

Abstract
Botulinum neurotoxin A (BoNT/A) is composed of three domains: a catalytic domain (LC), a translocation domain (HN) and a receptor-binding domain (HC). Like most bacterial toxins BoNT/A is an amphitropic protein, produced in a soluble form that is able to interact, penetrate and/or cross a membrane to achieve its toxic function. During intoxication BoNT/A is internalized by the cell by receptor-mediated endocytosis. Then, LC crosses the membrane of the endocytic compartment and reaches the cytosol. This translocation is initiated by the low pH found in this compartment. It has been suggested that LC passes in an unfolded state through a transmembrane passage formed by HN. We report here that acidification induces no major conformational change in either secondary or tertiary structures of LC and HN of BoNT/A in solution. GdnHCl-induced denaturation experiments showed that the stability of LC and HN increases as pH drops, and that HN further stabilizes LC. Unexpectedly we found that LC has a high propensity to interact with and permeabilize anionic lipid bilayers upon acidification without the help of HN. This property is downplayed when LC is linked to HN. HN thus acts as a chaperone for LC by enhancing its stability but also as a moderator of the membrane interaction of LC.
Introduction
Botulinum neurotoxin (BoNT) secreted by Clostridium botulinum is the causative agent of botulism, a neuroparalytic disease. There are seven serologically distinct BoNT isoforms designated as A to G [1]. These toxins are considered to be the most potent among bacterial, animal, plant toxins and chemical compounds. As there is no available specific cure or vaccine against botulism, BoNTs are also considered as a potential biological weapon [2]. It is also a remarkable cure for numerous neuromuscular and neuronal diseases [3]. BoNTs are synthesized as a single soluble polypeptide chain of 150 kDa. This inactive precursor is cleaved either by a clostridial or tissue protease generating two polypeptides chains linked through a disulfide bound. The light chain (LC, 50 kDa) corresponds to the catalytic domain with a zinc endopeptidase activity [3–5]. The heavy chain (HC, 100 kDa) encompasses two functional domains: the translocation domain at the N-terminus (HN) and the receptor-binding domain at the C-terminus (HC) [4,6,7]. The three domains are critical for cell intoxication by BoNTs. HC allows recognition of neuronal cells by a double anchorage to a ganglioside and a protein receptor [8–11]. Following binding to its receptors, BoNT is internalized through clathrin-dynamin mediated endocytosis of recycling neurosecretion vesicles [12]. Inside the endocytic compartments, HN assists the translocation of LC into the cytosol, upon acidification [13–15]. LC cleaves SNARE proteins involved in neurotransmitter release leading to the inhibition of acetylcholine release at the neuromuscular junction, resulting in flaccid paralysis [16].
The translocation, a key step of the intoxication, is still poorly understood. This step raises the issue of the physico-chemical and structural properties used by amphitropic proteins to interact and cross biological membranes. Among bacterial toxins, several mechanisms can be found such as conformational change and oligomerization for the pore-forming toxins [17]. Translocating toxins like BoNT/A have a domain, HN, dedicated to the membrane crossing of their catalytic domain HN. HN may act as a channel that would provide a way through the membrane for an unfolded LC; this is the case for the anthrax toxin [18]. For the diphtheria toxin, HN acts like a chaperone that helps LC to achieve a partially folded state necessary for membrane binding and penetration [19].
The details of LC membrane-crossing mechanism remain largely unknown. In the past decade based on electrophysiology measurements, it has been proposed that HN could ensure a dual channel and chaperone activity. According to this model, LC would unfold through the channel formed by HN to reach the cytosol [20,21]. An alternative model suggests that HN could constitute a hydrophilic “cleft” that would allow the hydrophilic regions of LC to cross the membrane [15,22]. The aims of this study were to characterize the capacity of LC and HN of BoNT/A to interact and penetrate a membrane and to determine how LC and HN of BoNT/A influence each other during the successive steps leading to membrane binding and penetration as pH drops. We investigated by spectroscopic methods the structure and stability of the domains in solution. We monitored the membrane binding and penetration capacities of the domains, isolated or linked together as in the native toxin. We found that acidification stabilizes the domains. Unexpectedly LC interacts with and permeabilizes model membranes more efficiently than HN, HN further enhances the stabilization of LC induced by acidic pH and acts as a moderator of LC in its interaction with the membrane.
Methods
Cloning, production, and purification of recombinant LC, HN and tandem LC-HN constructs of BoNT/A
Cloning of the Tm fragment, corresponding to the sequence encoding for fragment C454/S-S877 of HN, has been described previously [23]. A synthetic gene encoding residues M1-S877 of both LC and HN domains of BoNT/A (named LC-HN) was purchased from Geneart (Regensburg, Germany) and cloned in a pCR-Script plasmid. The sequence was optimized for the expression in Escherichia coli. A potential Shine Delgarno sequence was also mutated. Two restriction sites for BamHI at the 5’ end and for AvaI at the 3’ end were introduced. The plasmid pCR-Script encoding for LC-HN was amplified and then digested by BamHI and AvaI. The fragment corresponding to LC-HN was cloned in the pQE-81L (Qiagen) plasmid digested also by BamHI and AvaI. LC was obtained by site directed mutagenesis on the pQE-81L-LC-HN plasmid in order to introduce a stop codon at position 426 of LC-HN. The primers used for mutagenesis were 5’-CCGGCCTGTTCGAATTCTAaAAACTGCTGTGTGTTCGTGGC-3’ and 5’-GCCACGAACACACAGCAGTTTtTAGAATTCGAACAGGCCGG-3’. All plasmids, whose sequence was verified, code for a recombinant protein with a His-tag at the N-terminal end. The E. coli strain M15 was used as the host for recombinant protein production.
Expression and purification of the Tm fragment of HN has been described previously [23]. The Tm protein corresponds to the activated form of the BoNT/A HN, i.e. HN following proteolytic cleavage between LC and HC, before residue C454 (S1 Fig).
