

Synopsis
Membrane fusion is one of the most important cellular process by which two initially distinct lipid bilayers merge their hydrophobic cores, resulting in one interconnected structure. Proteins, called SNARE, play a central role in the fusion process which is also regulated by several accessory proteins. In order to study the SNARE-mediated membrane fusion, the in vitro protein reconstitution assay involving ensemble fluorescence resonance energy transfer FRET has been used over a decade. In this mini-review, we describe several single-molecule-based FRET approaches that have been applied to this field to overcome the shortage of the bulk assay in terms of protein and fusion dynamics.
Keywords: SNARE, membrane fusion, single molecule, FRET
Introduction
SNARE (soluble NSF attachment protein receptors)-mediated membrane fusion is a fundamental cellular process involved in many important life activities, such as egg fertilization, protein trafficking, and neurotransmitter release [1]. In order to communicate with each other, molecules called neurotransmitters are used for messages across the synaptic cleft between two neurons at the synapse. Synaptic vesicles in the axon bulb containing neurotransmitters are released in the submillisecond time scale by exocytosis into the synaptic cleft. As summarized in Table 1, this calcium-triggered fast neurotransmitter release is mediated by SNARE proteins and is tightly regulated by a number of accessory proteins, such as synaptotagmin, complexin, and Munc18 [2].
Table 1.
Key proteins involving in the SNARE-mediated membrane fusion for fast neurotransmitter release.
| Name | Family | Location | Function |
|---|---|---|---|
| synaptobrevin | SNARE | Synaptic vesicle | To form SNARE complex, which is known as the minimum fusion machinery |
| SNAP-25 | SNARE | Target membrane | |
| syntaxin | SNARE | Target membrane | |
| complexin | Free | Dual-functional regulator | |
| synaptotagmin | Synaptic vesicle | Calcium sensor | |
| Munc18 | SM | Free | Docking and priming regulator |
Although many proteins involved in the membrane fusion have been identified, the detailed mechanism still remains ambiguous. Traditionally, most studies rely on two major techniques, in vivo knockout and in vitro reconstitution methods [2,3]. For the scope of this review, we will only focus on the advancement of in vitro assays.
The in vitro protein reconstitution assay involving ensemble fluorescence resonance energy transfer (FRET) has been used to study the SNARE-mediated membrane fusion for a decade. Despite the great success of ensemble FRET for studying SNARE-mediated membrane fusion, it falls short in revealing the fast and transient fusion dynamics. In order to overcome limitations of conventional methods, more advanced assays are expected to study membrane fusion. Since single-molecule techniques have many advantages over ensemble or bulk measurements, it naturally becomes a good candidate of the substantial technique [4]. By watching single molecules, people can see many details such as different fusion states that would have been averaged out in the ensemble measurement.
FRET is a popular biological technique because of its sensitive distance range and for its compatibility to observe real-time reactions in biologically relevant conditions. The FRET signal is sensitive to 2-10 nanometer-scale distances between donor and acceptor fluorophores [5]. This range helps us understand molecular interactions, such as protein-protein, protein-DNA and antibody-antigen. Fluorophores commonly used for FRET measurements are small organic dyes, and with control, they do not purturb the biological function and are stable under most biologically relevant conditions. Two major ways of utilizing single-molecule FRET (smFRET) technique have been applied on SNARE-mediated membrane fusion through monitoring SNARE protein interactions [6,7,8,9,10] or fusion of lipid molecules [11,12,13,14,15,16]. Most recent application of the smFRET approach has been the use of labeled content [17] and the combination of the lipid marker and the labeled content [18].
