapolis, MN, USA), which was found to detect only the active enzyme by immunofluorescence, was applied to the slides for a 1-hour incubation at room temperature, then the slides were washed three times for 10 minutes in PBST. For negative control slides, the same volume of PBST was applied to sections as that of primary antibody used on test sections, followed by the same wash volume. Goat anti-mouse IgG conjugated to AlexaFluor488 (1:1000; Invitrogen, Carlsbad, CA, USA) in 1.5% NGS was applied for 1 hour at room-temperature incubation. After washing with PBST three times for 5 minutes, 0.4 mg/mL DAPI was applied to sections for 5 minutes to counterstain the nuclei. Prolong Gold was used in coverslipping the slides.
CellTracker TM DiI staining was also performed on 7-μm-thick OCT sections of naïve control and NM-treated rabbit corneas. Slides of these were fixed in cold 2% paraformaldehyde for 15 minutes, then incubated with 5 μM Perchlorate for 20 minutes at room temperature. This was followed by three 10-minute washes in PBS. Fifty microliters of Prolong Gold Antifade Mountant with DAPI were added to the sections before coverslipping. An Olympus Epi-fluorescent microscope was used to collect fluorescent images.
Protein Isolation
Frozen corneas were first ground with a pulverizer gun and then were ground in liquid nitrogen with a mortar and pestle. The powdered corneas were added to extraction buffer (25 mM Trizma Base, pH 7.4, 200 mM NaCl, 10 mM EDTA), 1% Triton X-100 containing a protease inhibitor cocktail (Roche, Nutley, NJ, USA) and furin inhibitors I and II (R&D Systems), following the manufacturer's instructions. Next, three cycles of polytron homogenization at 20,000 rpm was performed on wet ice. Each homogenization was for 30 seconds with a 0.5-mm probe. Corneal extracts were then centrifuged at 12,000g for 30 minutes at 4°C, then placed on ice for the collection of supernates. These were quantified by using the BCA Protein Assay Reagent (Pierce Chemical, Rockford, IL, USA), then stored at −80°C until use.
Western Analysis
Pooled corneal extract (10 μg per sample) in 1x sample buffer (63 mM Tris HCl, pH 6.8, 10% Glycerol, 2% SDS, 0.0025% Bromophenol Blue) were analyzed on 7.5% SDS-polyacrylamide gels and then transferred onto nitrocellulose membrane (Bio-Rad, Hercules, CA, USA) by 100-volt transblotting for 1 hour. Nonspecific binding was blocked by soaking the blot at 4°C in 5% bovine serum albumin, 0.02% NaN3 overnight. Blots were washed three times, 10 minutes each, with TBST (50 mM Tris, pH 7.6, 150 mM NaCl, [05% Tween-20]), and were incubated with a mouse monoclonal antibody against human ADAM17 amino acids 215 to 315 (1 μg/mL, ab57484; Abcam, Cambridge, MA, USA) for 1 hour at room temperature. Blots were washed 3 × 10 minutes with TBST, then incubated with goat anti-mouse IgG conjugated to horseradish peroxidase antibody (1:20,000; BioRad) incubation for 1 hour at room temperature. Proteins were detected by using chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA).
Statistics
All data from the epithelial–stromal attachment/detachment study, as well as the ADAM17 activity assays, were derived from at least three biological replicates, but often five replicates. Data were analyzed by using either one-way analysis of variance (ANOVA) followed by Duncan's multiple comparison tests or the unpaired Student's t-test. The NM plus inhibitor hydroxamate samples were compared to NM-exposed samples that were not treated with hydroxamate. A P value ≤ 0.05 (marked as *) was considered statistically significant.
Results
CEES and NM Injury in Corneal Organ Cultures
The phenotype of CEES and NM injury of organ-cultured corneas has previously been demonstrated to be comparable to that of in vivo SM-exposed corneas, with separation of the epithelial–stromal junction followed by repair. The differences in the effect of each vesicant are that the separations are extensive with SM, moderate to extensive with NM, and mild to moderate with CEES.10 For the current work, 20 nmol CEES or 100 nmol NM was applied in a volume of 20 μL to the central corneas of rabbit organ cultures (Fig. 1). The agents remained on the cornea for 2 hours, mimicking the time it takes to feel the exposure, then were washed off. Medium was then changed, applying it as a wash over the central cornea, using a volume that brought it up to the corneal–scleral junction, keeping the cornea exposed to air. Injury was assessed 24 hours after the start of the exposure and demonstrated the phenotypic histologic hallmarks of ocular mustard exposure (Fig. 2). This was assessed across the entire diameter of the corneas, and included the following: (1) hyperplasia of the epithelial layer, apparent by the increase in the number and depth of epithelial cells pushing down into the stroma. We refer to this as “downward hyperplasia” and it was the first microscopic indication of injury. Although unexposed corneas showed some degree of downward hyperplasia (Fig. 2A), it was never as extensive as in the mustard-exposed corneas, where it could be quite pronounced at 24 hours after CEES (Figs. 2B, 2D) or NM exposure (Figs. 2C, 2E); (2) basal cell nuclei rising up toward the top of the basal epithelial cells (yellow arrows in Figs. 2D, 2E, 2H); and (3) epithelial–stromal separation (best seen in Fig. 2C, where the epithelial stromal detachment is 68.8% for this cornea, i.e., 31.2% attachment). The CEES exposures resulted in minimal epithelial–stromal separation, but the corneas exposed to NM underwent a 45% to 90% epithelial–stromal separation (average separation = 65%). The cell layer attachment of three corneas exposed to NM (Fig. 2F) was only 10% to 55% (average remaining attachment = 35%), which was a statistically significant difference when compared to the unexposed corneas.
Figure 1.