We have chosen to work with 1–425 LC since 1–448 LC was reported to precipitate upon storage [24]. 1–425 LC was shown to have proper structure and enzymatic activity [25,26]. LC of BoNT/A expression was performed by inoculation of 1 L of Terrific Broth medium (Difco, Detroit, USA) with 15 mL of an overnight culture performed at 28°C. Induction was started at an optical density at 600 nm (OD600) of 0.8 by addition of 1 mM of isopropyl-1-thio-b-D-galactopyranoside and carried out overnight at 16°C. The cultures were centrifuged at 5,000 g for 45 min and the pellet was solubilized in a 20 mM Tris/HCl, 500 mM NaCl, 10 mM imidazole buffer at pH 7.9. Recombinant proteins were extracted by biochemical lysis of the bacteria with 10 mg of lysozyme for 1 hour followed by mechanical lysis using a cell disruption apparatus (Constant Systems Ltd, UK). LC was purified from the soluble fraction of the lysate using a His-Trap HP 5 mL column and an Äkta purifier system (GE Healthcare, USA). The protein was eluted with a 20 mM Tris/HCl, 500 mM NaCl, 250 mM imidazole buffer at pH 7.9. The sample was then subjected to a dialysis in a 10 mM Tris/HCl, 20 mM NaCl buffer at pH 7.8 before cation exchange chromatography using a Hi-TrapQ HP 5 mL column (GE Healthcare, USA). The protein was eluted with a 10 mM Tris/HCl, 150 mM NaCl buffer at pH 7.8.
The expression of LC-HN was performed by inoculation of 1 L of Terrific Broth medium with 25 mL of an overnight culture performed at 37°C. At an OD600 of 0.3, the culture was placed at 15°C. The induction was started at an OD600 of 0.6 by addition of 1 mM of isopropyl-1-thio-b-D-galactopyranoside and carried out overnight at 15°C. The culture was centrifuged at 5,000 g for 45 min and the pellet was solubilized in a 20 mM Na2HPO4/NaH2PO4, 500 mM NaCl, 10 mM imidazole buffer at pH 8. Recombinant proteins were extracted by biochemical lysis with 10 mg of lysozyme for 1 hour followed by mechanical lysis using a cell disruption apparatus (Constant Systems Ltd, UK). LC-HN protein was purified from the soluble fraction of the lysate using a His-Trap HP 5mL column and an Äkta purifier system. The protein was eluted with a 20 mM Na2HPO4/NaH2PO4, 500 mM NaCl, 50 mM imidazole buffer at pH 8. The sample was then subjected to a size exclusion chromatography using a Sephacryl S200 26/10 300 mL column (GE Healthcare, USA) with a 20 mM sodium phosphate, 150 mM NaCl, buffer at pH8.
For all proteins, purification buffer was exchanged by dialysis with a 5 mM phosphate/citrate buffer at pH 7 and were kept at -20°C for LC-HN and HN and at -80°C for LC. Protein concentration was performed by measuring the absorbance of the protein at the ultraviolet wavelength of 278 nm and using Beer-lambert’s law. Molar extinction coefficient of each protein was determined from the protein sequence on the Protein Calculator web resource (http://protcalc.sourceforge.net).
Coomassie blue staining of the proteins following SDS-PAGE showed that they had been purified nearly to homogeneity. The catalytic activity of LC and the protein LC-HN was assayed using a fluorogenic peptide mimic of the substrate of BoNT/A [25].
Lipid vesicles
Lipids were purchased from Avanti Polar Lipids (Alabaster, USA). Suspensions of large unilamellar vesicles (LUV) were prepared in 5 mM phosphate/citrate buffer at pH 7 with egg phosphatidylcholine (EPC, 840051) and egg phosphatidic acid (EPA, 840101) at a 9/1 molar ratio for anionic vesicles. After evaporation of the chloroform used for their solubilization, lipids were suspended in phosphate/citrate buffer and then subjected to 5 cycles of freezing in liquid nitrogen followed by thawing at room temperature. This suspension was then filtered twice on a 0.4 μm filter and twice on a 0.2 μm filters using a homemade extruder. The size of the LUV was checked by dynamic light scattering at 830 nm by using DynaPro-TC-04 DLS equipment (Protein Solutions, Wyatt Technology, Santa Barbara, CA, USA) and found to be between 140 nm and 160 nm.
Experimental buffers
Proteins were kept in 5 mM phosphate/citrate buffer at pH 7 and diluted in a series of phosphate/citrate buffers with varying pH. Permeabilization experiments were performed in 5 mM phosphate/citrate, 100 mM NaCl buffer of various pH. The concentration of protein was checked by measuring the absorbance at 278 nm. The pH of the diluted protein samples was checked before and after each experiment.
Circular dichroïsm (CD) spectropolarimetry
CD experiments were performed on a CD spectropolarimeter model 215 (Aviv, USA). The scans were recorded at a scan rate of 1 nm/s (step, 1 nm; integration time, 1s) with a time constant of 100 ms and a bandwidth of 1 nm. Each spectrum was an average of 5 scans and 3 scans for near-UV and far-UV CD spectra, respectively. A rectangular quartz Suprasil cell of 10 mm path length and a cylindrical quartz Suprasil cell of 0.2 mm path length (104B-QS and 121.000-QS, Hellma, Germany) were used for recording CD activity in near-UV and far-UV regions, respectively. LC, HN and LC-HN were diluted to 1 mg/mL for each protein (corresponding to 10 μM for LC-HN and 20 μM LC and HN). The protein concentration (absorbance at 280 nm, Beer-Lambert’s law) and pH were also checked after measurement. A 5 mM phosphate/citrate pH 5 dialysis buffer was used as blank in far-UV and near-UV regions and its spectrum was subtracted from protein CD spectra. Data were normalized to the molar peptide bond concentration and path length and expressed as Mean Residue Ellipticity per amino acid ([θ] degree·cm2·dmol-1.aa-1) for far-UV CD and as Mean Residue Ellipticity ([θ] degree.cm2.dmol-1) for near-UV CD.
