

Abstract
Uterine fibroids are benign, smooth muscle tumors that occur in approximately 70%–80% of women by age 50 yr. The cellular and molecular mechanism(s) by which uterine fibroids (UFs) develop are not fully understood. Accumulating evidence demonstrates that several genetic abnormalities, including deletions, rearrangements, translocations, as well as mutations, have been found in UFs. These genetic anomalies suggest that low DNA damage repair capacity may be involved in UF formation. The objective of this study was to determine whether expression levels of DNA damage repair-related genes were altered, and how they were regulated in the pathogenesis of UFs. Expression levels of DNA repair-related genes RAD51 and BRCA1 were deregulated in fibroid tissues as compared to adjacent myometrial tissues. Expression levels of chromatin protein enhancer of zeste homolog 2 (EZH2) were higher in a subset of fibroids as compared to adjacent myometrial tissues by both immunohistochemistry and Western blot analysis. Treatment with an inhibitor of EZH2 markedly increased expression levels of RAD51 and BRCA1 in fibroid cells and inhibited cell proliferation paired with cell cycle arrest. Restoring the expression of RAD51 and BRCA1 by treatment with EZH2 inhibitor was dependent on reducing the enrichment of trimethylation of histone 3 lysine 27 epigenetic mark in their promoter regions. This study reveals the important role of EZH2-regulated DNA damage-repair genes via histone methylation in fibroid biology, and may provide novel therapeutic targets for the medical treatment of women with symptomatic UFs.
Keywords: BRCA1, DNA damage repair, EZH2, fibroid, H3K27me3, RAD51
INTRODUCTION
Uterine fibroids (UFs) are very common, benign smooth muscle neoplasms that occur with a cumulative incidence of approximately 70% and 80% in White and African American women, respectively, by age 50 yr [1, 2]. Several genetic abnormalities, including deletions in 7q, trisomy of chromosome 12, rearrangements of HMGA2 gene, translocations of the HMGA2 and RAD51B loci, as well as mutations in exons 1 and 2 of the MED12 gene, are involved in fibroid pathogenesis [3–12].
DNA damage and repair are linked to many diseases, including tumor formation. DNA damage induced endogenously or from exogenous sources has the potential to result in mutations and genomic instability, if not properly repaired, eventually leading to tumorigenesis. Increasing evidence shows a link between low DNA repair capacity levels and an increased risk for tumorigenesis [13–16].
Enhancer of zeste homolog 2 (EZH2) is a catalytic core protein in the polycomb repressor complex 2 (PRC2), which catalyzes the trimethylation of histone 3 lysine 27 (H3K27me3) [17, 18], and mediates subsequent gene silencing of target genes that are involved in fundamental cellular processes, such as cell fate decision, cell cycle regulation, senescence, cell differentiation, and cancer [19–22]. Recent findings implicate EZH2 deregulation as an important driver of tumor development and progression, and that inactivation of EZH2 may be therapeutically effective in many tumors [23–28].
Although genetic abnormalities have been well described in human UFs, little is known about the DNA repair system related to epigenetic abnormalities in this common disease [2, 29, 30]. We hypothesize that a dysfunctional DNA damage repair system regulated by epigenetic machinery may be involved in UF formation. The aim of this study was to investigate whether the expression of DNA damage repair genes are deregulated in human UFs, and determine the molecular mechanism(s) underlying altered DNA repair capacity levels.
MATERIALS AND METHODS
Cell Line and Primary Cell Cultures
The immortalized human UF cell line (HuLM), which expresses both estrogen and progesterone receptors, was a generous gift from Dr. Darlene Dixon (National Institute of Environmental Health Sciences, Research Triangle Park, NC) [31]. Primary human UF and myometrial cells were generated from a human UF and adjacent myometrial tissue specimen. Isolation of the primary cell population from tissues was performed as previously described [32]. Briefly, a portion (∼1 cm3) of fresh UF tissue or a portion (∼0.5 cm3) of fresh myometrial tissue was washed in culture medium to remove blood and then chopped into small pieces under sterile conditions, transferred into a 15-ml screw cap tube, and suspended in Hanks Balanced Salt Solution containing 1× antibiotic-antimycotic (Thermo Fisher Scientific) and 300 U/ml collagenase type 4 (Worthington Biochemical Corp.). Suspended tissue pieces were incubated at 37°C for at least 12 h to obtain individual cells and/or clumps of cells. The cell suspension was passed through a 100-μm pore-sized sterile nylon filter and the suspension of individual cells was plated out and incubated at 37°C, allowing the cells to attach to the 100-mm sterile tissue culture-treated plate containing smooth muscle cell basal medium (SmBM; catalog no. CC-3181; Lonza) containing 5% fetal bovine serum (FBS) and supplemented with SmBM singlequots (catalog no. CC-4149). This SmBM singlequot contains hEGF, insulin, hFGF-B, and gentamicin/amphotericin-B.
Patients and Tumor Specimens
The study was approved by Augusta University's Institutional Review Board. Fibroid tissues were consistently collected from peripheral parts of large intramural fibroid lesions (≥5 cm in diameter) with care to avoid areas of apparent necrosis, bleeding, or degeneration. Myometrium was collected at least 2 cm away from the closest fibroid lesion. Patients underwent the informed consent process, and documented informed consent forms were collected and stored. Only those patient records that indicated that the patient had not used any hormonal treatment for at least 3 mo prior to the surgery date were included. All tissues used in this study were collected during the secretory phase of menses. The menstrual phase designation of the endometrium was performed by a board-certified pathologist based on standard morphological and histological criteria [33]. These fibroids have a white, pear-shaped appearance.
For gene and protein expression analysis, myometrium and matched fibroids from African American women were used. For immunohistochemical analysis, histological sections from human UF and myometrial tissue samples were used.
Cell Treatment
Human fibroid primary cells were cultured for 2 days in medium in the presence or absence of DNA methyltransferase (DNMT) inhibitor 5-Aza-dC (2 μM; Sigma), HDAC inhibitor trichostatin A (TSA; 2 μM; Sigma), and EZH2 inhibitor 3-deazaneplanocin A (DZNep; 1, 5 μM; Cayman), respectively. The concentration of inhibitors was determined in accordance with previous studies [34–36]. Dimethyl sulfoxide (DMSO) was used as a vehicle control.
Cell Number and Cell Viability
Cell number was determined using a hemocytometer. Trypan blue staining was used to distinguish nonviable from viable cells.
Cell Cycle Analysis
Cell cycle distribution was determined by flow cytometric analysis according to our previously described methodology with a slight modification [35, 37]. UF primary cells starting at 2 × 105 per 100-mm dish were grown for 4 days in the presence or absence of DZNep (1 and 5 μM). The medium containing freshly prepared DZNep was changed every other day. UF primary cells were also grown in DMSO vehicle, and served as a control. After treatment, cells were fixed in 5 ml of 70% ethanol overnight. The cells were then washed with PBS and resuspended in 0.25 ml of PBS and DNA extraction buffer containing 0.2 M Na2HPO4 with the addition of 0.1 M citric acid (pH 7.8). After 5 min, cells were resuspended in DNA staining solution containing 40 μg/ml propidium iodide (PI; Sigma) and 0.2 mg/ml DNase-free RNase (Thermo Fisher Scientific), and kept at 4°C overnight. The same ratio between PI concentration and cell number was adjusted across samples. Cell cycle was run with a Becton Dickinson FACSCalibur, and the software for both acquisition and analysis was CellQuest Pro.
Apoptosis Analysis
Apoptosis was determined by using an apoptosis and necrosis quantitation kit plus (catalog no. 30065; Biotium). UF primary cells starting at 2 × 105 per 100-mm dish were grown for 4 days in the presence or absence of DZNep (1 and 5 μM). Medium containing freshly prepared DZNep was changed every other day. UF primary cells were also grown in DMSO vehicle and served as a control. After treatment, cells were detached using trypsin without EDTA, and subjected to binding with CF488A-annexin V and ethidium homodimer III (EthD-III). After incubating at room temperature for 15 min, the cells were fixed in 2.5% formaldehyde fixative (pH 7.4) containing 1.25 mM calcium chloride, and run by flow cytometry. Annexin and EthDIII assays were run on the Becton Dickinson FACSCalibur and the software for both acquisition and analysis was CellQuest Pro.