Schematic diagram of the rabbit corneal organ culture system. The light gray at the bottom of the cornea represents ∼2 mm of sclera remaining after dissection. Medium (∼2 mL) is added only as high as the top of the scleral rims of the corneas placed in a pyrex culture dish.
Figure 2.

(A–C) Composites of overlapping low-magnification H&E-stained micrographs, representing sections across the diameter of a naive unexposed cornea and corneas exposed to CEES and NM. (D, E) High magnification of a region of a CEES-exposed cornea and of an NM-exposed cornea, showing the downward hyperplasia of the epithelial cells after mustard exposure, as well as rising of the nuclei in the basal epithelia in the NM-exposed sample. (F) Histogram representation of epithelial–stromal attachment derived from analysis of four unexposed corneas in culture for 24 hours, and from four corneas exposed to NM for 2 hours, followed by washing the cornea with medium, replacing medium, and incubating for an additional 22 hours at 37°C. The y-axis units are percentage epithelial–stromal attachment. The cornea in (C) has 68.8% of the epithelium detached from the cornea, that is, 31.2% epithelial–stromal integrity. Data are expressed as means ± SD and analyzed by using two-sided Student's t-tests. A value of P < 0.05 was considered statistically significant. *P < 0.05. (G, H) Sections of unexposed and NM-exposed corneas at high magnification, stained at 24 hour post exposure with DiI (red) to indicate lipid membranes and DAPI (blue) to show the nuclei. Note how the nuclei have risen in the cell by comparing (H) to (G).
Nitrogen Mustard Exposure Induces ADAM17 Activation in Rabbit Corneas
The epithelial–stromal junction is maintained by anchoring complexes. These are composed of three basic structures: the hemidesmosomes, the anchoring filaments, and the anchoring fibrils.11 Transmembranous collagen XVII is one of the hemidesmosomal components that provides a strong attachment to the anchoring filaments, supporting the integrity of the epithelial–stromal border. We postulated that, because ADAM17 (i.e., TACE or TNF-α converting enzyme) is one of the enzymes capable of cleaving collagen XVII,5 mustard-induced corneal microblistering is likely, in part owing to activation of ADAM17. This activation would result in cleavage of collagen XVII and would facilitate separation of the epithelium from the stroma. We reasoned that inhibiting this enzyme could help preserve epithelial–stromal integrity. To assess the effect of NM on ADAM17 activation, the anti-human ADAM17 antibody in the InnoZyme TACE activity kit would need to detect rabbit ADAM17, since there were no commercially ADAM17 activity assay kits for detection of the rabbit enzyme. A phylogenetic analysis12 indicated that ADAM17 does not share a great deal of sequence identity with other ADAMs. As described in Materials and Methods, the human and rabbit ADAM17 showed 95.5% similarity, with the transmembrane domain being 100% conserved. The next most conserved region (98.8% identity) was the peptidase M12B domain located in the catalytic region (aa 223–474 in human ADAM17).
With this degree of cross-species identity, the InnoZyme TACE activity kit for human ADAM17 was tested and was found capable of assessing rabbit ADAM17 activity. The enzyme's activity in unexposed organ-cultured rabbit corneal extracts (n = 3) was compared to the activity assessed from extracts of three corneal cultures exposed to NM for 2 hours, followed by washing and 22 hours of incubation at 37°C. Two important facts were determined from this: one was that organ culturing the corneas for 24 hours did not appreciably activate ADAM17. The other was that 22 hours after a 2-hour exposure to NM, the corneal organ cultures showed a significantly elevated ADAM17 activity, on the order of 75 to 80 times greater than that of unexposed corneas (Fig. 3A). Next, other corneas (n = 3 for each time point) were used to determine how fast ADAM 17 was induced by NM. To one set of corneas, NM was applied then immediately removed (0 minute); to another set of corneas, NM was allowed to remain on the corneas for 5 minutes; and to a third set, the NM was allowed to remain on the corneas for 10 minutes before washing the corneas. Each set was immediately assayed for ADAM17 activity. When NM was applied to the corneas and immediately removed (0 minute), ADAM17 was slightly activated, increasing to 40 ng/mL as compared to 33 ng/mL for the unexposed corneas (Fig. 3B). However, the set of corneas exposed to NM for 10 minutes showed significantly increased levels of active ADAM17.
Figure 3.

ADAM17 activity in unexposed and NM-exposed corneas. (A) Six corneas were placed in organ culture at 37°C. Three were unexposed for 2 hours, while the other three were exposed to 100 nmol NM for 2 hours. All corneas were washed and were given a change of medium, then incubated for 22 additional hours at 37°C before protein extraction and assaying for ADAM17 activity. Data were compared for exposed and unexposed corneas and are expressed as means ± SEM, analyzed by using two-sided Student's t-tests. A value of P < 0.05 was considered statistically significant (*P < 0.05). (B) Nine corneas were placed in organ culture. To three corneas, 100 nmol NM was applied and immediately washed off. To three others, NM was applied for 5 minutes, then immediately washed off; and to three others, 100 nmol NM was applied for 10 minutes and immediately washed off. After this washing, proteins were immediately extracted from the corneas and subjected to ADAM17/TACE activity assays. Data are presented as means ± SEM and analyzed by ANOVA followed by pairwise comparison to the 0-minute NM exposure samples in (B) using Duncan's test. The 10-minute NM exposures were statistically significant (*P < 0.05). Note that the unexposed cornea value in (A) expressed 33 ng/mL active ADAM17, and the 0-minute sample (NM applied and immediately washed off) in (B) showed 40 ng/mL active ADAM17. For both (A) and (B), assessments were made by using the InnoZyme TACE activity assay kit. The y-axis represents ng/mL of active ADAM17.