Small angle X-ray scattering (SAXS) experiments
SAXS samples were prepared in 5 mM phosphate/citrate, 150 mM NaCl pH 7 or pH 4. The initial protein concentration of the samples was 10 mg/ml. Prior to data collection, these samples were injected on the online HPLC system of the SWING Beamline at Synchrotron SOLEIL (Gif-sur-Yvette) to eliminate aggregates. Synchrotron radiation X-ray scattering data were collected at 17°C following standard procedures of the SWING Beamline and were processed with FOXTROT (from SWING beamline) and PRIMUS (Primary analysis and Manipulation with Small Angle Scattering DATA) from the ATSAS package (http://www.embl-hamburg.de/biosaxs/software.html). The scattering curves were corrected from scattering due to the buffer and Guinier plots corresponding to each curve were generated (S2 Fig). The pair-distribution functions were computed using the program GNOM from the ATSAS package, allowing determining the radius of gyration and the maximal distance of the molecule.
Analytical ultracentrifugation (AUC)
The protein samples in 5 mM phosphate/citrate, 150 mM NaCl pH 7 or pH 4 were centrifuged at 36,000 rpm in a Beckman Coulter XL-I analytical ultracentrifuge at 20°C. Detection of the protein concentration as a function of radial position and time was performed by optical density measurements at a wavelength of 280 nm. Sedimentation velocity analysis was performed by continuous size distribution analysis c(s) with Sedfit 12.0 [27]. The following parameters were calculated using Sednterp 1.09 (http://www.jphilo.mailway.com/download.htm) and used to analyze experimental data: partial specific volume 0.738 mL.g−1, viscosity 0.01016 Poise and density 1.0044 g.mL−1. The sedimentation coefficients were corrected for viscosity and expressed as s (value at 20°C in water).
Fluorescence spectroscopy
Tryptophan fluorescence measurements were performed with LC, HN and LC-HN in the absence or presence of LUV. Proteins were diluted to a final concentration of 1 μM in a 5 mM phosphate/citrate buffer with varying pH. Experiments with LUV were carried out using a protein/lipid molar ratio of 1/1000. After 2 hours of incubation, fluorescence measurements were performed with an FP-750 spectrofluorimeter (Jasco, Tokyo, Japan). Excitation wavelength was 295 nm. A bandwidth of 5 nm was used for both excitation and emission beams. The emission spectra were recorded from 310 to 370 nm at a scan rate of 125 nm/min. The fluorescence intensity ratio at 360 nm over 320 nm (rFI360/320) represents the average of three values obtained from emission spectra that were corrected for blank measurements [23,28,29].
FRET experiments
LUV containing dansyl (N-(5-DimethylAminoNaphthalene-1-SulfonYL) were prepared from EPC, EPA, and dansyl-DHPE (N-(5-dimethylaminonaphthalene-1-sulfonyl)-1,2-DiHexadecanoyl-sn-glycero-3-PhosphoEthanolamine, D57, Molecular probes, Life technologies, USA) at a 9/1/0.5 molar ratio. LUV were then prepared as described above. A rectangular Quartz Suprasil cell of 10 mm path length was used for recording the fluorescence transfer between protein tryptophan donors and the dansyl acceptor. LC, HN or LC-HN were diluted to a final concentration of 0.5 μM and mixed with LUV at protein/lipid molar ratio of 1/100 in a 5 mM phosphate/citrate buffer at various pH. Control samples without protein were prepared. After 2 hours of incubation fluorescence measurements were carried out with an FP-750 spectrofluorimeter (Jasco, Tokyo, Japan). Excitation and emission wavelengths were 292 nm and 520 nm, respectively. Bandwidths of 5 nm were used for both excitation and emission beams. The emission spectra were recorded from 500 to 540 nm at a scan rate of 125 nm/min. Each emission value at 520 nm is the average calculated from 3 emissions scans. The fluorescence intensity of the dansyl at the plateau was normalized to its initial fluorescence in the absence of protein [23,28]. Results represent the average of four independent measurements for each protein.
LUV permeabilization assay
LUV composed of EPC/EPA at 9/1 molar ratio were prepared as described above, in a 5 mM phosphate/citrate, 100 mM NaCl pH 7 buffer containing 50 mM sulforhodamine B (a self-quenching concentration). After extrusion, unincorporated dye was removed by size exclusion chromatography on a PD10 column (Amersham Biosciences, USA) equilibrated with 5 mM phosphate/citrate, 50 mM or 100 mM NaCl pH 7 buffer. Dye efflux was monitored on a Jasco FP-750 spectrofluorimeter by measuring the increase in fluorescence after adding of 0.1 μM of protein to a suspension of LUV at 100 μM lipids in 5 mM phosphate/citrate, 100 mM NaCl buffer at different pH with stirring. Excitation and emission wavelengths were 569 nm and 586 nm, respectively. Bandwidths of 5 nm were used for both excitation and emission beams [23].
Fluorescence was normalized with the following equation:
| (1) |
where F0 is the fluorescence level before protein addition and Fmax the level after addition of Triton X-100 at the end of each assay [23] (S3 Fig).