Monitoring Proteins
Many biologically relevant protein conformation and protein-protein interactions occur within the sensitivity range of FRET measurement. SNARE and its accessory proteins that are site-specifically conjugated with fluorescent dyes may be used to provide unique structural information [19]. There are number of conventional techniques to study the structure and conformation of SNARE proteins: X-ray crystallography, variety of nuclear magnetic resonance techniques, electron paramagnetic resonance, electron microscopy and circular dichroism to name the few. However, there are many instances where structural determination is challenging using these conventional methods for various reasons. Some examples include instability of protein complex at high concentration, fluctuation of the structure between multiple conformations and the averaging out of the active complex signal by being only a fraction of the large ensemble.
The smFRET approach provides an alternative to conventional methodologies. High sensitivity of the state-of-the-art camera allows detection down to a single fluorophore. Because signals from individual molecules or complexes are independently recorded, synchronization is not necessary and transient processes (such as domain conformational change) may be studied without being averaged out. Here, we discuss the overview of smFRET measurement applications on fluorescent-dye-labeled SNARE proteins by categorizing them into three groups depending on the type of data obtained: stoichiometry, intramolecular conformation and intermolecular orientation.
(i) Stoichiometry
One of the fundamental information obtained from single molecule detection is the localization of the molecule on the surface. Although FRET signal is not required, precise localization (within diffraction limited spot of ∼200 nm without fitting or within several nanometer with fitting) allows identification of the target protein and co-localization of different proteins of interest. By measuring the resident on and off times from the fluorescence intensity time trace, an accurate binding constant of the protein may be obtained [7]. However, perhaps the most useful information that may be obtained from localizing individual molecules is the stoichiometry of the complex. Each individual fluorophore photobleaches with a sharp drop of intensity. The number of drops within the fluorescence intensity time trace reveals the number of molecules. The cytoplasmic domain of syntaxin form fusion inactive dimer complex in micromolar affinity and also inactive tetramer with ten micromolar to millimolar affinity [20]. Photobleaching analysis of individual fluorescence intensity time profile has was used to ensure the dye-labeled-syntaxin molecules are in the active monomeric state within the surface deposited lipid bilayer [8].
The minimum number of SNARE complexes necessary for membrane fusion is of a fundamental interest. Van den Bogaart and colleagues also applied a similar photobleaching analysis to quantitate the number of synaptobrevin and acceptor SNARE complex (syntaxin and SNAP-25) reconstituted in the vesicle embedded in the agarose gel matrix [21]. Using vesicle samples containing, on the average, one synaptobrevin in one and one acceptor complex in another set of vesicles, they have concluded that single SNARE complex is sufficient for inducing lipid mixing.
(ii) Intramolecular conformation
The neurotransmitter release process takes place within several milliseconds, and it is important to study the protein conformation dynamic in the similar time scale. In one of the earliest work to apply smFRET on SNARE proteins, syntaxin 1a was dually labeled with Alexa 488 (donor) and Alexa 594 (acceptor) at several combinations of protein sites and the multiple physical parameters (intramolecular distance from FRET, fluorescence lifetime and anisotropy) have simultaneously been quantified in the presence and absence of interacting partners [10]. While contradictory from a NMR study, dynamic switching between opening and closing of N-terminal three helix bundle of syntaxin 1a with a relaxation time of 0.8 ms was observed from free diffusing syntaxin molecules [22]. Once the four-helix bundle of SNARE complex is formed, its unusal stability is well known [23,24]. In a different way, however, the conformational dynamic of two helices of SNAP-25 in the context of 1:1 binary acceptor complex was not known until recently [8].
Weninger and colleagues labeled SNAP-25 helices with Cy3 (donor) and Cy5 (acceptor) and observed, to their surprise, a significant conformational flexibility of helices bound to syntaxin molecule embedded in the lipid bilayer represented by the mid-FRET efficiency signal [8]. The real-time analysis of the FRET efficiency profile showed a transient motion of helices between two stable parallel and anti-parallel conformations. Completion of four-helix bundle by addition of synaptobrevin stabilizes the binding SNAP-25 helices. Interestingly, addition of cytosolic fragment of synaptotagmin 1 to the binary acceptor complex also stabilized the conformational dynamic to the similar extent as synaptobrevin, which authors suggest its role in “setting the stage” for the efficient tran-SNARE formation. Binding to the SNARE binary complex also stabilized the conformational dynamic of synaptotagmin itself even in the absence of Ca2+, but the effect is more pronounced in the presence of Ca2+ [25,26].