Based on the sequence identity between human and rabbit ADAM17, two monoclonal antibodies against human ADAM17 were purchased for use on the rabbit cornea sections. One antibody was from Abcam and was generated against a recombinant peptide representing amino acids 215 to 315 in the catalytic domain of the human enzyme. While this antibody was useful for probing Western blots, it only detected the appropriate-sized band for the rabbit inactive proenzyme (Fig. 4A). This assessment is based on the following: human ADAM17 migrates on polyacrylamide gels as a band somewhat larger than its predicted size owing to six posttranslational N-glycosylations. This form is usually referred to as the 130-kDa form of the molecule, which is the molecular weight of the product observed in Figure 4A. This form should predominate in uninjured corneas, as was used here. The human active ADAM17 migrates at approximately 90 to 95 kDa, but no such band appeared on Western blots of NM-exposed corneas. We were unable to find any antibody that would detect convincingly 90- to 95-kDa active form bands on Western blots. The intense 50-kDa band on the Western blot is most likely the IgG in the rabbit corneal extract reacting with the secondary antibody. An antibody against ADAM17 from R&D Systems, generated against a recombinant peptide representing human ADAM17 amino acids 18 to 675 (i.e., the entire extracellular domain of the enzyme), while not ideal for Western blots, worked for immunofluorescence analysis but only detected active ADAM17 (Fig. 4B)
Figure 4.

(A) An anti-human ADAM17 monoclonal antibody generated in mice was used as a probe on Western blots of proteins extracted from unexposed rabbit corneal organ cultures. The 130-kDa band is the molecular weight of the inactive N-glycosylated proform, normally seen for ADAM17. The faint 95-kDa band agrees with the size of the enzyme lacking the pro domain, that is, the activated form that is not prevalent in the unexposed sample. Because of the similarity of rabbit and mouse species, the 50-kDa band likely represents cross-reactivity with the goat anti-mouse IgG secondary antibody. (B) Immunofluorescence of corneas using an anti-human ADAM17 ectodomain antibody. This visualizes active ADAM17 immunoreactivity, detected with a goat anti-mouse IgG conjugated to AlexaFluor488. Nuclei are stained with DAPI. Immunodetection of ADAM17 was low in unexposed corneas, but in NM-exposed corneas, it was highly detectible (arrows) in the region of the basement membrane zone, where the enzyme would be concentrated for cleavage of collagen XVII to release cells.
In exposed corneas, the intense fluorescent signal was at the basement membrane zone, the expected site for the immunoreactivity, that is, the basement membrane zone where the enzyme would need to be positioned in order to degrade collagen XVII (Fig. 4B). This antibody did not appreciably detect the proform of ADAM17 in unexposed corneas. However, the specificity of the antibody for recognizing only the active form of ADAM17 in rabbit tissue by immunofluorescence worked to our advantage as a useful tool to gauge injury in corneal sections. The attenuation or lack of this fluorescent signal always corresponded with better histologic appearance of the corneal epithelial–stromal junction.
ADAM17 Inhibitors
Since there are no FDA-approved therapies for mustard injury, our goal in the last several years has been to identify potential drugs that could be developed into effective ocular vesicant therapies. Since ADAM17 activity was shown to be upregulated after mustard exposure, inhibiting it might attenuate mustard injury or might facilitate wound healing after injury. The enzyme's catalytic activity is dependent on Zn+2. Hydroxamates are good candidate inhibitors of ADAM17 because they complex Zn+2.13–15 Therefore, three olvanil compounds previously reported to be effective topical pretreatments for sulfur mustard exposure of skin16 were resynthesized to include hydroxamate moieties (see Supplementary Material). These were designated NDH4385, NDH4409, and NDH4417. In addition, a hydroxamate known to be an inhibitor of matrix metalloproteinases (MMPs), which has been used as an anticancer drug (Abbott Laboratories ABT-518),17 was also synthesized by using a modified protocol (see Supplementary Material). This product was designated NDH4450. The hydrophilicity (cLogP) for three of the hydroxamates was below 5.0, suggesting that at least three would likely be well absorbed by tissue (Fig. 5A). The structures of these drugs are shown to the right of the cLogP values, and their synthesis is provided in the Supplementary Material.
Figure 5.

Structure and activity of hydroxamate drugs. (A) Structures of the four hydroxamates are shown on the right. The lipophilicity of each compound is indicated by the cLogP value (increased value indicates increased lipophilicity). The IC50 is the concentration of inhibitor (in nanomoles) necessary to decrease ADAM17 activity to 50% of its initial value when no inhibitor is added, as determined by the graph in (B). (B) Dose-response curves for NM-exposed corneas plus and minus different concentrations of inhibitors. Since the maximal ADAM17 activity with no inhibitor was 118 ng/mL, the IC50s were the concentrations of inhibitors yielding 59 ng/mL ADAM17 activity, read off the graph. There was no correlation between lipophilicity and IC50 (note that inhibitor concentrations are plotted on a log scale). The linear range of ADAM17 activity for all hydroxamates fell in the range of 0 to 3 nmol of drug, applied four times over the course of 22 hours post NM exposure. Data were analyzed by using one-way ANOVA followed by Duncan's multiple comparison tests. Dose-response studies (0.1, 0.3, 1, 3, 10, 30, and 100 nmol) of NM plus inhibitor samples were compared among data from the same inhibitor treatment. A value of P < 0.05 was considered statistically significant (*P < 0.05; color of * corresponds to the color of the line indicating each inhibitor as shown at the bottom).