Chemical protein denaturation by guanidine hydrochloride (GdnHCl)
Stock solutions of 5 mM phosphate/citrate and 7 M GdnHCl were prepared, their pH were adjusted using HCl and NaOH, and filtered. The refractive index of each solution was measured to check the precise GdnHCl concentration (R-5000 hand refractometer (Atago, Japan)). A range of solutions with GdnHCl concentrations varying from 0 to 6.8 M for each studied pH were prepared by dilution of stock solutions in 5 mM phosphate/citrate at the same pH as the stock solution [30]. LC, HN and LC-HN proteins were diluted to a final concentration of 1 μM in 1.5 mL with the complete set of GdnHCl solutions. Samples were incubated overnight before fluorescence measurements on a Jasco FP-750 spectrofluorimeter. Excitation wavelength was 295 nm with a bandwidth of 5 nm for both excitation and emission beams. The emission spectra were recorded from 310 to 370 nm at a scan rate of 125 nm/min. Fluorescence intensity ratio at 360 nm over 320 nm (rFI360/320) were determined. These wavelengths are related to tryptophan fluorescence intensities in apolar (320 nm) and solvated (360 nm) environments, respectively, and the values of rFI360/320 are therefore very sensitive to weak polarity changes in the tryptophan surroundings. Each data point represents the average of three values obtained from emission spectra that were corrected for blank measurements. rFI was plotted as a function of GdnHCl concentration to obtain the GdnHCl-unfolding curves [31]. Curves were analyzed assuming a two-state mechanism and were fitted with the following equation:
| (2) |
where ΔG0 is the free energy in the absence of chaotropic agent, D the denaturant concentration and m the coefficient of dependence of free energy on the denaturant concentration. m is related to the variation of the solvent-accessible surface area between the folded and unfolded states. D1/2, the denaturant concentration required to achieve half denaturation was deduced from the expression of free energy at half denaturation:
| (3) |
Hence,
| (4) |
Results
Three protein constructs were used in this study: LC and HN, corresponding to the isolated catalytic and translocation domains of BoNT/A, and LC-HN, corresponding to a truncated BoNT/A toxin lacking the HC domain (Fig 1). In the LC-HN protein construct, LC and HN are linked through a disulfide bond as in the native toxin. The peptide chain linking the domains is cleaved by incubation with trypsin. Cleavage of the peptide chain and integrity of the inter-domain disulfide bond were checked by SDS-PAGE under reducing and non-reducing conditions, and Coomassie blue staining (S1 Fig).
Fig 1. Structure of the recombinant LC, HN and LC-HN proteins of BoNT/A.

Ribbon representations of LC, HN and LC-HN predicted from the structure of BoNT/A (Protein Data Bank file: 3BTA). The N- and C-terminal ends are indicated (NH2 and COOH) for each protein. The Trp residues are represented as blue spheres for the three proteins and indicated in C as W43, W118 and as W460, W606, W706 and W717 for HN.
LC and HN of BoNT/A do not undergo any major conformational change upon acidification in solution
The structure of the proteins constructs was investigated by circular dichroïsm (CD) in the far- and near-UV as a function of the pH. In the far-UV (180–260 nm) the CD signal measured for a protein is due mostly to the peptide bonds, so the spectra are indicative of its secondary structure. The far-UV spectra of the three proteins display π-π and n-π characteristics of a α-helical content (Fig 2). Analysis of the secondary structure of the three proteins from these CD data is in agreement with the crystal structure of BoNT/A (Fig 1 and Table 1) [4]. The protein CD signal in the near-UV (250–320 nm) arises from aromatic amino acid side chains constrained in a tertiary structure. LC spectra show a strong negative signal around 280 nm that may be attributed to Tyr or Trp side chains and two smaller signals at 262 nm and 268 nm that may be attributed to Phe side chains. The spectra of HN display some weak signals in the absorption regions of Phe, Tyr and Trp side chains as observed previously [23]. LC-HN near-UV spectra seem to combine signals from both LC and HN.
Fig 2. Far and near-UV CD spectra of LC, HN and LC-HN proteins of BoNT/A.

Left, Far-UV and right Near-UV at various pH, pH 7 (black curve); pH 6 (pink curve); pH 5 (green curve); pH 4 (blue curve) and pH 3 (red curve).
Table 1. Comparison of the secondary structure of the three proteins based on the far-UV CD spectra at pH 7 (BestSel) with the secondary structure observed in the crystal structure of BoNT/A (3BTA) (STRIDE PDB).
| BestSel | STRIDE PDB | |||||||
|---|---|---|---|---|---|---|---|---|
| LC | HN | LC-HN | LC + HN isolated | LC | HN | LC-HN | LC + HN isolated | |
| alpha | 28.3 | 51.8 | 39.6 | 40.05 | 30.2 | 50.2 | 40.7 | 40.2 |
| beta | 17.3 | 7 | 14.2 | 12.15 | 14 | 1.4 | 9.7 | 7.7 |
| Others1 | 54.4 | 41.2 | 46.2 | 47.8 | 55.8 | 48.4 | 49.6 | 52.1 |
1Others: turns and random coils
Upon acidification no remarkable change could be observed in the far-UV spectra for the three proteins. However, a moderate increase of the negative peak intensity around 280 nm in the near-UV region can be seen at pH 3 for LC and LC-HN. This suggests an increase of the tertiary structure constraints surrounding one or several aromatic residues within the LC chain, as it is shared by both proteins.
Altogether, the data show that LC, HN and LC-HN are folded in the range of pH 3–7 with secondary and tertiary structures compatible with those found in the crystal structure of the toxin (pdb 3BTA). Acidification does not trigger any visible conformational change, except around pH 3 within the LC and LC-HN proteins. The data suggest a moderate gain of tertiary structure within LC without any indication of partial unfolding.