In a recent study, a similarly dual-labeled neuronal SNAP-25 and widely expressed SNAP-29 were microinjected into live cells to investigate the conformational dynamic of the protein by combining smFRET and single particle tracking [27]. From this proof of concept study, they verified the promiscuous nature of SNAP-25 binding, which results in folding, to non-neuronal origin syntaxin molecules.
(iii) Intermolecular interaction and orientation
Structural determination of multicomplex systems that may bind to one another in more than one conformation or that are weakly interacting is challenging. To obtain the accurate structural information on such system, FRET pair fluorophores may be conjugated to different interacting partner proteins. Using this strategy, the relative orientations and stabilities of syntaxin, synaptobrevin and SNAP-25 has been investigated through smFRET [6,8].
With careful calibration, FRET efficiency signal may be converted into the distance information [28,29,30]. Taking the full advantage of this, Choi and colleagues obtained 34 smFRET derived distances to elucidate the relative arrangement of synaptotagmin 1 with respect to the SNARE complex [26]. In conjunction with previously known crystal structures, they have fitted these measured distances to show synaptotagmin interacts with the central region of the SNARE complex without obstructing the complexin binding site.
(iv) Caveats
The smFRET measurements from labeled SNARE protein provides dynamic structural information unobtainable by conventional ensemble methods. A typical strategy to label proteins with fluorescent dyes is done through forming covalent linkage to naturally existing or cloned cysteine residue(s). However, special care must be taken in selecting the site of dye conjugation. First, the labeling site of the dye needs to be chosen such that the native function of the protein is not perturbed [9]. High-resolution crystal structure will greatly assist in this process, but with or without such information, a series of control biochemical assays must be done to ensure the functionality. Second, the labeled fluorescent dye should be free of interactions with nearby amino acid residues; otherwise it may result in unpredictable photoemission. Both enhancement as well as quenching of dye is known to happen. This will potentially skew the conversion of FRET efficiencies to distances. A further review on this topic may be found elsewhere [31].
Monitoring Fusion
The application of smFRET has a great advantage in monitoring different stages of membrane fusion events. Compared to the ensemble fusion assay, where averaged FRET signal from the entire population is obtained, instead, the FRET efficiency value from each pair of vesicles may be collected to identify different stages of fusion such as docking, hemifusion and full fusion. The importance of such detailed information may be exemplified in the experiment of fast vesicle aggregation induced by C2AB/Ca2+ [14]. Using the ensemble lipid mixing assay (Figure 1A), upon addition of Ca2+ into the system, the sudden increase of FRET signal has been observed. This indicates a high degree of lipid mixing, which may be interpreted as an efficient fusion. However, upon testing the same system using the single vesicle-vesicle lipid mixing assay (Figure 1B), the FRET efficiency distribution peaks at ∼0.3, which signifies the aggregation of vesicles [11]. The ensemble assay is incapable of distinguishing between the aggregation and the fused membranes, therefore additional supplemental experiments may be necessary for an accurate interpretation of the data. In order to watch fusion process, labeled lipid or content molecules are mainly used as fusion reporters for smFRET.
Figure 1.