RNA Extraction, cDNA Synthesis, Primer PCR Assays, and SYBR Green Real-Time PCR
RNA was isolated from UFs and myometrium from fibroids uteri (MyoF) as well as from corresponding UF and MyoF primary cells using Trizol reagent (Invitrogen), and reverse transcribed into the first-strand cDNA using superscript III cDNA Transcription Kit. The first-strand cDNA synthesis reaction was primed using random hexamers (Invitrogen). Complementary DNA was loaded onto the DNA damage signaling pathway H96 plate (no. 100-34132; Bio-Rad Inc.). Messenger RNA levels of over 80 genes were detected at the same time using SsoAdvanced Universal SYBR Green Supermix on a Bio-Rad CFX96 real-time PCR system. Data were analyzed using Bio-Rad CFX manager software.
SYBR Green real-time PCR (q-PCR) reactions were set up containing 1× Power SYBR Green Master Mix (Applied Biosystems), 250 nM forward and reverse primers, in a 20-μl reaction. All experiments were performed in replicates. Each biological sample was run in triplicate for each individual experiment. All assays were carried out in 96-well format. Real-time fluorescent detection of PCR products was performed with the CF96X Real-Time PCR System (Bio-Rad) using the following thermocycling conditions: 1 cycle of 95°C for 10 min; 40 cycles of 95°C for 30 sec, and 60°C for 1 min. The primer sequences were designed using Primer Express software (Applied Biosystems) as shown in Table 1. 18S ribosome RNA was used as an endogenous control for gene expression. For data analysis, the comparative method (ΔΔCt) was used to calculate relative quantities of nucleic acid sequence.
TABLE 1.
Primer sequences.

Cell Infection
Immortalized human uterine leiomyoma cells (HuLM) were cultured in 60-mm dishes at 30%–40% confluence. Cells were transduced with adenovirus (Ad)-EZH2 (enhancer of EZH2 adenovirus) and Ad-GFP at varying multiplicity of infection values (0–100 plaque forming unit [pfu]/cell). After 6 h, transduction was stopped by changing the media to smooth muscle growth medium-2 (Lonza # CC3182) supplemented with growth factors and 5% FBS. Protein lysates were prepared on Day 5 of transduction for measurement of EZH2, H3K4me3, and H3K27me3 levels. RNA was isolated on Day 3 of transduction for measurement of RAD51, BRCA1, p21, and EZH2 expression.
Immunohistochemistry
Histological sections from human UF and myometrial tissue samples were used for immunostaining. Tumor tissue was fixed in 10% buffered formalin and embedded in paraffin. Slides were placed on the Leica Bond Max IHC stainer. All steps, with the exception of dehydration, clearing, and coverslipping, were performed on the Bond Max. Slides were deparaffinized. Heat-induced antigen retrieval was performed on the Bond Max using their Epitope Retrieval 2 solution for 20 min. Sections were incubated with mouse anti-EZH2 (catalog no. EZH2-L-CE; Leica Microsystems) and diluted 1:200 for 30 min (Table 2). The Bond Refine Polymer detection system was used for visualization. Slides were then dehydrated, cleared, and coverslipped. EZH2 expression was recorded as percentage of EZH2-expressing cells with nuclear expression.
TABLE 2.
List of antibodies used for Western blot, immunohistochemistry, and ChIP assay analysis.

Protein Extraction from Human Tissue Samples
UFs and adjacent myometrial tissue (MyoF) samples were pulverized to a fine powder in liquid nitrogen using the Cellcrusher tissue pulverizer (Cell Crusher Limited), and homogenized in RIPA buffer containing protease inhibitors. Cell lysates were briefly sonicated and clear lysates were obtained from supernatant by centrifuging protein lysates at 12 000 rpm for 15 min at 4°C.
Western Blot Analysis
Total proteins from fibroid primary cells were treated with the vehicle (DMSO) or DZNep (0.25, 1, and 2.5 μM) and then extracted by lysing and sonication in RIPA buffer containing protease and phosphatase inhibitor cocktails. Protein concentrations were determined using the Bradford protein assay kit (Bio-Rad). Equal amounts of total proteins (20 μg) were subjected to SDS-PAGE. Proteins were transferred to a nitrocellulose membrane for 90 min at 100 V. Membranes were blocked for 1 h at room temperature in Tris-buffered saline (TBS) containing 5% nonfat powdered milk and probed with primary antibody in TBS at concentrations from 1:500 to 1:10 000 overnight, in accordance with the manufacturer's instructions for each antibody (Table 2). Membranes were washed with TBS containing 0.05% Tween-20. In all cases, a secondary antibody labeled with horseradish peroxidase (Santa Cruz Biotechnology) was used at concentrations from 1:5000 for 1 h at room temperature, and immunoreactive bands were detected by using SuperSignal West Pico Chemi-luminescent Substrate (Thermo Fisher Scientific) and recorded on photosensitive film. The relative intensities of immunoreactive bands were detected by Western blot analysis, quantified by densitometry using NIH ImageJ software (National Institutes of Health), and normalized with βactin levels.
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation (ChIP) assay was performed as previously described [38, 39]. Primary cells from UFs were grown in media in the presence or absence of DZNep (0.25 and 2.5 μM) for 3 days. The concentration range of DZNep was determined for ChIP assay according to the in vitro studies previously mentioned in this article. Cells (1 × 107) were incubated with 1% formaldehyde for 10 min to crosslink histones to DNA. After washing with cold PBS, cell pellets were resuspended in a cell lysis buffer (10 mM Tris [pH 8.0], 10 mM NaCl, 0.2% NP40). Nuclei were resuspended in nuclei lysis buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 1% SDS), and sonicated for 25 min. The soluble chromatin fraction was collected, and 5 μl of antibody for H3K27me3, H3K4me3 (Table 2), or normal rabbit IgG was added. After incubation, chromatin-antibody complexes were collected using A/G magnetic beads (Millipore). After washing, immunoprecipitated DNA was treated with proteinase K at 62°C for 2 h. DNA was extracted with a QIAquick PCR Purification kit (Qiagen) and analyzed by SYBR Green real-time PCR. The primer pairs used for ChIP assays are shown in Table 1.
Statistical Analysis
All values are expressed as means ± SE. Comparisons between two groups were done using the unpaired Student t-test. Differences between groups were examined by ANOVA and Newman-Keuls post hoc test for multiple comparisons. Values of P < 0.05 were considered statistically significant.
RESULTS
DNA Repair-Related Genes Are Deregulated in Fibroid Tissues as Compared to Adjacent Myometrium
In order to determine if and which DNA repair-related genes are deregulated in fibroid tissues as compared to adjacent myometrial tissue, we performed quantitative real-time PrimePCR array using the Bio-Rad predesigned assay specifically for the DNA damage signaling pathway. Results indicated that expression levels of RAD51 and BRCA1 are markedly downregulated in fibroid tissues as compared to adjacent myometrial tissues (data not shown). To verify our results of the PrimePCR Array and confirm the altered expression of DNA repair genes in human UFs, we validated the mRNA levels of RAD51 and BRCA1 using quantitative PCR in fibroid and adjacent myometrial tissues from five patients. Consistent with the PrimePCR array, expression levels of RAD51 and BRCA1 were downregulated in three of five and four of five fibroid tissues, respectively, as compared to adjacent myometrial tissues (Fig. 1, A and B). The expression of RAD51 and BRCA1 was increased in one of five UFs as compared to adjacent myometrium tissue, suggesting that increased expression of BRCA1 and RAD51 in UF is not a universal phenomenon among patients with UFs.
FIG. 1.

Expression levels of DNA damage repair genes RAD51 and BRCA1 in fibroids (F) and adjacent myometrial tissues (MyoF) as well as primary cells. Each experiment was conducted in replicates. A) Expression of RAD51 was determined by real-time PCR in fibroid and adjacent myometrial tissues. *P < 0.05 compared with the respective control (n = 5). B) Expression of BRCA1 was measured by real-time PCR in fibroid and adjacent myometrial tissues. *P < 0.05 compared with the control (n = 5). Expression of RAD51 (C) and BRCA1 (D) was determined by real-time PCR in primary cells from a fibroid and adjacent myometrial tissue. The cells from a fibroid and from myometrium tissue were plates in 6 × 60-mm dishes, including three dishes for primary fibroid cells and three dishes for primary myometrium cells. Three biological samples from both fibroid and myometrium samples were subjected to RNA isolation, cDNA synthesis, and q-PCR analysis. *P < 0.05 compared with the respective control.