Small Angle X-ray scattering (SAXS) and analytical ultracentrifugation (AUC) provide information on the shape of a protein and the formation of multimers (Fig 3). SAXS gives accurate information about the conformation in solution of macromolecules. The SAXS curves of LC-HN at pH 7 and pH 4 are similar indicating that acidification does not trigger any change in the global fold of LC-HN (Fig 3A). The normalized Kratky plots are characteristic of folded proteins (Fig 3B). The Inverse Fourier transform of the scattering intensity yields the distance distribution function, P(r). Two important parameters can be extracted from this curve: the radius of gyration (Rg) and the maximum particle diameter (Dmax) of the protein Rg (Fig 3C). The Rg of LC-HN at pH 7 and pH 4 are close, confirming that LC-HN does not undergo important shape modification upon acidification. Consistently, AUC experiments showed similar sedimentation coefficients (s) for LC-HN at neutral and acid pH (Fig 3C), suggesting that the overall shape of the protein is not modified by the acidification. In summary, SAXS and AUC data pointed out that the global shape of the LC-HN protein remains the same and that no oligomerization occurred upon acidification.
Fig 3. SAXS and analytical ultracentrifugation analysis of LC-HN at pH 7 and pH 4.

A, comparison of the superposition of the logarithm of the SAXS intensities as a function of the amplitude of the diffusion vector [f/f0 = 4πsinθ/λ], for LC-HN at pH 7 (red curve) and pH 4 (blue curve). B, Superposition of the dimensionless Kratky plots of LC-HN at pH 7 (red curve) and pH 4 (blue curve). This representation reveals the type of structure: compact, partially folded or unfolded. C, summary of shape describing parameters obtained from AUC and SAXS: sedimentation coefficient (s, in Svedberg), frictional ratio (f/f0), radius of gyration (Rg).
Acid pH stabilizes LC and HN of BoNT/A
We investigated the stability of LC, HN and LC-HN as a function of pH by monitoring their unfolding in the presence of increasing concentrations of the chaotropic agent GdnHCl from 0 to 6.8 M (Fig 4). Chaotropic denaturation was preferred to thermal denaturation to avoid possible aggregation. Unfolding was monitored by measuring the ratio of the fluorescence signal at 360 nm and 320 nm of the Trp (rFI360/320 that reflects the polarity of Trp environment) as a function of pH. LC contains two Trp at positions 43 and 118 and HN contains four Trp at position 460, 606, 707, and 717 (Fig 1). A rFI360/320 that shifts toward high values means that Trp become exposed to the solvent, due to the unfolding of the protein. All proteins exhibit a typical GdnHCl-induced denaturation from the folded state to the unfolded state at neutral pH (Fig 4). The denaturation curves show only a slight change in the fluorescence of the Trp between 0 and 2 M of GdnHCl. This indicates minor structural changes. A pH drop does not notably change the curves at these concentrations of denaturing agent. In contrast, at higher GdnHCl concentrations, a clear denaturation transition is observed for all three proteins (Fig 4 for pH 7 and pH 4). The rFI360/320 signal of LC-HN is almost identical to the signal of HN indicating that HN is dominating the LC-HN signal. This may be due to the fact that HN contains four Trp as compared to LC that contains only two. In addition, HN stabilizes LC (see below), further reducing the contribution of LC to the change of rFI360/320. Interestingly, the denaturation curves are shifted towards higher GdnHCl concentrations for all the proteins as pH drops (Fig 4 for pH 7 and pH 4). Thermodynamic parameters were extracted by applying a fitting procedure to the experimental data [31]. One or two folding intermediates may appear during denaturation as revealed by slight changes in the slope of the curves. However, fitting according to a three-state model could not be achieved and a two-state model was used as a best approximation. At neutral pH, 2.17 M, 4.25 M and 3.45 M of GdnHCl is needed to achieve half denaturation of LC, HN and LC-HN, respectively (Fig 4, inset tables). These values of D1/2 increase as pH drops (shown Fig 4 for pH 4). At pH 4, these D1/2 concentrations are 3.27 M, 4.76 M and 4.85 M (Fig 4, inset tables), respectively. This is consistent with the increase of ΔG0 values extracted from the curves upon acidification. At pH 7 the ΔG0 are 3.7 kcal.mol-1, 3.3 kcal.mol-1 and 3.5 kcal.mol-1 for LC, HN and LC-HN respectively, whereas at pH 4 the values rise to 4.3 kcal.mol-1, 6.1 kcal.mol-1 and 5.7 kcal.mol-1, respectively. Interestingly, while the stability of LC slightly increases with the pH drop from 7 to 4 with a gain in ΔG0 of 0.6 kcal.mol-1, that of HN is considerably increased with almost a doubling of its ΔG0. The situation for LC-HN is close to that of HN suggesting that the presence of HN increases the stability of LC, especially at acidic pH. Altogether, the results show that LC is less stable than HN, and that acidic pH strongly increase the stability of HN, which then stabilizes LC within the LC-HN protein.
Fig 4. Effect of pH on the stability of LC, HN and LC-HN proteins. GdnHCl-induced denaturation of LC, HN and LC-HN followed by Trp fluorescence at pH 7 and pH 4.

A, B, C, denaturation curves of LC, HN and LC-HN respectively, at pH 7 (open symbols) and pH 4 (closed symbols). D, superposition of the denaturation curves of LC (red squares), HN (green circles) and LC-HN (blue triangles) at pH 4. Inset, table of thermodynamic parameters ΔG0 and D1/2, for each protein at pH 7 and pH 4.
The isolated LC of BoNT/A interacts with a membrane without requiring the presence of HN
We characterized previously the interaction of HN of BoNT/A with negatively charged lipid vesicles [23]. Here, we investigated the capacity of the isolated LC to interact with a negatively charged membrane (LUV) in the absence of HN, as a function of pH. The interaction was monitored by three fluorescence methods: intrinsic fluorescence of the Trp of LC, FRET from the Trp of LC towards dansyl groups linked to the phospholipid head-groups of LUV and fluorescence dye recovery upon LUV permeabilization (Fig 5).
Fig 5. Membrane interaction of LC of BoNT/A as a function of pH.