Dose Response of Hydroxamates for Inhibition of ADAM17 Activity
Sets of corneal organ cultures (n = 3) were used to test specific doses (0.3, 1, 3, 10, 30, or 100 nmol) of each of the four hydroxamates (i.e., three corneas for each of six concentrations of each of four hydroxamates = 72 corneal organ cultures; experimental design as described in Materials and Methods). Figure 5B shows the dose response for activity of the enzyme. Twenty-four hours after NM exposure, when no hydroxamate was applied to corneas, the assayed extracts all contained an average of 118 ng/mL active ADAM17, defining the half-maximal inhibitory concentration (IC50) for each hydroxamate as 59 ng/mL. The concentration of inhibitor in nanomoles (x-axis) that corresponded to the 50% ADAM17 activity level was determined from the plotted graph. NDH4417 had the lowest IC50, being 0.52 nmole of drug. NDH4385, NDH4409, and NDH4450 required 1.0 nmol or more to reach half-maximal inhibition of ADAM17, as indicated by the IC50s in Figure 5A. Overall, according to the statistical analysis, the most effective ADAM17 inhibitor was NDH4417 (Fig. 5B, green line). ADAM17 activity showed a linear dose-dependent inhibition up to the 3-nmol concentration, with NDH4417. Doses of 10 nmol NDH4417 or greater did not improve inhibition, and in fact hampered it. For NDH4385 (Fig. 5B, blue line), inhibition leveled off at doses higher than 3 nmol, and at the 10-nmol doses of NDH4450 (purple) and NDH4409 (red line), inhibition was slightly less effective than the 3-nmol dose. In all, NDH4417 was the best inhibitor of ADAM17 activity.
Assessment of Healing of Mustard-Exposed Corneal Cultures Treated With Hydroxamates
To evaluate whether inhibition of ADAM17 correlated with better healing after NM exposure, the four hydroxamates were individually applied to three to five sets of corneas at 2 hours post NM exposure. The 3-nmol dose for the hydroxamates was used, delivered in 20 μL four times over the course of the 22 hours after the 2-hour exposure. This dose for each was chosen because it was in the linear range of inhibition for each hydroxamate, being optimal for NDH4417. In addition, this dose gave the best spread for the levels of inhibition of the four hydroxamates, and it would therefore give a range of ADAM17 inhibition that should affect the resulting degree of epithelial–stromal separation. Our first measure of attenuated injury was from calculating the preservation of epithelial–stromal integrity at 24 hours post NM exposure from the histology. Figure 6A demonstrates that NDH4385, a treatment that gave the least ADAM17 inhibition at 3-nmol doses after NM exposure, did not effectively help retain epithelial–stromal integrity. When compared to NM-exposed corneas with no subsequent treatment (compare to Fig. 2C), there was little difference between an exposed cornea receiving no therapy and one receiving four 3-nmol doses of NDH4385 over a 24-hour period. With NDH4409 (Fig. 6B) and NDH4450 (Fig. 6D) there was some improvement in epithelial–stromal integrity as observed by more areas where the cell layers remained attached. As indicated by the ADAM17 activity assays, corneas treated with 3-nmol doses of NDH4417 four times over a 22-hour period showed the highest level of epithelial–stromal integrity at 24 hours post NM exposure (Fig. 6C). Higher magnifications of other H&E-stained corneas, untreated or treated with the hydroxamates, are shown in Figure 7. These are accompanied by immunofluorescence analyses of sections reacted with the R&D ADAM17 antibody, which detects the active enzyme on sections. The series of corneal sections in Figure 7A across the top of the figure represent corneas that receive no hydroxamate (indicated as “no drug”). The two corneas in the left two panels were not exposed to NM and showed 100% epithelial–stromal integrity in the H&E section and no green fluorescence in the companion section, suggesting ADAM17 activity was minimal. The two panels on the right side of Figure 7A were exposed to NM and received no hydroxamate. The H&E section showed approximately 85% detachment (15% attachment) of the epithelium at 24 hours after NM exposure. Sections of these exposed but untreated corneas reacted with the ADAM17 antibody, showing intense green fluorescence at the epithelial–stromal junction across most of the cornea. Figures 7B through 7E follow the same setup, with two unexposed cornea sections that were treated with a hydroxamate on the left side, and NM-exposed corneas treated with the same hydroxamate on the right side. The H&E-stained sections in Figure 7B indicate that treatment with NDH4385 after NM exposure has some small ability to prevent total cell layer separation 24 hours later. The immunofluorescence (IF) images of both the unexposed and NM-exposed samples in Figure 7B show green fluorescence at the epithelial–stromal junction, albeit less in the unexposed corneas. This suggests that NDH4385 alone may slightly activate ADAM17 activity at this dose. Comparing the NM-exposed corneas receiving no treatment and those receiving NDH4385, however, it is clear that ADAM17 immunofluorescence does indicate that the enzyme was somewhat attenuated with NDH4385 treatment. In the Figure 7C panels are corneas treated with NDH4409. Here the unexposed corneas appear normal, but after NM exposure, the H&E and IF samples have the typical appearance of epithelial and stromal layers that are likely to detach. In the Figure 7D panels, the corneas were treated with NDH4417, the best ADAM17 inhibitor as evaluated by ADAM17 activity assays. These corneas showed a good outcome. The nuclei did not rise in the basal epithelial cells of the NM-exposed corneas treated with NDH4417, and the immunofluorescent signal for ADAM17 was attenuated compared to that of exposed corneas receiving treatment with other hydroxamates. The NDH4450-treated corneas (Fig. 7E, panels) have some green fluorescence in both the unexposed and NM-exposed corneas. In the unexposed cornea, this fluorescence was close to the basal cell nuclei, unlike the NM-exposed cornea, where the fluorescence was distant from the basal cell nuclei. This was supported by the H&E panels of the unexposed and NM-exposed corneas, which showed nuclei higher in the basal epithelial cells after NM exposure. The fluorescence in unexposed corneas suggests NDH4450 may also activate ADAM17 to some degree, and therefore it would not be an ideal therapy with this dosing regimen. The best result was clearly obtained with NDH4417 (Fig. 7D). At 24 hours post NM exposure, the basal epithelial cell nuclei remained in place in the H&E sections and the ADAM17 antibody produced a minor, patchy fluorescent signal at the epithelial–stromal border, much attenuated compared to NM-exposed corneas receiving no hydroxamate treatment.