Next, we determined whether expression levels of DNA repair genes, including RAD51 and BRCA1, are also downregulated in fibroid primary cells as compared to adjacent myometrial (MyoF) primary cells. We isolated fibroid and myometrial primary cells from a patient and measured mRNA levels of RAD51 and BRCA1. To avoid patient-to-patient variability, we used primary myometrium and fibroid cells from the same utero-same patient to compare. We consistently used low-passage primary cells to ensure mimicry under in vivo conditions and to avoid the introduction of any in vitro artifact. As shown in Figure 1, C and D, expression levels of RAD51 and BRCA1 were downregulated 70% and 55%, respectively, in cultured fibroid primary cells as compared to myometrial (MyoF) primary cells.
Inhibition of EZH2 Increased the Expression of RAD51 and BRCA1 in Human Primary Fibroid Cells
To determine the mechanism underlying the reduced expression of RAD51 and BRCA1 in human fibroid primary cells, we treated the cells with three epigenetic drugs: DNMT inhibitor (5-Aza-dC), histone deacetylase inhibitor (TSA), and EZH2 inhibitor (DZNep), respectively. As shown in Figure 2, A and B, treatment with DNMT inhibitor 5-Aza-dC markedly increased expression levels of RAD51 and BRCA1 in cultured fibroid primary cells. In contrast, treatment with TSA decreased expression levels of RAD51 and BRCA1, respectively. Interestingly, treatment with EZH2 inhibitor showed a robust increase in the expression of RAD51 (1.8-fold, P < 0.05) and BRCA1 (2.8-fold, P < 0.05), suggesting that EZH2 may play a dominant role in regulating DNA repair-related gene expression in human fibroid cells.
FIG. 2.
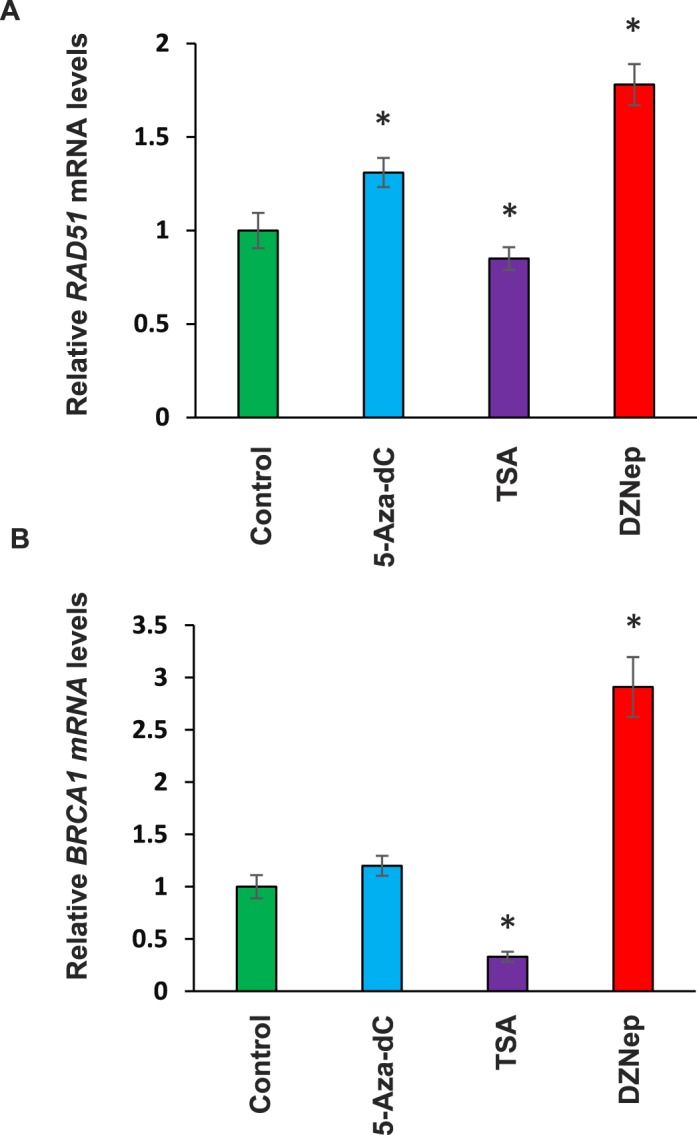
Restoring of RAD51 and BRCA1 in primary cells from fibroids treated with EZH2 inhibitor. A) Expression of RAD51 in primary cells from fibroid tissues was determined by real-time PCR in the presence or absence of 5-Aza-dC, TSA, and DZNep, respectively. *P < 0.05 compared with the respective control. B) Expression of BRCA1 in primary cells from fibroid tissues was determined by real-time PCR in the presence or absence of 5-Aza-dC, TSA, and DZNep, respectively. *P < 0.05 compared with the control.
Expression of EZH2 Is Upregulated in Human Fibroid Tissues as Compared to Adjacent Myometrial Tissues
We performed both immunohistochemistry and Western blot analysis to determine the relative protein expression of EZH2 in UFs as compared to adjacent MyoF myometrial tissues. As shown in Figure 3A, more intense and nuclear-positive cells for EZH2 staining were observed in two fibroid tissues as compared to two matched adjacent myometrial tissues. Quantitative analysis demonstrated that EZH2-positive cells were significantly higher in fibroid tissues as compared to matched myometrial samples (Fig. 3B). Next, we detected expression levels of EZH2 in fibroid tissues as well as in adjacent myometrial tissues by Western blot analysis (n = 8). As shown in Figure 3C, expression levels of EZH2 were upregulated in a subset of fibroid tissues (five of eight) as compared to matched adjacent myometrial tissues. Our results suggest that 70% of UF lesions (7 of 10 patients) exhibit elevated expression of EZH2.
FIG. 3.

Expression of EZH2 expression in fibroid tissues and adjacent myometrium. A) Immunohistochemistry of EZH2 expression in fibroid and adjacent tissues. Arrows indicate EZH2-positive cells (magnification ×400). B) Semiquantitative analysis of EZH2-positive cells in myometrium and fibroids from fibroid and matched myometrium samples (n = 2). C) Western blot analysis of EZH2 in fibroid and adjacent tissues from African American women (n = 8). Total lysates from fibroids and adjacent myometrial tissues were extracted and subjected to Western blot analysis using antibody against EZH2. βactin was used as an endogenous control. Asterisks indicate the upregulation of EZH2 in fibroid tissues as compared to adjacent myometrium tissues. EZH2 protein bands were quantified and normalized to βactin, and relative values were used to generate data graphs (C, bottom panel). F, uterine fibroid; M, adjacent myometrial tissue.
EZH2 Increases the Levels of H3K27me3 in Human Fibroid Primary Cells
To determine whether overexpression of EZH2 increases the level of bivalent silencing mark H3K27me3 in UF cells, we introduced EZH2 into HuLM using an adenoviral transduction approach. As shown in Figure 4A, EZH2 protein expression was much higher in transduced HuLM cells as compared to nontransduced cells. Moreover, increased expression of EZH2 markedly elevated expression levels of H3K27me3 (Fig. 4B), suggesting that EZH2 mediates the production of the silencing mark H3K27me3 in UF cells.
FIG. 4.

EZH2 directly targets epigenetic mark H3K27me3 in fibroid primary cells. A) Introduction of EZH2 into HuLM cells by adenoviral transduction showed a dose-dependent increase in the expression levels of EZH2. B) Expression levels of H3K27me3 were markedly upregulated in Ad-EZH2 transduced cells in comparison to control in a dose-dependent manner. *P < 0.05 compared with the control. C) Protein lysates from fibroid primary cells treated with different concentrations of EZH2 inhibitor (DZNep) for 3 days were subjected to Western blot analysis using antibody against H3K27me3 and HeK4me3, respectively. βactin was used as an endogenous control. D) Densitometry quantification of H3K27me3 levels for C. E) Densitometry quantification of H3K4me3 levels for C.
DZNep is an S-adenosyl-L-homocysteine hydrolase inhibitor, which chemically inhibits EZH2 histone methyltransferase activity. To further confirm the role of EZH2 in the production of the repressive H3K27me3 mark in human fibroid primary cells, we treated cells with DZNep and examined the levels of H3K27me3 and H3K4me3, respectively. As shown in Figure 4, C–E, H3K27me3 levels were markedly downregulated (70% inhibition) in a dose-dependent manner in fibroid primary cells treated with DZNep. However, H3K4me3 levels were not changed following treatment with DZNep.