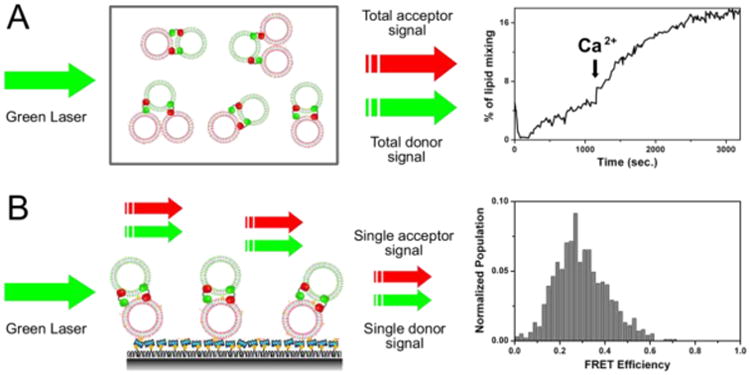
Schematics of the bulk fusion assay (A) and the single-vesicle lipid mixing assay (B) for the mixture of protein free vesicles (35% PS & 65% PC, DiD or DiI labeled), C2AB (cytoplasmic domain of synaptotagmin) and 1 mM calcium. The single-vesicle FRET histogram is plotted by compiling FRET signals from over one thousand vesicles. Y-axis is normalized population, where we divided the distribution by the total number of vesicles measured and X-axis is FRET efficiency value.
(i) Labeling lipid
When membrane fusion occurs between two membranes, lipid molecules in these membranes undergo two-dimensional diffusion and completely get mixed together. This process, called lipid mixing can be used as marker for membrane fusion [32]. Actually lipid mixing is good marker to observe membrane fusion because labeling membrane is relatively easy with in vitro systems and lipid mixing process is fast enough so it does not hamper time resolution of many microscopy systems [33].
First in vitro lipid mixing experiment on SNARE-driven membrane fusion used a FRET pair consisting of NBD and rhodamine [34]. Two groups of vesicles were prepared containing v-SNARE proteins (SNARE proteins located on synaptic vesicle, v-vesicle) and t-SNARE proteins (SNARE proteins on target membrane, t-vesicle), respectively. V-vesicles were labeled with NBD and rhodamine so that NBD fluorescence is quenched by nearby rhodamine via FRET. When t-vesicles were added to v-vesicles, vesicle fusion and substantial lipid mixing caused dilution of labeled v-vesicle lipids with unlabeled t-vesicle lipids. As the average inter-dye distance between NBD and rhodamine increased, fluorescence signal from NBD recovered. This NBD fluorescence recovery was recorded in bulk solution, which worked as the evidence that SNARE proteins can work as the minimal fusion machinery in vitro system.
The ensemble lipid mixing assay is a convenient tool for recording overall fusion kinetics in the solution. However, it records signal from a large number of vesicles and therefore, does not give information about kinetics of each fusion step, which is an important component to understand detailed fusion mechanism. To observe vesicle fusion with single vesicle resolution, several methods including single vesicle-bilayer fusion [35,36,37] and single vesicle-vesicle fusion systems were invented [11].
The single vesicle-bilayer fusion assay with the geometry similar to the real synaptic fusion, has been developed by several groups [35,36,37]. They were able to record single vesicles fusing to the planar lipid bilayer. However, these studies based on planar bilayers suffered from results contradictory to the known SNARE mechanism. For example, SNAP-25 was not required in the acceptor t-SNARE complex [36,37] or the calcium dependent fusion was observed in the absence of the calcium sensor protein, synaptotagmin [35], which might be induced by the substrate surface. Recently, several groups recovered SNAP-25 dependence of single-vesicle fusion assays in planar bilayers by directly incorporating polyethylene glycol (PEG), a synthetic polymer to mimic surface effect, into the fusion system [38,39].
Single vesicle-vesicle fusion system utilizes specifically attached acceptor dye (DiD) labeled v-vesicles on the polymer passivated and functionalized imaging surface. After immobilization is complete, donor dye (DiI) labeled t-vesicles are added and membrane fusion between t- and v- vesicles are measured using TIR microscope. What is notable in this approach is, from calculated FRET efficiency values from single v- and t-vesicle complexes, docked (FRET<0.25) and fused (FRET>0.6) vesicle complexes are clearly distinguishable. Comparing with bulk fusion method, this technique can also distinguish intermediate states between docked and fully fused vesicles (e.g. hemifusion), quantify the kinetics of transitions between individual intermediate states, and detect post-fusion pathway such as the kiss-and-run event. Such information may be obtained at a certain time point as well as from real-time fluorescence time traces from hundreds of vesicle-vesicle interactions in parallel. Through this assay, accessory proteins, including complexin [13], Munc18 [15], and membrane-anchored synaptotagmin 1 [16] have been shown to work with SNARE proteins to accelerate the lipid mixing process, but in unique manners.