Figure 6.

Overlapping H&E-stained micrographs (across the diameter of the central cornea) for corneas exposed to NM for 2 hours, then treated four times over the course of the subsequent 22 hours with ADAM17 inhibitors at 3 nmol/dose. (A) NDH4385 shows extensive epithelial–stromal detachment after 22 hours of hydroxamate treatment. (B) NDH4409 demonstrated approximately 40% detachment. (C) With NDH4417 treatment after NM exposure, the corneal epithelial–stromal junction appeared nearly perfectly intact. Separation was on the order of 18% or less for N = 3 corneas. (D) NDH4450-treated corneas showed the epithelium to be partially detached.
Figure 7.

Higher magnifications of regions of H&E-stained corneas (first and third images in each panel) and corresponding ADAM17 immunofluorescence patterns (second and fourth images in each panel) for unexposed and NM-exposed corneas, plus and minus hydroxamate treatments. (A) Sections of unexposed cornea are on the left; NM-exposed corneas are on the right (no hydroxamate therapy was applied to these). (B) Left: Unexposed corneas treated with NDH4385. Right: NM-exposed corneas that received NDH4385 treatment. (C) Left: Unexposed corneas treated with NDH4409. Right: NM-exposed corneas that received NDH4409 treatment. (D) Left: Unexposed corneas treated with NDH4417. Right: NM-exposed corneas treated with NDH4417. (E) Left: Unexposed corneas that received NDH4450 treatment. Right: NM-exposed corneas that received NDH4450 treatment.
Further support for NDH4417 being the best of the four hydroxamates for preserving epithelial–stromal integrity after mustard exposure came from comparing graphs of the degree of epithelial–stromal separation in unexposed, NM-exposed, and NM-exposed/hydroxamate-treated corneas, with those derived from ADAM17 activity assays (Fig. 8). The percentage separation of cell layers was calculated for unexposed corneas plus and minus hydroxamates, as well as corneas exposed to NM, plus and minus hydroxamate treatments. Unexposed corneas showed no separation of the cell layers, while the average of the three NM-exposed corneas was 85% separation. The remaining histograms in Figure 8A show unexposed and exposed corneas treated with hydroxamates. The percentage separation was nearly zero when unexposed corneas were treated with NDH4409 or NDH4417. However, NDH4450 caused a small level (∼3%) of epithelial–stromal separation when applied to naïve unexposed corneas for 22 hours, and NHD4385 caused approximately 7% separation. There was no significant difference between NM-exposed corneas and NM-exposed corneas treated with NDH4385. In contrast, there was a significant difference between NM-exposed cornea and corneas exposed to NM followed by treatment with NDH4409, NDH4417, and NDH4450. Epithelial stromal separation was reduced to approximately 30% when NDH4450 and NDH4409 were used after NM exposure. Even better was that NM-exposed corneas treated with NDH4417 retained most of their epithelial–stromal integrity, having only approximately 16% separation of the cell layers.
Figure 8.

Decreased epithelial–stromal separation (i.e., preservation of epithelial–stromal integrity) roughly approximates inhibition of ADAM17 activity. (A) Histograms indicating epithelial–stromal separation after 22 hours of hydroxamate treatment, applied to unexposed corneas and to NM-exposed corneas. Separation is calculated as described in Materials and Methods. (B) Identically exposed and treated corneas were extracted for ADAM17 activity assays. Data from (A) and (B) are presented as means ± SEM. Corneas exposed to NM then treated with inhibitors (NDH 4385, NDH 4409, NDH 4417, and NDH 4450) were compared to NM-exposed samples that were not treated with any hydroxamate. One-way ANOVA followed by Duncan's multiple comparison tests compared to NM-exposed alone samples were used as statistical analysis of the data. A P value < 0.05 was defined as statistically significant (a P value < 0.05 was marked with *). Comparing the corresponding samples from both sets of histograms demonstrates that, as ADAM17 activity decreases, epithelial–stromal separation decreases, that is, preservation of cell layer integrity increases. NDH4417 was most effective at ameliorating separation.
ADAM17 activity measured by the assay kit also demonstrated significant differences, and approximated the epithelial–stromal separation pattern (compare Fig. 8B to Fig. 8A). The enzyme activity assay also roughly agreed with the immunofluorescent signals in the rightmost panels of Figure 7D, where NDH4417 gave the least immunofluorescent signal, and NDH4409 (Fig. 7C) gave the second least immunofluorescence. Overall, NDH4417 was the hydroxamate that best inhibited NM damage to the epithelial–stromal junction, and best inhibited ADAM17 activity. These data strongly suggest that ADAM17 activity is an important contributor to epithelial–stromal separation from mustard exposure.
Discussion
When considering how to identify good therapies for ocular sulfur mustard injury, it is clear that epithelial cell culture models fall short, as they cannot reveal how effectively a drug will preserve or improve the integrity of the major region that mustard compounds affect in the cornea, the epithelial–stromal junction. In the experiments described here, rabbit corneas were put in organ culture, wounded with a mustard compound, then treated with hydroxamates to determine whether the inhibition of ADAM17 would ameliorate the injury. Other zinc-dependent enzymes are likely also affected by these, but our focus was on how each hydroxamate affected ADAM17 by using a specific activity assay kit. The hydroxamates were used at a concentration that would result in a range of healing based on their IC50s, which was calculated from Figure 5.