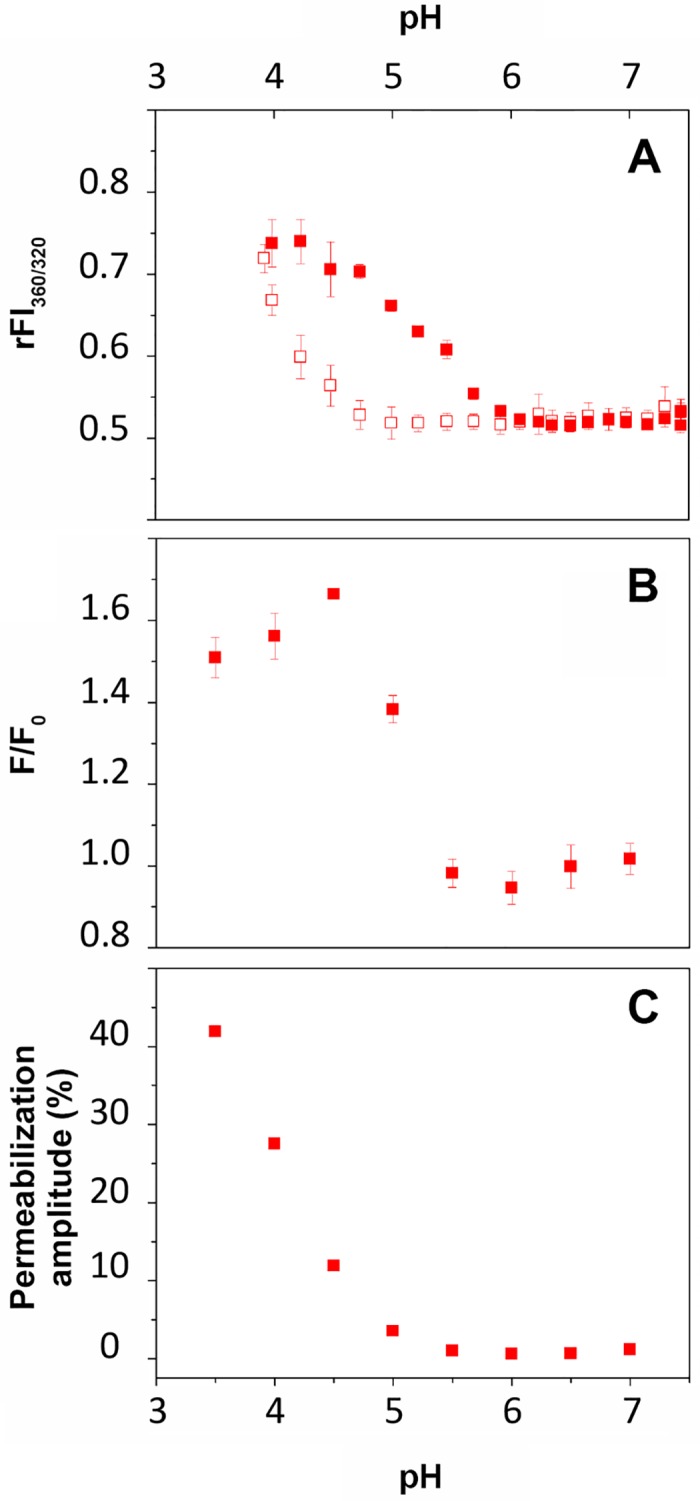
A, ratio of Trp fluorescence intensities at 360 nm and 320 nm in solution in absence (open squares) and in presence of LUV (closed squares). B, FRET from LC protein (0.5 μM) to dansyl-DHPE-containing LUV (50 μM). C, LUV (100 μM) permeabilization induced by LC (0.1 μM), expressed as percent of permeabilization amplitude reflected the maximum rate of permeabilization achieved for each pH.
In the presence of LUV, the rFI360/320 increases as pH drops from 6 to 4.2 (Fig 5, top). In the absence of LUV, a similar transition occurs but at more than one pH unit lower. These results indicate that the environment of the Trp of LC in solution become more polar as pH decreases. The presence of membrane shifts the transition to higher pH values due to the partition of the protein from solution to the membrane.
When the Trp of the protein (donors) are close to the dansyl groups in the membrane (acceptor) FRET is detected as an increase of acceptor fluorescence. In the presence of LUV, the Trp intrinsic fluorescence transition we detected correlates nicely with an increase of the FRET signal from pH 6 to 4.5. The increase of the FRET signal upon acidification reveals a closer proximity between the Trp and the polar headgroups of the lipid bilayer and demonstrates the interaction of the protein with the membrane. Together, these data indicate that LC interacts progressively with the LUV as pH drops from 6 to about 4.
LC exhibits a pH-dependent capacity to permeabilize the LUV as shown by its ability to induce the release of sulforhodamine B entrapped at self-quenching concentration in the LUV. The efficiency of permeabilization is expressed as the maximum percentage of permeabilization induced by LC at a given pH (Fig 5, bottom and Fig 6, top). The permeabilization phenomenon starts below pH 5.5, 0.5 pH units lower than the appearance of the FRET signal. Altogether, the data suggest that the interaction of LC with the lipid bilayer involves at least two steps: partition of LC into the interfacial region of the membrane (FRET signal without permeabilization) followed by a deeper penetration into the bilayer, which induce leakage of the dye contained in the LUV (permeabilization).
Fig 6. Comparison of permeabilization of, and membrane binding to LUV by LC, HN and LC-HN proteins of BoNT/A.