To determine whether overexpression of EZH2 alters expression levels of RAD51 and BRCA1, real-time PCR analysis was performed in HuLM cells infected with 20 pfu of Ad-EZH2 or Ad-GFP, respectively for 3 days. As shown in Figure 5, infection of HuLM cells with Ad-EZH2 tremendously elevated EZH2 expression of RNA levels. The expression of both RAD51 and BRCA1 genes was downregulated in cells introduced with EZH2 as compared to GFP control. In addition, cell cycle arrest-related genes, including p21, were also downregulated, as shown in Figure 5D.
FIG. 5.

Overexpression of EZH2 decreases expression levels of DNA damage repair and cell cycle arrest-related genes. Overexpression of EZH2 was achieved by adenoviral transduction approach in HuLM cells. The expression of EZH2 (A), RAD51 (B), BRCA1 (C), and p21 (D) was determined by real-time PCR after infection of HuLM cells with Ad-EZH2 (20 pfu) and Ad-GFP (20 pfu) for 3 days. *P < 0.05 compared with the control.
EZH2 Inhibition Decreases Fibroid Cell Proliferation More Efficiently
Since EZH2 expression is higher in human fibroid tissues, we then determined whether fibroid primary cells and myometrium show different proliferative responses after treatment with DZNep. As shown in Figure 6A, DZNep treatment significantly decreased (30% inhibition, P < 0.05) fibroid primary cell proliferation via cell count while having no effect on myometrial primary cells. In addition, at 1 μm of DZNep treatment, no visible cell death was observed in either fibroid or MyoF primary cells.
FIG. 6.

DZNep treatment efficiently inhibited fibroid primary cell proliferation by increased expression of P21 and decreased protein expression of PCNA. A) Fibroid (F) and MyoF primary cells were grown in media containing either DZNep (0.25 and 1 μM) or vehicle (DMSO) for 5 days. Cell proliferation was assessed by cell counting. Trypan blue staining was used to distinguish nonviable from viable cells. P < 0.05 compared with the respective control. B) Real-time PCR was performed to determine expression levels of p21 in fibroid primary cells treated with DZNep or vehicle. C) Real-time PCR was performed to determine expression levels of p21 in MyoF primary cells treated with DZNep or vehicle. D) Western blot analysis was performed to examine expression levels of PCNA in fibroid primary cells treated with varying concentrations of DZNep. E) Western blot analysis was performed to examine expression levels of PCNA in MyoF primary cells treated with varying concentrations of DZNep. *P < 0.05 compared with the control.
In addition, p21 expression was increased (1.9-fold, P < 0.05) in DZNep-treated fibroid primary cells as compared with vehicle-treated fibroid cells, as shown in Figure 6B. In MyoF primary cells, p21 expression was increased 1.3-fold as compared to the vehicle-treated group (Fig. 6C). Moreover, PCNA expression was lower (55% inhibition) in DZNep-treated fibroid primary cells as compared with vehicle-treated fibroid primary cells, as shown in Figure 6D. However, only a 20% reduction of PCNA expression was observed in MyoF primary cells treated with DZNep, as shown in Figure 6E. These data suggest that EZH2 inhibition by DZNep exhibited a more inhibitory effect on fibroid primary cell proliferation as compared to DZNep inhibition on myometrial primary cell proliferation.
To determine whether inhibition of EZH2 by DZNep impairs cell phase distribution, we performed cell cycle analysis in the fibroid primary cells treated with DZNep and vehicle (DMSO). The representative cell phase distribution in vehicle- and DZNep (1 μM, 5 μM) -treated cells is shown in Figure 7, A, B, and C, respectively. Our data indicate that treatment of cells with DZNep significantly changed the cell phase distribution in a dose-dependent manner by identification of G2/M accumulation (*P < 0.05). Treatment of fibroid cells with 5 μM DZNep exhibited more accumulation of G2/M cells as compared to 1 μM treatment (Fig. 7D, #P < 0.05). These data suggest that inhibition of EZH2 by DZNep decreased cell cycle progression in a dose-dependent manner.
FIG. 7.

DZNep treatment changes cell phase distribution. Fibroid cells (2.5 × 105) were grown in a 100-mm dish. The procedure for processing the cells for cell cycle analysis appears in the Materials and Methods section. Representative cell cycle analysis in fibroid cells treated with DMSO (A), 1 μM DZNep (B), and 5 μM DZNep (C) for 4 days. D) Comparison of cell phase distribution in fibroid cells treated with DMSO, 1 μM DZNep, and 5 μM DZNep, respectively. Each column represents the mean of three independent experiments. *P < 0.05; #P < 0.05.
The apoptotic ratios induced by DZNep were assessed by flow cytometry through annexin-V-EthD-III staining. In live cells, phosphatide serine is located on the cytoplasmic surface of the cell membrane. However, in apoptotic cells, phosphatidyl serine is translocated from the inner to the outer leaflet of the membrane, so that it is susceptible to annexin-V staining. EthD-III is a nucleic acid dye, and, therefore, could only permeate into late-apoptotic and dead cells. Early- and late-apoptotic cells and dead cells could be distinguished using a combination of annexin V/EthD-III staining. The results indicate that the percentages of apoptotic cells (annexin-V+ EthD-III+ and annexin-V+ EthD-III−) for control and 5 μM DZNep, were 14.33 ± 2.36 and 12.12 ± 1.56, respectively (Fig. 8). No significant difference in the percentages of apoptotic cells was observed between control and DZNep treatment groups. These results suggest that DZNep treatment suppressed fibroid cell proliferation through cell cycle arrest rather than the apoptotic pathway.
FIG. 8.

Measurement of cell apoptosis in response to DZNep treatment. Primary fibroid cells were grown in a 100-mm dish in the presence or absence of DZNep (5 μM) for 3 days. DMSO treatment served as a control. Cells were stained with both annexin-V and EthD-III. Apoptosis was measured by flow cytometry. Double-negative staining representing living cells is shown in quadrant Q4; positive staining for annexin-V and negative staining for PI represent the early apoptotic stage shown in quadrant Q3; double-positive staining representing the late apoptotic stage is shown in quadrant Q2; and dead cells are presented in quadrant Q1. Each column represents the mean of three independent experiments.
RAD51 and BRCA1 Are Epigenetic Targets of EZH2
To determine whether RAD51 and BRCA1 are directly regulated by EZH2, the enrichment of the bivalent marks of H3K27me3 and H3K4me3 in the promoter regions of both RAD51 and BRCA1 were examined by ChIP assay in human fibroid primary cells (Fig. 9A). We detected the enrichment of both epigenetic marks in the promoter regions of RAD51 and BRCA1 due to the fact that PcG/TrxG group response elements (PRE/TRE) switching occurs during development, suggesting that PcG and TrxG act as counterparts and may target the same genes in their promoter regions [40]. Following DZNep treatment at 0.25 and 2.5 μM, the enrichment of H3K27me3 in the promoter regions of RAD51 was markedly decreased, as shown in Figure 9B (70% reduction at 2.5 μM treatment, P < 0.05). In addition, the enrichment of H3K4me3 in the promoter regions of RAD51 slightly increased in response to DZNep treatment without a significant difference (Fig. 9C). Similarly, the enrichment of H3K27me3 in both distal and proximal promoter regions of BRCA1 were also significantly decreased in response to DZNep treatment (2.5 μM, 90% and 75% reduction, P < 0.05; Fig. 9D). However, the change of enrichment of H3K4me3 was not observed in both distal and proximal promoter regions of BRCA1 in response to DZNep treatment (Fig. 9E). This is consistent with the increased expression of RAD51 and BRCA1 after treatment with DZNep in fibroid primary cells. These data suggest that inhibition of EZH2 upregulated the expression of RAD51 and BRCA1, specifically through epigenetic mark H3K27me3.
FIG. 9.

DZNep treatment restores expression levels of RAD51 and BRCA1 through epigenetic mark H3K27me3. A) Location of regions analyzed by ChIP/PCR along human RAD51 and BRCA1 promoters. The position of the transcriptional start site was designated as +1. Short horizontal lines indicate the regions analyzed by ChIP/PCR. B) ChIP/quantitative PCR was performed with anti-H3K27me3 antibody in the RAD51 promoter region in fibroid primary cells in the presence or absence of DZNep. C) ChIP/quantitative PCR was performed with anti-H3K4me3 antibody in the RAD51 promoter region in fibroid primary cells in the presence or absence of DZNep. D) ChIP/quantitative PCR was performed to determine the enrichment of H3K27me3 in the distal and proximal promoter regions of BRCA1 in the presence or absence of DZNep. E) ChIP/quantitative PCR was performed to determine the enrichment of H3K4me3 in the distal and proximal promoter regions of BRCA1 in the presence or absence of DZNep. *P < 0.05 compared with the control.