Since contracting in vivo SM exposure studies to approved facilities, such as MRI Global or Battelle, are cost prohibitive, we first perform tests on rabbit corneal organ cultures, moving only the best candidates forward to the SM studies on in vivo animal eyes. An advantage of this is that, using NM as the vesciant, the mechanisms of mustard injury can be identified, and less effective drug candidates can be assessed before proceeding to the animal studies. We have used this approach to demonstrate that NM causes corneal injuries analogous to those of SM, differing only in degree.10 It induces the loss of the corneal epithelium, as SM does, and the microscopic and ultrastructural phenotypes are the same. Here we used NM at a dose that, at 24 hours post exposure produced a range of 50% to 85% epithelial–stromal separation without therapeutic intervention, averaging approximately 65%. Other advantages of the organ culture system are that (1) purchasing rabbit eyes from a company that sells rabbits for food is in line with efforts to decrease the number of animals required for validating potential drugs; (2) cultures are easy to set up, and can be used for experiments that run for up to 1 to 2 weeks; (3) application of a vesicant to the corneal organ cultures is simple compared to applying it to live animal corneas; and (4) the expense of running corneal organ culture experiments is a fraction of that for in vivo animal exposures. Hundreds of organ cultures can easily be done to narrow down potential therapies to the best candidate drug before performing the in vivo rabbit cornea exposures. Disadvantages are that (1) rabbits are the main host for generating polyclonal antibodies, and therefore very few antibodies are generated specifically against rabbit proteins; (2) any contribution to healing provided by intact corneal nerves is lost by the dissection to set up the cultures; and (3) the lack of an immune system may result in the cultures not responding in a perfectly normal fashion. Still, the organ culture model is an effective first approach for screening potential drugs for their ability to preserve epithelial–stromal integrity before live animal testing, as shown by experiments investigating the therapeutic efficacy of doxycycline,10,18 silibinin, and dexamethasone18 after mustard exposure. The preliminary organ culture experiments have allowed these drugs to move forward to Battelle-contracted in vivo rabbit SM eye exposure studies, similar to those contracted by Israeli scientists studying ocular mustard injury.19,20
Mustard compounds induce visible blisters in skin,21–23 but microbullae in the cornea,24 in the same plane of the skin and corneal structure as does bullous pemphigoid25 and junctional epidermolysis bullosa.26 This involves disruption of the anchoring complex that holds the corneal or skin epithelial layer firmly to the stromal or dermal layer below.23 Therefore, molecules that “tack down” the epithelium of skin to the dermis11 and the corneal epithelium to the stroma,27 such as α6β4 integrin and collagen XVII, molecules defective and/or nonfunctional in bullous pemphigoid4,28 and in certain forms of epidermolysis bullosa,29 are disrupted by mustards. Anchoring complex components such as α6β4 integrin and collagen XVII are cleaved after injury to allow the basal epithelial cells to migrate for wound closure. Collagen XVII is cleaved by ADAM17,5,6 and other ADAMs, and α6β4 integrin is cleaved by MMP9.30 We have previously shown that MMP9 is activated by mustard exposure and that inhibiting it improves healing of ocular vesicant injury,10 and here we showed that using a hydroxamate to inhibit ADAM17 is also effective. While ADAM17 is not the only enzyme able to cleave collagen XVII,5,6 its definitive role as a collagen XVII sheddase has been verified in ADAM17-deficient mice.6 As compared to age-matched normal murine keratinocytes, shedding of collagen XVII is ∼40% reduced in ADAM17-deficient cells. Therefore, it made sense to investigate the impact of inhibiting ADAM17 on corneal healing after mustard injury. The hydroxamates have the ability to inhibit many Zn+2-containing metalloproteinases, and it is possible the improvement seen in corneal histology could be due to Zn+2 inhibiting these other enzymes, and not just ADAM17. However, our goal here was to concentrate on the role that ADAM 17 plays in mustard injury.
ADAM17 activation was detectable within minutes of applying NM to the corneas. ADAM17 is an enzyme that plays an important role in healing by releasing cytokines such as TNF-α,14,15,31 TGF-α, heparin-binding EGF-like growth factor (HB-EGF), and other EGF receptor ligands necessary to initiate the healing process.8,32–35 It was important that we consider how to inhibit ADAM17 without totally impeding the initial steps in healing. Because ADAM17 activity was found to be high even a full day after exposure, and this was harmful to the integrity of the tissue, we were able to allow the wound healing functions of ADAM17 to begin and proceed unimpeded for 2 hours, and then inhibit the enzyme at later time points to reduce the microblistering from adhesion complex disruption, a later event. This regimen was effective in the organ-cultured corneas and identified NDH 4417 as a potential therapeutic for preserving epithelial–stromal integrity.
Since hydroxamates bind the catalytic zinc (II) in the active site of MMPs and ADAMs,36 we selected three compounds that were not MMP inhibitors but were effective topical anti-inflammatory pharmaceuticals in responding to 0.75 μmol 12-O-tetradecanoylphorbol-13-acetate or to SM exposure in the mouse ear injury model16,37,38 and had them synthesized with internal hydroxamate moities (see Supplementary Material and Laskin et al.39). These were designated NDH4385, NDH4409, and NDH4417. In addition, Abbott Pharmaceutical's clinically effective hydroxamate ABT-518 is known to possess anti-inflammatory activity,40,41 and was synthesized by using a modification of the published Abbott pathway42 and designated NDH4450. Because the IC50s were determined as different for each hydroxamate, this allowed us to use all of the compounds at the same concentration and achieve a broad range of corneal healing from identical exposures. Because of the complexity of sulfur mustard alkylation of cell products, the mechanisms responsible for the pathology of mustard injury are not clearly understood. Therefore, work demonstrating that inhibition of MMP9 improves healing of mustard injury10,19 and the work presented here, indicating that ADAM17 inhibition also contributes to healing, offer the beginnings of an arsenal of potential therapies for mustard exposure. Studies with hydroxamate NDH4417 will move forward toward evaluating its potential to improve recovery of rabbit eyes exposed in vivo to SM. A final point to make was that the goal of the work presented here is an endeavor to identify therapies for initial mustard exposures, such as those after a terrorist attack. The experiments assessing NDH4417 have been directed toward developing a drug for a “fast response” situation. We have no evidence that treatment with NDH4417 would have an impact on the long-term effects of SM exposure, such as corneal conjunctivalization or limbal stem cell deficiency. However, in the wake of having no therapy at all for SM exposure, even a drug that reduces injury in the short term would be desirable.