(ii) Labeling content
The fundamental assumption of using lipid mixing to study membrane fusion is that there is a direct correlation between lipid and content mixing. However, through simultaneous detection of lipid and small content indicators of DNA-mediated membrane fusion, Boxer and coworkers revealed that efficient lipid mixing (> 90%) of both outer and inner leaflets can occur without content mixing (< 2%) [40]. This study calls into question liposome “fusion” assays that rely on lipid-mixing indicator alone to assess the fusion. This study strongly suggests that future vesicle fusion experiments have to employ content-mixing indicators in addition to lipid-mixing indicators.
Even prior to the wide spread of ensemble lipid-mixing assay, Rothman and coworkers utilized radioactive probe labeled DNA to report that SNARE proteins constitute the minimal machinery for membrane fusion [41]. However, in this assay, the content mixing signal was measured after vesicles were lysed by detergent, leaving a possibility that docked vesicles without fusion could still contribute to the final readout. To observe the SNARE-mediated membrane fusion, fluorescent-dye-based small indicators have previously been substituted for lipid-dye through the ensemble approach. However, when large number of transmembrane domain-containing SNARE proteins are reconstituted into vesicles, membranes are destabilized, causing the indicator molecules to leak out of the vesicle over time [42]. Unless a very low protein to lipid ratio (< 1:1000) was used [21,43], this leakage problem makes the data analysis challenging.
Approaches involving smFRET have also been used to detect the content mixing. For single-vesicle fusion assays utilizing planar bilayers, small content indicators showed that SNAP-25 was not required in the acceptor t-SNARE complex for fusion, which is contradictive to known physiology and biochemistry [36,44].
A more promising result comes from a vesicle-vesicle content mixing assay [17]. In this assay, a large Cy3/Cy5 dual-labeled DNA probe has been encapsulated inside surface immobilized v-vesicle. The soluble t-vesicles contains the complementary DNA, and upon two cavities of vesicles inter-connect through fusion, two DNA strands may hybridize to induce a conformational change. Through smFRET detection of this conformational change, authors are able to detect the fusion pore expansion, the very late step of membrane fusion process, for the first time.
Outlook
The single molecule FRET methodologies are still evolving outside of the SNARE applications. By using increased number of fluorophores, 3-color, 4-color and other variations of FRET measurements are possible [45,46,47,48]. This enables monitoring of multiple relative distances simultaneously. Although previously have been done, multiparameter measurements to include parameters such as anisotropy and fluorescence lifetime may benefit in observing the assembly of complexes and domain flexibility [10]. In order to increase the number of fluorophores, novel labeling schemes, such as incorporation of unnatural amino acids and short amino acid tags, to allow orthogonal site-specific labeling is necessary [49]. In order to achieve physiologically relevant micromolar concentrations of labeled protein, the use of nanofabricated device such as zero-mode waveguide or tightly focused excitation beam of STED microscopy may broaden the concentration range of the study [50,51].
Studying membrane fusion based on protein-reconstituted vesicles has achieved tremendous successes, but certainly has areas of development In order to understand how lipid mixing steps are related to the pore expansion step, simultaneous lipid- and content-mixing detection assays are desirable. With such assays, the exact roles of multiple accessory proteins that all seemingly promote lipid-mixing (or inhibit) may be identified. The single vesicle lipid- and content-mixing assays described above are blind to the conformations of SNARE protein. Understanding how SNARE proteins and its accessory proteins coordinate themselves during these observed fusion intermediate steps not only will deepen our understanding of the system, but to discover new drug targets. Because the size of common vesicles (50 ∼ 100 nm in diameter) is smaller than the diffraction limit, direct optical observation is impossible. In order to overcome this issue, utilization of new super resolution imaging techniques are demanded, such as stochastic optical reconstruction microscopy (STORM) [52], may become an ultimate tool to study protein-protein interactions during the SNARE-mediated membrane fusion process.