DISCUSSION
RAD51 family members are strikingly similar to bacterial RecA and Saccharomyces cerevisiae Rad51, and are known to be involved in the homologous recombination and repair of DNA. Several studies have identified that stabilization of the cancer genome in homologous recombination-deficient cells can be achieved by overexpression of the recombinase RAD51 [41]. This protein can interact with the ssDNA-binding protein RPA and RAD52, and is thought to play a role in homologous pairing and strand transfer of DNA. RAD51 protein has also been found to interact with BRCA1 and BRCA2, which may be important in the cellular response to DNA damage [42]. It is also reported that endogenous levels of RAD51 and BRCA2 are required for homologous recombination and regulated by homeostatic rebalancing [43]. BRCA1 is a nuclear phosphoprotein that plays a role in transcription, DNA repair of double-stranded breaks, recombination, maintaining genomic stability, and also acts as a tumor suppressor gene [44–46]. For women who have a BRCA mutation, the risk of developing breast or ovarian cancer is greatly increased [47–49]. It has also been recognized that RAD51 and BRCA1 work in a coordinated fashion in responding to DNA damage [50–52], suggesting that deregulation of RAD51 and BRCA1 might simultaneously increase susceptibility to tumorigenesis. In this study, we found that expression levels of both RAD51 and BRCA1 were downregulated in fibroid tissues as compared to adjacent myometrial tissues. The tissue findings are also consistent with our human fibroid primary cell data showing that expression levels of RAD51 and BRCA1 were lower in fibroid primary cells as compared to adjacent myometrial primary cells. These findings indicate that low DNA repair capacity due to decreased expression levels of DNA repair-related genes might contribute to the pathogenesis of UF tumorigenesis. It should also be mentioned here that all patients' samples were collected from African American women; therefore, we are limited in asserting any generalizations to those of non-African American heritage. Since fibroid tissues with small lesions (≤5 cm) were not available, we used only fibroid lesions larger than 5 cm in this study. It has been reported that the size of lesions may impair the UF extracellular matrix metabolism [53]; therefore, an altered UF environment may lead to alteration of DNA repair gene expression.
EZH2 is highly expressed in a wide range of cancers, including breast, prostate, bladder, colon, lung, pancreatic, sarcoma, and lymphoma [54–57]. Overexpression of EZH2 is frequently correlated with advanced stages of human cancer progression and an overall poor prognosis [24, 58]. In this study, we reported, for the first time, that expression levels of EZH2 were higher in fibroid tissues as compared to adjacent myometrium in the same patient by both immunohistochemistry and Western blot analysis. While the mechanism underlying increased levels of EZH2 in fibroid tissues is largely unknown, increasing evidence shows that EZH2 is regulated at transcriptional and posttranscriptional levels in other types of tumors. Transcriptional factors, E2Fs, bind to the EZH2 promoters and transactivate its expression [59]. EZH2 is transcriptionally induced by estradiol and estrogenic endocrine disruptors, bisphenol-A, and diethylstilbestrol [60]. It has been reported that intracellular estradiol levels were increased by intrinsic aromatase activity, and aromatase inhibitors have been shown effective in treating estrogen dependent UFs [61, 62]. In addition, UFs also exhibit increased estradiol receptors and specific polymorphisms of the estradiol receptor [63–66]. These reports highlight the fact that UFs provide a high intracellular estrogen environment, which may increase the expression of EZH2. EZH2 expression can also be transcriptionally activated by a fusion oncoprotein EWS-FLI1 in Ewing sarcoma [67]. Micro-RNA-101 targets EZH2 in ostersarcoma and endometrial cancer cells [68, 69].
It is well recognized that polycomb group proteins can alter chromatin structure such that epigenetic silencing of genes takes place, bind to specific regions of gene promoters, and direct posttranslational modifications at certain histone sites, thereby silencing gene expression [70]. In the present study, we initially determined which epigenetic machineries are involved in the regulation of DNA repair genes. We treated fibroid cells with DNMT inhibitor (5-Aza-dC), histone deacetylase inhibitor (TSA), and EZH2 inhibitor (DZNep). As shown in Figure 2, although treatment with 5-Aza-dC increased expression levels of RAD51, DZNep treatment showed a robust increase in the expression of both RAD51 and BRCA1, suggesting that EZH2 may be involved in the downregulation of DNA repair genes. Moreover, enrichment of H3K27me3 in the promoter region of RAD51 was markedly decreased in fibroid primary cells following treatment with EZH2 inhibitor, which was concurrently associated with an increased expression of RAD51. Our data suggest that DZNep treatment in fibroid cells restores RAD51 and BRCA1 expression through decreased enrichment of H3K27me3 in their promoter regions mediated by EZH2. This finding was consistent with a previous observation in breast tumor-initiating cells (BTIC) of EZH2-mediated epigenetic repression of the DNA damage repair gene RAD51 [71]. This was identified as a mechanism that could promote the expansion of the cells, and may contribute to cancer progression [71]. In addition, it has also been found that EZH2 decreases the expression of five RAD51 paralog proteins involved in homologous recombination repair of DNA double-strand breaks. EZH2 overexpression impaired the formation of RAD51 repair foci at sites of DNA breaks, indicating that EZH2 may contribute to breast tumorigenesis by specific downregulation of RAD51-like proteins and by impairment of homologous recombination [72]. We identified another EZH2-targeted gene, BRCA1, in fibroid tissues, consistent with the previous finding, which showed that EZH2 is involved in malignant biological behavior through alterations of BRCA1 expression [73].
EZH2 is considered the mandatory histone methyltransferase required to establish H3K27me3 epigenetic marks; however, to date, there is no documented evidence regarding a correlation of EZH2 expression with H3K27me3 levels in human UFs. To address this issue, we determined levels of H3K27me3 in fibroid primary cells in context with EZH2 expression. We demonstrated that EZH2 overexpression was positively correlated with increased H3K27me3 expression, and that EZH2 directly regulated the levels of epigenomic mark H3K27me3 in fibroid HuLM cells. Our data are consistent with the concept that wild-type PRC2 showed robust expression of H3K27me3 and that PRC2 loss showed complete loss of H3K27me3 in tumor cells [21]. In colorectal cancer, tumor tissues also exhibit higher expression of EZH2 and H3K27me3 as compared to their normal counterparts [74]. However, in other types of cancer, the studies regarding correlation between H3K27me3 levels and EZH2 expression are contradictory. For instance, in triple-negative breast cancers, EZH2 and H3K27me3 show an inverse correlation [26, 75]. Although the rationale as to why EZH2 levels are not correlated with H3K27me3 expression is largely unknown, accumulating evidence shows that EZH2 functions independently of H3K27me3 in some types of cancers [76]. For example, in contrast to functioning as a gene silencer, EZH2 interacts with estrogen receptor α- and βcatenin and upregulates C-MYC and CYCLIN D expression in breast cancer [77]. Growing evidence also shows that EZH2's activity and stability are regulated by posttranslational modifications, and that these modifications are critical for the biological functioning of PRC2 [23]. Cha et al. initially reported that Akt phosphorylates EZH2 at serine 21 and suppresses its methyltransferase activity by impeding EZH2 binding to H3, leading to a decrease of H3K27me3 and derepression of silenced genes [78]. Bredfeldt et al. found that, in response to both 17b-estradiol and xenoestrogen diethylstilbestrol, rapid signaling from membrane-associated ER phosphorylates EZH2 at S21, reducing levels of H3K27me3 in hormone-responsive cells [79]. This study suggests that activation of nongenomic signaling via environmental exposure to xenoestrogen in an early stage reprogrammed the expression profile of estrogen-response genes in uterine myometrial cells, which leads to an increased risk of UF development. In this article, we first compared expression levels of DNA repair genes between myometrium from fibroid uteri and matched fibroids (i.e., myometrium and fibroids from the same patients). Low levels of DNA repair gene expression in fibroid cells were due to EZH2 by gain and loss of functional analysis. Given the evidence that developmental environmental exposure to xenoestrogen increases the incidence of UF development, our study suggests that early-life exposure to xenoestrogen may impair the DNA damage repair system, leading to an increased risk of UF development. In addition, there are several other posttranslational phosphorylations, including EZH2 at T345, T372, T487, Y641, and S734, which have been identified [23]. Moreover, EZH2 at S75 has been found to be glycosylated by O-linked N acetylglucosamine transferase, and EZH2 stability is also modulated by ubiquitination [23]. These studies provide multiple potential mechanisms to modify the activity of polycomb protein EZH2.