Supplementary Material
Acknowledgments
The authors thank Lakshmi Raman who passed away on December 19, 2010. She was a friend and colleague who participated in many conversations about this work, providing insights. We also greatly appreciate the help of Linda Everett in assisting with submission of the manuscript.
Supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases U54AR055073, National Institute of Environmental Health Sciences P30ES005022, and National Eye Institute EY009056.
Disclosure: A. DeSantis-Rodrigues, None; Y.-C. Chang, None; R.A. Hahn, None; I.P. Po, None; P. Zhou, None; C.J. Lacey, None; A. Pillai, None; S.C. Young, None; R.A. Flowers II, None; M.A. Gallo, None; J.D. Laskin, None; D.R. Gerecke, None; K.K.H. Svoboda, None; N.D. Heindel, None; M.K. Gordon, None
References
- 1. Balali-Mood M,, Hefazi M. Comparison of early and late toxic effects of sulfur mustard in Iranian veterans. Basic Clin Pharmacol Toxicol. 2006; 99: 273–282. [DOI] [PubMed] [Google Scholar]
- 2. Etezad-Razavi M,, Mahmoudi M,, Hefazi M,, Balali-Mood M. Delayed ocular complications of mustard gas poisoning and the relationship with respiratory and cutaneous complications. Clin Experiment Ophthalmol. 2006; 34: 342–346. [DOI] [PubMed] [Google Scholar]
- 3. Kadar T,, Horwitz V,, Sahar R,, et al. Delayed loss of corneal epithelial stem cells in a chemical injury model associated with limbal stem cell deficiency in rabbits. Curr Eye Res. 2011; 36: 1098–1107. [DOI] [PubMed] [Google Scholar]
- 4. Diaz LA,, Ratrie H,, III, Saunders WS,, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera: immunolocalization of this protein to the hemidesmosome. J Clin Invest. 1990; 86: 1088–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franzke CW,, Bruckner-Tuderman L,, Blobel CP. Shedding of collagen XVII/BP180 in skin depends on both ADAM10 and ADAM9. J Biol Chem. 2009; 284: 23386–23396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franzke CW,, Tasanen K,, Schacke H,, et al. Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J. 2002; 21: 5026–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Loffek S,, Hurskainen T,, Jackow J,, et al. Transmembrane collagen XVII modulates integrin dependent keratinocyte migration via PI3K/Rac1 signaling. PLoS One. 2014; 9: e87263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin J,, Yu FS. ERK1/2 mediate wounding- and G-protein-coupled receptor ligands-induced EGFR activation via regulating ADAM17 and HB-EGF shedding. Invest Ophthalmol Vis Sci. 2009; 50: 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flicek P,, Amode MR,, Barrell D,, et al. Ensembl 2012. Nucleic Acids Res. 2012; 40: D84–D90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gordon MK,, Desantis A,, Deshmukh M,, et al. Doxycycline hydrogels as a potential therapy for ocular vesicant injury. J Ocul Pharmacol Ther. 2010; 26: 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerecke DR,, Gordon MK,, Wagman DW,, Champliaud MF,, Burgeson RE. Hemidesmosomes anchoring filaments and anchoring fibrils: components of a unique attachment complex. : Yurchenco PD,, DE Birk,, Mecham RE, Extracellular Matrix Assembly and Structure. Orlando, FL: Academic Press; 1994: 417–439. [Google Scholar]
- 12. Edwards DR,, Handsley MM,, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008; 29: 258–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McGeehan GM,, Becherer JD,, Bast RC,, Jr, et al. Regulation of tumour necrosis factor-alpha processing by a metalloproteinase inhibitor. Nature. 1994; 370: 558–561. [DOI] [PubMed] [Google Scholar]
- 14. Mohler KM,, Sleath PR,, Fitzner JN,, et al. Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature. 1994; 370: 218–220. [DOI] [PubMed] [Google Scholar]
- 15. Mohler KM,, Torrance DS,, Smith CA,, et al. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993; 151: 1548–1561. [PubMed] [Google Scholar]
- 16. Casillas RP,, Kiser RC,, Truxall JA,, et al. Therapeutic approaches to dermatotoxicity by sulfur mustard, I: modulaton of sulfur mustard-induced cutaneous injury in the mouse ear vesicant model. J Appl Toxicol. 2000; 20 (suppl 1): S145–S151. [DOI] [PubMed] [Google Scholar]
- 17. Raja SN,, Surber BW,, Du J,, Cross JL. Synthesis of [3H]ABT-518, a matrix metalloproteinase inhibitor (MMPI) labeled in the phenyl rings. J Labelled Comp Radiopharm. 2009; 52: 98–102. [Google Scholar]
- 18. Tewari-Singh N,, Jain AK,, Inturi S,, et al. Silibinin, dexamethasone, and doxycycline as potential therapeutic agents for treating vesicant-inflicted ocular injuries. Toxicol Appl Pharmacol. 2012; 264: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horwitz V,, Dachir S,, Cohen M,, et al. The beneficial effects of doxycycline, an inhibitor of matrix metalloproteinases, on sulfur mustard-induced ocular pathologies depend on the injury stage. Curr Eye Res. 2014; 39: 803–812. [DOI] [PubMed] [Google Scholar]