Acknowledgments
We thank Dr. Taekjip Ha, Dr. Yeon-Kyun Shin, and Dr. Tae-Young Yoon for continuous support, and Dr. Zengliu Su for illustration preparation.
Funding: The work on single vesicle FRET assay in this review was supported by National Institutes of Health (R21 GM074526) and Howard Hughes Medical Institute through Dr. Taekjip Ha at University of Illinois.
Abbreviations used
- FRET
fluorescence resonance energy transfer
- SNARE
soluble NSF attachment protein receptors
- TIR
total internal reflection
References
- 1.White JM. Membrane fusion. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 2.Jahn R, Lang T, Sudhof TC. Membrane fusion. Cell. 2003;112:519–533. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 3.Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoon TY, Shin YK. Progress in understanding the neuronal SNARE function and its regulation. Cell Mol Life Sci. 2009;66:460–469. doi: 10.1007/s00018-008-8372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stryer L. Fluorescence energy transfer as a spectroscopic ruler. Ann Rev Biochem. 1978;47:819–846. doi: 10.1146/annurev.bi.47.070178.004131. [DOI] [PubMed] [Google Scholar]
- 6.Weninger K, Bowen ME, Chu S, Brunger AT. Single-molecule studies of SNARE complex assembly reveal parallel and antiparallel configurations. Proc Natl Acad Sci U S A. 2003;100:14800–14805. doi: 10.1073/pnas.2036428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowen ME, Weninger K, Ernst J, Chu S, Brunger AT. Single-molecule studies of synaptotagmin and complexin binding to the SNARE complex. Biophys J. 2005;89:690–702. doi: 10.1529/biophysj.104.054064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT. Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP-25 complex. Structure. 2008;16:308–320. doi: 10.1016/j.str.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Augustine GJ, Weninger K. Kinetics of complexin binding to the SNARE complex: correcting single molecule FRET measurements for hidden events. Biophys J. 2007;93:2178–2187. doi: 10.1529/biophysj.106.101220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margittai M, Widengren J, Schweinberger E, Schroder GF, Felekyan S, Haustein E, Konig M, Fasshauer D, Grubmuller H, Jahn R, Seidel CAM. Single-molecule fluorescence resonance energy transfer reveals a dynamic equilibrium between closed and open conformations of syntaxin 1. Proc Natl Acad Sci U S A. 2003;100:15516–15521. doi: 10.1073/pnas.2331232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon TY, Okumus B, Zhang F, Shin YK, Ha T. Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A. 2006;103:19731–19736. doi: 10.1073/pnas.0606032103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su Z, Ishitsuka Y, Ha T, Shin YK. The SNARE complex from yeast is partially unstructured on the membrane. Structure. 2008;16:1138–1146. doi: 10.1016/j.str.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon TY, Lu X, Diao J, Lee SM, Ha T, Shin YK. Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat Struct Mol Bio. 2008;15:707–713. doi: 10.1038/nsmb.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diao J, Yoon TY, Su Z, Shin YK, Ha T. C2AB: a molecular glue for lipid vesicles with negatively charged surface. Langmuir. 2009;25:7177–7180. doi: 10.1021/la901676e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diao J, Su Z, Lu X, Yoon TY, Shin YK, Ha T. Single-vesicle fusion assay reveals Munc18-1 binding to the SNARE core is sufficient for stimulating membrane fusion. ACS Chem Neurosci. 2010;1:168–174. doi: 10.1021/cn900034p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HK, Yang Y, Su Z, Hyeon C, Lee TS, Lee HW, Kweon DH, Shin YK, Yoon TY. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science. 2010;328:760–763. doi: 10.1126/science.1187722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diao J, Su Z, Ishitsuka Y, Lu B, Lee KS, Lai Y, Shin YK, Ha T. A single vesicle content-mixing assay for SNARE-mediated membrane fusion. Nat Commun. 2010;1:54. doi: 10.1038/ncomms1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kyoung M, Srivastava A, Diao J, Zhang Y, Vrljic M, Grob P, Nogales E, Chu S, Brunger AT. Ca2+-triggered kinetic control of SNARE-dependent synaptic vesicle fusion by accessory factors. 2011 submitted. [Google Scholar]