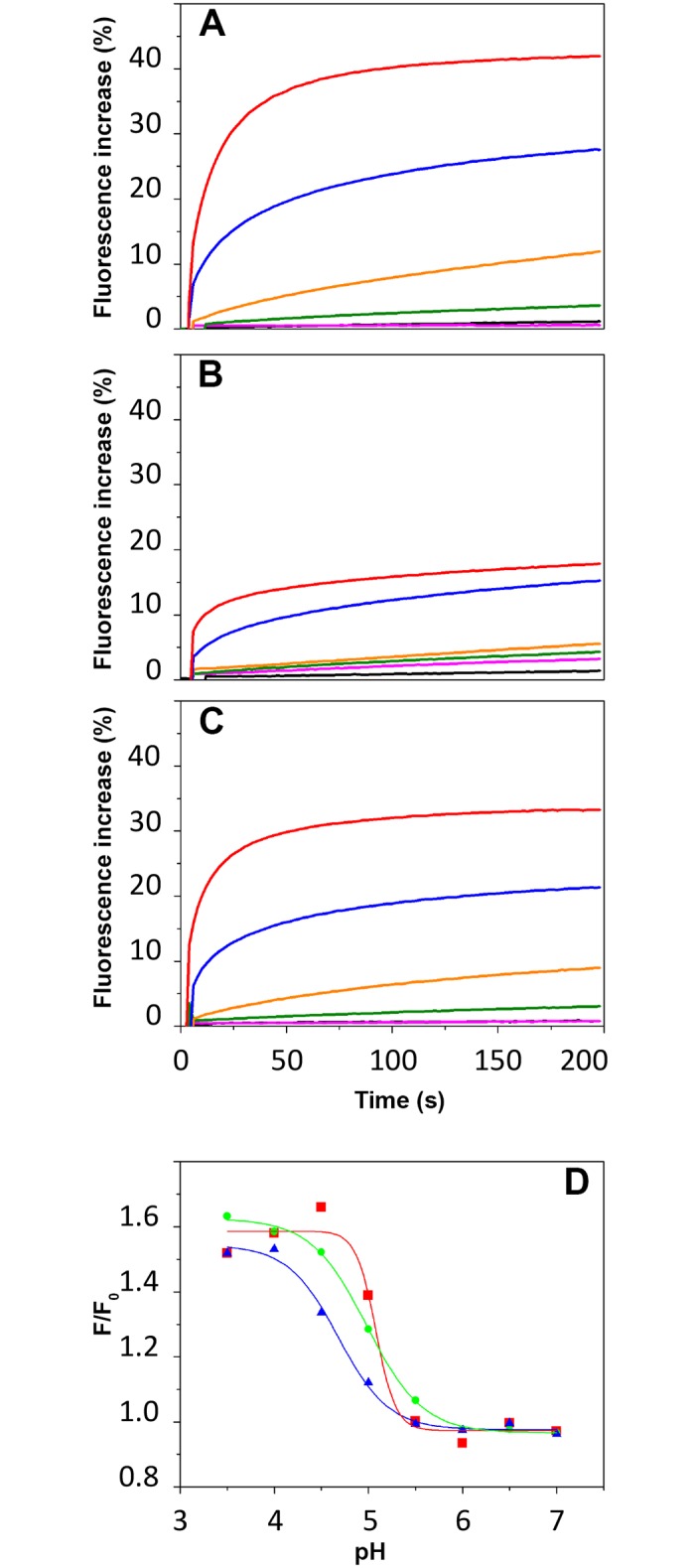
A, B, C, Kinetics of SRB leakage from LUV (100 μM) after addition of LC (A), HN (B) and LC-HN (C) (0.1 μM) at pH 7 (black), pH 6 (pink), pH 5 (green), pH 4.5 (orange), pH 4 (blue) and pH 3.5 (red). S3 Fig shows absence of SRB leakage in the absence of proteins and 100% SRB release after Triton X-100 addition. D, FRET from LC (red), HN (green) and LC-HN (blue) at 0.5 μM to dansyl-DHPE-containing LUV at 50 μM.
No permeabilization was observed with negative LUV in the presence of increased NaCl concentrations (200 mM instead of 100 mM). Similarly, no permeabilization was observed with LUV made of neutral phospholipids only (S4 Fig). This indicates that permeabilization involves electrostatic interactions between the proteins and negative charges on the membrane.
It is noteworthy that this partition-permeabilization process can occur in the absence of HN.
HN of BoNT/A moderates the propensity of LC to interact with membranes
Permeabilization and FRET experiments were also performed with the isolated HN and the protein LC-HN in order to compare the behavior of both domains isolated or linked by the native inter-domain disulfide bond. Fig 6A–6C show the kinetics of dye release from LUV in the presence of LC, HN and LC-HN at various pHs from 7 to 3.5. The results are expressed as the percentage of fluorescence increase after addition of the protein to the LUV, with 100% corresponding to the maximum release obtained by triton X-100. The three proteins show a capacity to destabilize the membrane as pH drops. HN starts to permeabilize the LUV below pH 5, while the phenomenon starts below pH 5.5 for LC and LC-HN. However, LC was about twice more efficient than HN in permeabilizing the LUV. While maximum dye release reached 40% for LC, it reached about 15% for HN and 30% for LC-HN. In addition, the transition observed by FRET for the interaction with the membrane (Fig 6D) is more cooperative for LC than for HN, an intermediate situation being found for LC-HN.
Altogether, the data suggest that HN moderates the propensity of LC to interact with and permeabilizes membranes at acidic pH.
Discussion
How does BoNT/A, a soluble protein, manage to interact with and further penetrate into a membrane is a key issue for its translocation. Here we designed a study using a combination of spectroscopic methods to probe the effect of the pH on the structural properties and membrane interaction of the LC and HN of BoNT/A, alone or covalently linked together.