- 20. Kadar T,, Dachir S,, Cohen L,, et al. Ocular injuries following sulfur mustard exposure--pathological mechanism and potential therapy. Toxicology. 2009; 263: 59–69. [DOI] [PubMed] [Google Scholar]
- 21. Hurst CG,, Newmark J,, Romano JAJ. Chemical terrorism. : Kasper D,, Fauci A,, Hauser S,, et al. , eds. Harrison's Principles of Internal Medicine 19/e. New York, NY: McGraw-Hill; 2015. [Google Scholar]
- 22. Momeni AZ,, Enshaeih S,, Meghdadi M,, Amindjavaheri M. Skin manifestations of mustard gas: a clinical study of 535 patients exposed to mustard gas. Arch Dermatol. 1992; 128: 775–780. [PubMed] [Google Scholar]
- 23. Petrali JP,, Oglesby-Megee S. Toxicity of mustard gas skin lesions. Microsc Res Tech. 1997; 37: 221–228. [DOI] [PubMed] [Google Scholar]
- 24. Petrali JP,, Dick EJ,, Brozetti JJ,, Hamilton TA,, Finger AV. Acute ocular effects of mustard gas: ultrastructural pathology and immunohistopathology of exposed rabbit cornea. J Appl Toxicol. 2000; 20( suppl 1): S173 –S. [DOI] [PubMed] [Google Scholar]
- 25. Razzaque AA. Diagnosis of bullous disease and studies in the pathogenesis of blister formation using immunopathological techniques. J Cutan Pathol. 1984; 11: 237–248. [DOI] [PubMed] [Google Scholar]
- 26. Fine JD,, Eady RA,, Bauer EA,, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008; 58: 931–950. [DOI] [PubMed] [Google Scholar]
- 27. Gipson IK,, Spurr-Michaud SJ,, Tisdale AS. Anchoring fibrils form a complex network in human and rabbit cornea. Invest Ophthalmol Vis Sci. 1987; 28: 212–220. [PubMed] [Google Scholar]
- 28. Giudice GJ,, Squiquera HL,, Elias PM,, Diaz LA. Identification of two collagen domains within the bullous pemphigoid autoantigen, BP180. J Clin Invest. 1991; 87: 734–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pohla-Gubo G,, Lazarova Z,, Giudice GJ,, et al. Diminished expression of the extracellular domain of bullous pemphigoid antigen 2 (BPAG2) in the epidermal basement membrane of patients with generalized atrophic benign epidermolysis bullosa. Exp Dermatol. 1995; 4: 199–206. [DOI] [PubMed] [Google Scholar]
- 30. Pal-Ghosh S,, Blanco T,, Tadvalkar G,, et al. MMP9 cleavage of the beta4 integrin ectodomain leads to recurrent epithelial erosions in mice. J Cell Sci. 2011; 124: 2666–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sakimoto T,, Yamada A,, Kanno H,, Sawa M. Upregulation of tumor necrosis factor receptor 1 and TNF-alpha converting enzyme during corneal wound healing. Jpn J Ophthalmol. 2008; 52: 393–398. [DOI] [PubMed] [Google Scholar]
- 32. Le Gall SM,, Auger R,, Dreux C,, Mauduit P. Regulated cell surface pro-EGF ectodomain shedding is a zinc metalloprotease-dependent process. J Biol Chem. 2003; 278: 45255–45268. [DOI] [PubMed] [Google Scholar]
- 33. Lee DC,, Sunnarborg SW,, Hinkle CL,, et al. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann N Y Acad Sci. 2003; 995: 22–38. [DOI] [PubMed] [Google Scholar]
- 34. Scheller J,, Chalaris A,, Garbers C,, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011; 32: 380–387. [DOI] [PubMed] [Google Scholar]
- 35. Sunnarborg SW,, Hinkle CL,, Stevenson M,, et al. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem. 2002; 277: 12838–12845. [DOI] [PubMed] [Google Scholar]
- 36. Puerta DT,, Cohen SM. A bioinorganic perspective on matrix metalloproteinase inhibition. Curr Top Med Chem. 2004; 4: 1551–1573. [DOI] [PubMed] [Google Scholar]
- 37. Janusz JM,, Buckwalter BL,, Young PA,, et al. Vanilloids, 1: analogs of capsaicin with antinociceptive and antiinflammatory activity. J Med Chem. 1993; 36: 2595–2604. [DOI] [PubMed] [Google Scholar]
- 38. Young SC,, Fabio KM,, Huang MT,, et al. Investigation of anticholinergic and non-steroidal anti-inflammatory prodrugs which reduce chemically induced skin inflammation. J Appl Toxicol. 2012; 32: 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Laskin JD,, Lacey CJ,, Pillai AN,, et al. Vanilloid fatty hydroxamates as therapeutic anti-inflammatory pharmaceuticals. Provisional patent, serial No. 61/453,274. 2011.
- 40. Hu J,, Van den Steen PE,, Sang QX,, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007; 6: 480–498. [DOI] [PubMed] [Google Scholar]
- 41. Muri EM,, Nieto MJ,, Sindelar RD,, Williamson JS. Hydroxamic acids as pharmacological agents. Curr Med Chem. 2002; 9: 1631–1653. [DOI] [PubMed] [Google Scholar]
- 42. Wada CK,, Holms JH,, Curtin ML,, et al. Phenoxyphenyl sulfone N-formylhydroxylamines (retrohydroxamates) as potent, selective, orally bioavailable matrix metalloproteinase inhibitors. J Med Chem. 2002; 45: 219–232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.