It has been shown that DZNep, a carbocyclic analog of adenosine, depletes cellular levels of the PRC2 components, notably EZH2, and inhibits levels of H3K27me3 [36, 80]. In this study, data relating to a loss of function showed that inhibition of EZH2 by DZNep in fibroid primary cells increased expression of RAD51 and BRCA1. To determine whether DZNep showed inhibitory effect on cell proliferation, we treated fibroid primary cells and adjacent myometrial cells with DZNep. We found that DZNep (1 μM) caused 30% inhibition in the proliferation of fibroid primary cells paired with cell cycle arrest. However, DZNep treatment did not exhibit an inhibitory effect on myometrial primary cells. In addition, DZNep treatment increased p21 expression and reduced PCNA expression, which suggest that DZNep could exhibit a more inhibitory effect on fibroid cell proliferation rather than on myometrial cells. This could possibly be due to low levels of DNA repair gene expression mediated by EZH2 in fibroid cells versus normal levels of DNA repair gene myometrial cells. DZNep, as a potent inhibitor of EZH2, has been shown to reduce the global H3K27me3 levels in many type of cells, including chondrosarcoma cell lines [36], insect HzAm1 cells [81], pancreatic ductal adenocarcinoma [82], skin cancer cells [83], etc. Importantly, EZH2-mediated H3K27me3 epigenetic regulation of certain genes is a common mechanism in human cancer cells. For instance, tumor suppressor Deleted in liver cancer 1 (DLC1) was identified as a target of repression by EZH2-mediated H3K27me3. Depletion of EZH2 by shRNA reduced H3K27me3 level at DLC1 promoter region and induced DLC1 gene re-expression. In addition, treating cancer cells with DZNep restores Dlc1 expression in different cancer cell lines [84]. Although DZNep, as a small EZH2 inhibitor, reduces the levels of H3K27me3, a recent study suggests that DZNep also acts on other histone marks, such as H4K20me3 [85]. However, regulation of DZNep-induced gene expression in UFs through other histone marks requires further investigation. One of the candidate histone marks is H3K9me3. Ostrup et al. reported that DZNep affected promoter enrichment of both H3K27me3 and H3K9me3 in one-to-two-cell-stage embryos [86]. DZNep induced a loss of H3K27me3 and H3K9me3 from a number of promoters, but did not prevent de novo acquisition of these marks on others, indicating gene-specific targeting of its action [86]. Therefore, a more specific effect by EZH2 will be achieved if a genetic approach is used to deplete EZH2 expression. Nevertheless, our data show that restoring DNA repair genes by treatment of cells with DZNep is at least through epigenetic mark H3K27me3. Our data are consistent with a previous study by Chang et al., showing that EZH2 expression-mediated downregulation of DNA damage repair leads to accumulation of recurrent RAF1 gene expression amplification in BTICs, which promote BTIC expansion [71].
ChIP assay demonstrated that the enrichment of H3K27me3 in the promoter region of both RAD51 and BRCA1 was decreased in response to DZNep treatment. These data suggest that low levels of DNA repair gene expression were associated with EZH2, and restoration of DNA damage repair-related genes (RAD51 and BRCA1) by inhibition of EZH2 was through the role of EZH2 canonical activity in fibroid primary cells. The H3K27me3-mediated regulation of RAD51 and BRCA1 in response to DZNep treatment seems to be specific, because we did not see a marked difference in H3K4me3 enrichment in the promoter region of RAD51 and BRCA1 between vehicle- and DZNep-treated groups. It should be mentioned that RAD51 is a crucial partner of BRCA1 for mediating the DNA damage repair by homologous recombination [50, 51]. In such instances, one may consider that the abnormal expression of both RAD51 and BRCA1 may increase susceptibility to the formation of DNA instability and abnormalities in myometrial tissues, leading to the pathogenesis of UFs.
In conclusion, our studies provide the first evidence showing that expression of DNA repair-related genes, including RAD51 and BRCA1, is dysregulated in fibroid tissues as compared with matched adjacent myometrial tissues. Importantly, expression levels of EZH2 are higher in fibroids as compared to matched adjacent myometrium. EZH2 regulates the expression of DNA damage repair genes RAD51 and BRCA1 through epigenetic mark H3K27me3 in their promoter regions. Our studies suggest that dysregulation of DNA damage repair genes by EZH2 may be attributable to UF development. The function of EZH2 as a transcriptional repressor has been explored as the basis for drug discovery. Further research aimed at a more comprehensive understanding of EZH2 function and mechanism, and the design of more effective and specific inhibitors for EZH2, are warranted, which, in turn, could lead to novel medical treatment options for women with symptomatic UFs.
ACKNOWLEDGMENT
We would like to thank Ms. Lelyand Elam for technical assistance. We also thank Walidah Walker, MPH for editing this manuscript.
Footnotes
This work was supported in part by an Augusta University Startup package, the National Institutes of Health grant HD04622811 to A.A., and the Augusta University intramural Grants Program to Q.Y.
REFERENCES
- Bulun SE. Uterine fibroids. N Engl J Med. 2013;369((14)):1344–1355. doi: 10.1056/NEJMra1209993. [DOI] [PubMed] [Google Scholar]
- Al-Hendy A, Salama S. Gene therapy and uterine leiomyoma: a review. Hum Reprod Update. 2006;12((4)):385–400. doi: 10.1093/humupd/dml015. [DOI] [PubMed] [Google Scholar]
- Heinonen HR, Sarvilinna NS, Sjoberg J, Kampjarvi K, Pitkanen E, Vahteristo P, Makinen N, Aaltonen LA. MED12 mutation frequency in unselected sporadic uterine leiomyomas. Fertil Steril. 2014;102((4)):1137–1142. doi: 10.1016/j.fertnstert.2014.06.040. [DOI] [PubMed] [Google Scholar]
- Rieker RJ, Agaimy A, Moskalev EA, Hebele S, Hein A, Mehlhorn G, Beckmann MW, Hartmann A, Haller F. Mutation status of the mediator complex subunit 12 (MED12) in uterine leiomyomas and concurrent/metachronous multifocal peritoneal smooth muscle nodules (leiomyomatosis peritonealis disseminata) Pathology. 2013;45((4)):388–392. doi: 10.1097/PAT.0b013e328360bf97. [DOI] [PubMed] [Google Scholar]
- Halder SK, Laknaur A, Miller J, Layman LC, Diamond M, Al-Hendy A. Novel MED12 gene somatic mutations in women from the Southern United States with symptomatic uterine fibroids. Mol Genet Genomics. 2015;290:505–511. doi: 10.1007/s00438-014-0938-x. : [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertsch E, Qiang W, Zhang Q, Espona-Fiedler M, Druschitz S, Liu Y, Mittal K, Kong B, Kurita T, Wei JJ. MED12 and HMGA2 mutations: two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod Pathol. 2014;27((8)):1144–1153. doi: 10.1038/modpathol.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DS, Klotzbucher M, Kugoh H, Cai SL, Mullen JP, Manfioletti G, Fuhrman U, Walker CL. Aberrant expression of HMGA2 in uterine leiomyoma associated with loss of TSC2 tumor suppressor gene function. Cancer Res. 2002;62((13)):3766–3772. [PubMed] [Google Scholar]
- Ingraham SE, Lynch RA, Surti U, Rutter JL, Buckler AJ, Khan SA, Menon AG, Lepont P. Identification and characterization of novel human transcripts embedded within HMGA2 in t(12;14)(q15;q24.1) uterine leiomyoma. Mutat Res. 2006;602((1-2)):43–53. doi: 10.1016/j.mrfmmm.2006.07.007. [DOI] [PubMed] [Google Scholar]
- El-Shennawy GA, Elbialy AA, Isamil AE, El Behery MM. Is genetic polymorphism of ER-alpha, CYP1A1, and CYP1B1 a risk factor for uterine leiomyoma? Arch Gynecol Obstet. 2011;283((6)):1313–1318. doi: 10.1007/s00404-010-1550-x. [DOI] [PubMed] [Google Scholar]
- Vikhliaeva EM. Molecular-genetic determinants of the neoplastic process and state-of-the-art treatment of patients with uterine leiomyoma [in Russian] Voprosy Onkologii. 2001;47((2)):200–204. [PubMed] [Google Scholar]
- Yang Y, Zhai XD, Gao LB, Li SL, Wang Z, Chen GD. Genetic polymorphisms of DNA repair gene XRCC1 and risk of uterine leiomyoma. Mol Cell Biochem. 2010;338((1-2)):143–147. doi: 10.1007/s11010-009-0347-3. [DOI] [PubMed] [Google Scholar]