- 19.Brunger AT, Weninger K, Bowen M, Chu S. Single-molecule studies of the neuronal SNARE fusion machinery. Ann Rev Biochem. 2009;78:903–928. doi: 10.1146/annurev.biochem.77.070306.103621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lerman JC, Robblee J, Fairman R, Hughson FM. Structural analysis of the neuronal SNARE protein syntaxin-1. A Biochem. 2000;39:8470–8479. doi: 10.1021/bi0003994. [DOI] [PubMed] [Google Scholar]
- 21.van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R. One SNARE complex is sufficient for membrane fusion. Nat Struct Mol Biol. 2010;17:358–364. doi: 10.1038/nsmb.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Lu J, Dulubova I, Rizo J. NMR analysis of the closed conformation of syntaxin-1. J Biomol NMR. 2008;41:43–54. doi: 10.1007/s10858-008-9239-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutton R, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 24.Strop P, Kaiser SE, Vrljic M, Brunger AT. The Structure of the yeast plasma membrane SNARE complex reveals destabilizing water-filled cavities. J Bio Chem. 2008;283:1113–1119. doi: 10.1074/jbc.M707912200. [DOI] [PubMed] [Google Scholar]
- 25.Vrljic M, Strop P, Ernst JA, Sutton RB, Chu C, Brunger AT. Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat Struct Mol Biol. 2010;17:325–331. doi: 10.1038/nsmb.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger K. Single molecule FRET-derived model of the synaptogagmin 1-SNARE fusion complex. Nat Struct Mol Biol. 2010;17:318–324. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakon JJ, Weninger KR. Detecting the conformation of individual proteins in live cells. Nat Methods. 2010;7:203–205. doi: 10.1038/nmeth.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kapanidis AN, Lee NK, Laurence TA, Doose S, Margeat E, Weiss S. Fluorescence-aided molecule sorting: analysis of structure and interactions by alternating-laser excitation of single molecules. Proc Natl Acad Sci USA. 2004;101:8936–8941. doi: 10.1073/pnas.0401690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, Weiss S. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys J. 2005;88:2939–2953. doi: 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruttinger S, Macdonald R, Kramer B, Koberling F, Roos M, Hildt E. Accurate single-pair Förster resonant energy transfer through combination of pulsed interleaved excitation, time correlated single-photon counting, and fluorescence correlation spectroscopy. J Biomed Opt. 2006;11:024012. doi: 10.1117/1.2187425. [DOI] [PubMed] [Google Scholar]
- 31.Brunger AT, Strop P, Vrljic M, Chu S, Weninger KR. Three-dimensional molecular modeling with single molecule FRET. J Struct Biol. 2011;173:497–505. doi: 10.1016/j.jsb.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Struck DK, Hoekstra D, Pagano RE. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 33.Schlessinger J, Axelrod D, Koppel DE, Webb WW, Elson EL. Lateral diffusion of surface immunoglobulin, Thy-1 antigen, and a lipid probe in lymphocyte plasma membranes. Science. 1977;195:307–309. doi: 10.1073/pnas.76.10.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 35.Fix M, Melia TJ, Jaiswal JK, Rappoport JZ, You D, Sollner TH, Rothman JE, Simon SM. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci USA. 2004;101:7311–7316. doi: 10.1073/pnas.0401779101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowen ME, Weninger K, Brunger AT, Chu S. Single molecule observation of liposome-bilayer fusion thermally induced by SNAREs. Biophys J. 2004;87:3569–3584. doi: 10.1529/biophysj.104.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu T, Tucker WC, Bhalla A, Chapman ER, Weisshaar JC. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 2005;89:2458–2472. doi: 10.1529/biophysj.105.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domanska MK, Kiessling V, Stein A, Fasshauer D, Tamm LK. Single Vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 2009;284:32158–32166. doi: 10.1074/jbc.M109.047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karatekin E, Di Giovanni J, Iborra C, Coleman J, O'Shaughnessy B, Seagar M, Rothman JE. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci USA. 2010;107:3517–3521. doi: 10.1073/pnas.0914723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan YH, van Lengerich B, Boxer SG. Effects of linker sequences on vesicle fusion mediated by lipid-anchored DNA oligonucleotides. Proc Natl Acad Sci USA. 2009;106:979–984. doi: 10.1073/pnas.0812356106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nickel W, Weber T, McNew JA, Parlati F, Sollner TH, Rothman JE. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci USA. 1999;96:12571–12576. doi: 10.1073/pnas.96.22.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dennison SM, Bowen ME, Brunger AT, Lentz B. Neuronal SNAREs do not trigger fusion between synthetic membranes but do promote PEG-mediated membrane fusion. Biophys J. 2006;90:1661–1675. doi: 10.1529/biophysj.105.069617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohya T, Miaczynska M, Coskun U, Lommer B, Runge A, Drechsel D, Kalaidzidis Y, Zeria M. Reconstitution of Rab- and SNARE-dependent membrane fusion by synthetic endosomes. Nature. 2009;459:1091–1097. doi: 10.1038/nature08107. [DOI] [PubMed] [Google Scholar]
- 44.Wang T, Smith EA, Chapman ER, Weisshaar JC. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1-5 ms resolution. Biophys J. 2009;96:4122–4131. doi: 10.1016/j.bpj.2009.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee NK, Kapanidis AN, Koh HR, Korlann Y, Ho SO, Kim Y, Gassman N, Kim SK, Weiss S. Three-color alternating-laser excitation of single molecules: monitoring multiple interactions and distances. Biophys J. 2007;92:303–312. doi: 10.1529/biophysj.106.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeRocco VC, Anderson T, Piehler J, Erie DA, Weninger K. Four-color single-molecule fluorescence with noncovalent dye labeling to monitor dynamic multimolecular complexes. Biotech. 2010;49:807–816. doi: 10.2144/000113551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee J, Lee S, Ragunathan K, Joo C, Ha T, Hohng S. Single-molecule four-color FRET. Angew Chem Int Ed. 2010;49:9922–9925. doi: 10.1002/anie.201005402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uphoff S, Holden SJ, Le Reste L, Periz J, van de Linde S, Heilemann M, Kapanidis AN. Monitoring multiple distances within a single molecule using switchable FRET. Nat Methods. 2010;7:831–836. doi: 10.1038/nmeth.1502. [DOI] [PubMed] [Google Scholar]
- 49.Ryu Y, Schultz PG. Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat Methods. 2006;3:263–265. doi: 10.1038/nmeth864. [DOI] [PubMed] [Google Scholar]
- 50.Uemura S, Aitken CE, Korlach J, Flusberg BA, Turner SW, Puglisi JD. Real-time tRNA transit on single translating ribosomes at codon resolution. Nature. 2010;464:1012–1017. doi: 10.1038/nature08925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eggeling C, Ringemann C, Medda R, Schwarzmann G, Sandhoff K, Polyakova S, Belov VN, Hein B, von Middendorff C, Schonle A, Hell SW. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457:1159–1162. doi: 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- 52.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]