- Mittal P, Shin YH, Yatsenko SA, Castro CA, Surti U, Rajkovic A. Med12 gain-of-function mutation causes leiomyomas and genomic instability. J Clin Invest. 2015;125((8)):3280–3284. doi: 10.1172/JCI81534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8((3)):193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- Ramos JM, Ruiz A, Colen R, Lopez ID, Grossman L, Matta JL. DNA repair and breast carcinoma susceptibility in women. Cancer. 2004;100((7)):1352–1357. doi: 10.1002/cncr.20135. [DOI] [PubMed] [Google Scholar]
- Benhamou S, Sarasin A. Variability in nucleotide excision repair and cancer risk: a review. Mutat Res. 2000;462((2-3)):149–158. doi: 10.1016/s1383-5742(00)00032-6. [DOI] [PubMed] [Google Scholar]
- Matta JL, Villa JL, Ramos JM, Sanchez J, Chompre G, Ruiz A, Grossman L. DNA repair and nonmelanoma skin cancer in Puerto Rican populations. J Am Acad Dermatol. 2003;49((3)):433–439. doi: 10.1067/s0190-9622(03)00918-6. [DOI] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419((6907)):624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Yoo KH, Hennighausen L. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int J Sci. 2012;8((1)):59–65. doi: 10.7150/ijbs.8.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24((2)):268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100((20)):11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Teckie S, Wiesner T, Ran L. PrietoGranada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, et al., editors. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46((11)):1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski IJ, Blewitt ME, de Graaf CA, McManus EJ, Bahlo M, Hilton AA, Hyland CD, Smyth GK, Corbin JE, Metcalf D, Alexander WS, Hilton DJ. Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLoS Biol. 2008;6((4)):e93. doi: 10.1371/journal.pbio.0060093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hung MC. Regulation and role of EZH2 in cancer. Cancer Res Treat. 2014;46((3)):209–222. doi: 10.4143/crt.2014.46.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106((2)):243–247. doi: 10.1038/bjc.2011.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Hung MC, Li LY. EZH2: a pivotal regulator in controlling cell differentiation. Am J Transl Res. 2012;4((4)):364–375. [PMC free article] [PubMed] [Google Scholar]
- Holm K, Grabau D, Lovgren K, Aradottir S, Gruvberger-Saal S, Howlin J, Saal LH, Ethier SP, Bendahl PO, Stal O, Malmstrom P, Ferno M, et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Molecular Oncol. 2012;6((5)):494–506. doi: 10.1016/j.molonc.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JZ, Yan Y, Wang XX, Jiang Y, Xu HE. EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol Sin. 2014;35((2)):161–174. doi: 10.1038/aps.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y. Targeting histone methyltransferase EZH2 as cancer treatment. J Biochem. 2014;156((5)):249–257. doi: 10.1093/jb/mvu054. [DOI] [PubMed] [Google Scholar]
- Akinyemi BO, Adewoye BR, Fakoya TA. Uterine fibroid: a review. Niger J Med. 2004;13((4)):318–329. [PubMed] [Google Scholar]
- Yang Q, Mas A, Diamond MP, Al-Hendy A. The mechanism and function of epigenetics in uterine leiomyoma development. Reprod Sci. 2016;23:163–175. doi: 10.1177/1933719115584449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney SA, Tahara H, Swartz CD, Risinger JI, He H, Moore AB, Haseman JK, Barrett JC, Dixon D. Immortalization of human uterine leiomyoma and myometrial cell lines after induction of telomerase activity: molecular and phenotypic characteristics. Lab Invest. 2002;82((6)):719–728. doi: 10.1097/01.lab.0000017499.51216.3e. [DOI] [PubMed] [Google Scholar]
- Halder SK, Osteen KG, Al-Hendy A. Vitamin D3 inhibits expression and activities of matrix metalloproteinase-2 and -9 in human uterine fibroid cells. Hum Reprod. 2013;28((9)):2407–2416. doi: 10.1093/humrep/det265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TC, Rogers AW, Dockery P, Lenton EA, Cooke ID. A new method of histologic dating of human endometrium in the luteal phase. Fertil Steril. 1988;50((1)):52–60. doi: 10.1016/s0015-0282(16)60008-0. [DOI] [PubMed] [Google Scholar]
- Yang Q, Liu S, Tian Y, Hasan C, Kersey D, Salwen HR, Chlenski A, Perlman EJ, Cohn SL. Methylation-associated silencing of the heat shock protein 47 gene in human neuroblastoma. Cancer Res. 2004;64((13)):4531–4538. doi: 10.1158/0008-5472.CAN-04-0956. [DOI] [PubMed] [Google Scholar]
- Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU. Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation. Am J Physiol Lung Cell Mol Physiol. 2012;303((11)):L1001–1010. doi: 10.1152/ajplung.00092.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard N, Bazille C, Lhuissier E, Benateau H, Llombart-Bosch A, Boumediene K, Bauge C. 3-Deazaneplanocin A (DZNep), an inhibitor of the histone methyltransferase EZH2, induces apoptosis and reduces cell migration in chondrosarcoma cells. Plos One. 2014;9((5)):e98176. doi: 10.1371/journal.pone.0098176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang QW, Liu S, Tian Y, Salwen HR, Chlenski A, Weinstein J, Cohn SL. Methylation-associated silencing of the thrombospondin-1 gene in human neuroblastoma. Cancer Res. 2003;63((19)):6299–6310. [PubMed] [Google Scholar]
- Yang Q, Tian Y, Ostler KR, Chlenski A, Guerrero LJ, Salwen HR, Godley LA, Cohn SL. Epigenetic alterations differ in phenotypically distinct human neuroblastoma cell lines. BMC Cancer. 2010;10:286. doi: 10.1186/1471-2407-10-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Dahl MJ, Albertine KH, Ramchandran R, Sun M, Raj JU. Role of histone deacetylases in regulation of phenotype of ovine newborn pulmonary arterial smooth muscle cells. Cell Prolif. 2013;46((6)):654–664. doi: 10.1111/cpr.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development. 2007;134((2)):223–232. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- Ward A, Khanna KK, Wiegmans AP. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat Rev. 2015;41((1)):35–45. doi: 10.1016/j.ctrv.2014.10.006. [DOI] [PubMed] [Google Scholar]
- Subramanyam S, Jones WT, Spies M, Spies MA. Contributions of the RAD51 N-terminal domain to BRCA2-RAD51 interaction. Nucleic Acids Res. 2013;41((19)):9020–9032. doi: 10.1093/nar/gkt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwood AC, Malysewich MJ, Cealic I, Mundia MM, Knapp J, Baker MD. Endogenous levels of Rad51 and Brca2 are required for homologous recombination and regulated by homeostatic re-balancing. DNA Repair. 2013;12((12)):1122–1133. doi: 10.1016/j.dnarep.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Bernier J, Poortmans P. Clinical relevance of normal and tumour cell radiosensitivity in BRCA1/BRCA2 mutation carriers: a review. Breast. 2015;24:100–106. doi: 10.1016/j.breast.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Carcangiu ML, Peissel B, Pasini B, Spatti G, Radice P, Manoukian S. Incidental carcinomas in prophylactic specimens in BRCA1 and BRCA2 germ-line mutation carriers, with emphasis on fallopian tube lesions: report of 6 cases and review of the literature. Am J Surg Pathol. 2006;30((10)):1222–1230. doi: 10.1097/01.pas.0000202161.80739.ac. [DOI] [PubMed] [Google Scholar]
- Paterson JW. BRCA1: a review of structure and putative functions. Dis Markers. 1998;13((4)):261–274. doi: 10.1155/1998/298530. [DOI] [PubMed] [Google Scholar]
- Narod S, Lynch H, Conway T, Watson P, Feunteun J, Lenoir G. Increasing incidence of breast cancer in family with BRCA1 mutation. Lancet. 1993;341((8852)):1101–1102. doi: 10.1016/0140-6736(93)92468-9. [DOI] [PubMed] [Google Scholar]
- Fuller S, Liebens F, Carly B, Pastijn A, Rozenberg S. Breast cancer prevention in BRCA1/2 mutation carriers: a qualitative review. BReast J. 2008;14((6)):603–604. doi: 10.1111/j.1524-4741.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- Lee EH, Park SK, Park B, Kim SW, Lee MH, Ahn SH, Son BH, Yoo KY, Kang D. Effect of BRCA1/2 mutation on short-term and long-term breast cancer survival: a systematic review and meta-analysis. Breast Cancer Res Treat. 2010;122((1)):11–25. doi: 10.1007/s10549-010-0859-2. Kohbra Research Group; Korean Breast Cancer Society. [DOI] [PubMed] [Google Scholar]
- Cousineau I, Abaji C, Belmaaza A. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: implications for sister chromatid cohesion, genome stability, and carcinogenesis. Cancer Res. 2005;65((24)):11384–11391. doi: 10.1158/0008-5472.CAN-05-2156. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Silver D, Cantor S, Livingston DM, Scully R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res. 1999;59((7 Suppl)):1752s–1756s. [PubMed] [Google Scholar]
- Yuan R, Fan S, Wang JA, Meng Q, Ma Y, Schreiber D, Goldberg ID, Rosen EM. Coordinate alterations in the expression of BRCA1, BRCA2, p300, and Rad51 in response to genotoxic and other stresses in human prostate cancer cells. Prostate. 1999;40((1)):37–49. doi: 10.1002/(sici)1097-0045(19990615)40:1<37::aid-pros5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wen Y, Polan ML, Qiao J, Chen BH. Increased expression of latent TGF-beta binding protein-1 and fibrillin-1 in human uterine leiomyomata. Mol Hum Reprod. 2007;13((5)):343–349. doi: 10.1093/molehr/gam007. [DOI] [PubMed] [Google Scholar]
- Cao W, Younis RH, Li J, Chen H, Xia R, Mao L, Chen W, Ren H. EZH2 promotes malignant phenotypes and is a predictor of oral cancer development in patients with oral leukoplakia. Cancer Prev Res (Phila) 2011;4((11)):1816–1824. doi: 10.1158/1940-6207.CAPR-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17((9)):2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647((1-2)):21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ, Jiang X, Tan J, Aau M, Lim CZ, Yu Q. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18((11)):1771–1779. doi: 10.1038/cdd.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7((3)):299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22((20)):5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhan A, Hussain I, Ansari KI, Bobzean SA, Perrotti LI, Mandal SS. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-A and diethylstilbestrol. J Mol Biol. 2014;426((20)):3426–3441. doi: 10.1016/j.jmb.2014.07.025. [DOI] [PubMed] [Google Scholar]
- Shozu M, Murakami K, Segawa T, Kasai T, Inoue M. Successful treatment of a symptomatic uterine leiomyoma in a perimenopausal woman with a nonsteroidal aromatase inhibitor. Fertil Steril. 2003;79((3)):628–631. doi: 10.1016/s0015-0282(02)04761-1. [DOI] [PubMed] [Google Scholar]
- Hilario SG, Bozzini N, Borsari R, Baracat EC. Action of aromatase inhibitor for treatment of uterine leiomyoma in perimenopausal patients. Fertil Steril. 2009;91((1)):240–243. doi: 10.1016/j.fertnstert.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Burns KA, Korach KS. Estrogen receptors and human disease: an update. Arch Toxicol. 2012;86((10)):1491–1504. doi: 10.1007/s00204-012-0868-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Burns KA, Arao Y, Luh CJ, Korach KS. Differential estrogenic actions of endocrine-disrupting chemicals bisphenol A, bisphenol AF, and zearalenone through estrogen receptor alpha and beta in vitro. Environ Health Perspect. 2012;120((7)):1029–1035. doi: 10.1289/ehp.1104689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer C, Juhasz-Boess I, Lattrich C, Ortmann O, Treeck O. Estrogen receptor beta gene polymorphisms and susceptibility to uterine fibroids. Gynecol Endocrinol. 2010;26((1)):4–9. doi: 10.3109/09513590903159573. [DOI] [PubMed] [Google Scholar]
- Villanova FE, Andrade PM, Otsuka AY, Gomes MT, Leal ES, Castro RA, Girao MJ, Nishimura E, Baracat EC, Silva ID. Estrogen receptor alpha polymorphism and susceptibility to uterine leiomyoma. Steroids. 2006;71((11–12)):960–965. doi: 10.1016/j.steroids.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, Quintanilla-Martinez L, Kovar H, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A. 2009;106((13)):5324–5329. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Zhang Y, Ren K, Zhao G, Yan K, Ma B. MicroRNA-101 inhibits the metastasis of osteosarcoma cells by downregulation of EZH2 expression. Oncology Rep. 2014;32((5)):2143–2149. doi: 10.3892/or.2014.3459. [DOI] [PubMed] [Google Scholar]
- Konno Y, Dong P, Xiong Y, Suzuki F, Lu J, Cai M, Watari H, Mitamura T, Hosaka M, Hanley SJ, Kudo M, Sakuragi N. MicroRNA-101 targets EZH2, MCL-1 and FOS to suppress proliferation, invasion and stem cell-like phenotype of aggressive endometrial cancer cells. Oncotarget. 2014;5((15)):6049–6062. doi: 10.18632/oncotarget.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Verrijzer P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr Opin Genet Dev. 2009;19((2)):150–158. doi: 10.1016/j.gde.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19((1)):86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler M, Varambally S, Cao Q, Chinnaiyan AM, Ferguson DO, Merajver SD, Kleer CG. The Polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia. 2005;7((11)):1011–1019. doi: 10.1593/neo.05472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Cai J, Ding H, Xu L, Yang Q, Wang Z. EZH2 participates in malignant biological behavior of epithelial ovarian cancer through regulating the expression of BRCA1. Cancer Biol Ther. 2014;15((3)):271–278. doi: 10.4161/cbt.27306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard A, Goossens-Beumer IJ, van Hoesel AQ, Horati H, Putter H, Zeestraten EC, van de Velde CJ, Kuppen PJ. Prognostic value of polycomb proteins EZH2, BMI1 and SUZ12 and histone modification H3K27me3 in colorectal cancer. Plos One. 2014;9((9)):e108265. doi: 10.1371/journal.pone.0108265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae WK, Yoo KH, Lee JS, Kim Y, Chung IJ, Park MH, Yoon JH, Furth PA, Hennighausen L. The methyltransferase EZH2 is not required for mammary cancer development, although high EZH2 and low H3K27me3 correlate with poor prognosis of ER-positive breast cancers. Mol Carcing. 2015;54:1172–1180. doi: 10.1002/mc.22188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science. 2012;338((6113)):1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, Zhang Y, Hong M, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27((14)):5105–5119. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310((5746)):306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol. 2010;24((5)):993–1006. doi: 10.1210/me.2009-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21((9)):1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YX, Denlinger DL, Xu WH. Polycomb repressive complex 2 (PRC2) protein ESC regulates insect developmental timing by mediating H3K27me3 and activating prothoracicotropic hormone gene expression. J Biol Chem. 2013;288((32)):23554–23564. doi: 10.1074/jbc.M113.482497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avan A, Crea F, Paolicchi E, Funel N, Galvani E, Marquez VE, Honeywell RJ, Danesi R, Peters GJ, Giovannetti E. Molecular mechanisms involved in the synergistic interaction of the EZH2 inhibitor 3-deazaneplanocin A with gemcitabine in pancreatic cancer cells. Mol Cancer Ther. 2012;11((8)):1735–1746. doi: 10.1158/1535-7163.MCT-12-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury SR, Balasubramanian S, Chew YC, Han B, Marquez VE, Eckert RL. (−)-Epigallocatechin-3-gallate and DZNep reduce polycomb protein level via a proteasome-dependent mechanism in skin cancer cells. Carcinogenesis. 2011;32((10)):1525–1532. doi: 10.1093/carcin/bgr171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au SL, Wong CC, Lee JM, Wong CM, Ng IO. EZH2-mediated H3K27me3 is involved in epigenetic repression of Deleted in liver cancer 1 in human cancers. Plos One. 2013;8((6)):e68226. doi: 10.1371/journal.pone.0068226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8((6)):1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrup O, Reiner AH, Alestrom P, Collas P. The specific alteration of histone methylation profiles by DZNep during early zebrafish development. Biochim Biophys Acta. 2014;1839((11)):1307–1315. doi: 10.1016/j.bbagrm.2014.09.013. [DOI] [PubMed] [Google Scholar]