The acidic state in solution of LC and HN, competent for membrane interaction, is characterized by increased stability without any major conformational change. Upon acidification, no significant secondary or tertiary structure change in solution can be observed for either LC or HN, alone or covalently linked by the native inter-domain disulfide bond (Fig 2). This observation is in agreement with previous studies that reported no major structural change upon acidification for LC [32,33], HN [23,34] or the entire toxin [35,36]. Here, in contrast with previous studies on the LC and the whole toxin, we investigated a wide range of pHs from 7 down to 3. Interestingly, a gain of tertiary constraints seems to occur for the LC-HN protein at low pH (Fig 2). This observation is in agreement with the increase of stability found for this protein as pH drops (Fig 4). Overall, the data suggest that LC and HN, isolated or linked together, do not undergo large pH-induced conformational changes. Nevertheless, our observations do not exclude local conformational changes, which would favor membrane partitioning. Indeed, Trp fluorescence changes can be seen as pH drops both for LC (Fig 5, top panel, open squares) and HN [23,34].
However, an important finding of the thermodynamic analysis is that the stability of the domains in solution is increased as pH drops. Indeed, GdnHCl-induced unfolding experiments showed that HN is more stable than LC and that the stability of HN is considerably increased at low pH, thus apparently contributing to an increased stability of LC within LC-HN.
LC in its acidic state interacts and permeabilizes anionic phospholipid membranes in the absence of HN. Tables 2 and 3 summarize the features of the acidic states of LC and HN in solution and in the presence of membranes. Considering LC alone, the data strongly suggest that the presence of the membrane displaces the equilibrium toward the acidic state of LC and its transition into a membrane-bound state (Fig 5A and 5B). Then, further acidification provokes permeabilization of the membrane by LC, indicative of a state in which LC destabilizes the lipid bilayer (Fig 5, bottom and Fig 6, top). The striking feature of this interaction is that membrane permeabilization by LC occurs without the help of another domain or component of the toxin or cellular machinery [37,38]. This finding infers the following question: what is the role of HN for membrane interaction and thereby for translocation?
Table 2. Features of LC and HN upon acidification in solution.
| Features in solution | LC | HN |
|---|---|---|
| Trp1 | Exposed from pH 4.7 to 3.8 | Buried from pH 6.2 to 3.82 |
| Secondary structure3 | Unchanged | Unchanged |
| Tertiary structure3 | Increased | Unchanged |
| Stability4 | Increased | Increased |
| Influence over the other domain | ? | Stabilization |
1Trp fluorescence
2not shown
3CD
4GdnHCl denaturation
Table 3. Features of LC and HN upon acidification in the presence of membranes (LUV).
| Features in presence of LUV | LC | HN |
|---|---|---|
| Trp1 | Exposed from pH 5.9 to 4.2 | Buried from pH 6.2 to 3.82 |
| Membrane binding3 | From pH 5.5 to 4.5 | From pH 5.5 to 3.5 |
| Membrane permeabilization4 | From 5 to 3.5 | From 4.5 to 3.5 |
| Influence over the other domain | ? | Moderates membrane interaction |
1Trp fluorescence
2Not shown
3FRET
4Fluorescent dye release from LUV
HN of BoNT/A moderates the propensity of LC to interact with membranes at acidic pH. The data obtained here with the LC-HN protein bring three observations: HN contributes to the stabilization of LC at neutral and acidic pH in solution, it reduces the cooperativity of the binding of LC with membranes and it reduces the efficiency of membrane permeabilization of the membrane by LC. It is likely that at a given pH the lipid bilayer destabilization by LC is reduced by the presence of HN. We found previously that this capacity of HN was due to its N-terminal belt that surrounds LC blocking its catalytic site [23]. One could hypothesize that the stabilization of LC by HN that we observed in solution, also occurs during interaction with the membrane, thereby limiting membrane penetration and destabilization by LC. Thus, HN would act as a chaperone-like domain, probably both through its belt and the wide interfacial region between the two domains. These two contact surfaces between the domains certainly represent major constraints over LC.
LC and HN of BoNT/A possess structural and physico-chemical properties distinct from that of other translocating toxins. This is inferred by a comparison with the mechanisms of action of other translocating toxins: monomeric (ABRTC) and multimeric (anthrax, binary and AB5 toxins). The enzymatic components of anthrax toxin need the help of the membrane protective antigen hepta/octamer that catalyzes the unfolding of these enzymatic components and offers them a protein pore for their passage through the membrane [39]. Ricin and AB5 toxins such as Shiga toxin and Cholera toxin hijack the ERAD system in the endoplasmic reticulum to translocate into the cytosol [37,38].
It has been shown by electron and atomic force microscopies that BoNT/B and E [40,41] form multimers. In our observations by SAXS and AUC, no multimers were found in solution whatever the pH. Multimerization may depend highly on experimental conditions. In any case, one cannot rule out yet whether multimers of BoNT must form to ensure translocation.
The catalytic domain (C) of the diphtheria toxin, which is also a RTC toxin, interacts and permeabilizes the membrane of LUV with much better efficacy with the help of the translocation domain (T) [19,42]. In this case, T acts as a chaperone stabilizing successively several partially folded states of the C domain. Further description of this process for BoNT/A would require the determination of the structure of translocation intermediates.
Both the C and T domains of diphtheria toxin, which correspond functionally to LC and HN, undergo remarkable conformational changes upon acidification (in solution and in presence of membrane) to achieve a molten globule state needed for the membrane interaction [43,44]. We show by CD and SAXS (Figs 2 and 3) that LC and HN of BoNT/A do not display any major conformational changes upon acidification in solution. This observation is supported by other works [32,33,35,36]. In addition, we found that LC has by itself a high propensity to bind (Fig 5B) and penetrate (Fig 5C) a membrane at acidic pH. The results suggest that the role of HN is to negatively regulate this propensity. We showed previously that the belt region of HN is responsible for the negative regulation of the interaction of HN with the membrane [23]. As a consequence, it is very likely that LC of BoNT/A interacts with both the membrane and HN during the first steps of its translocation. This is compatible with the cleft model proposed earlier by Montecucco [15,22], who showed also that LC is able to bind phospholipids. Single channel conductance measurements showed that LC blocks the ion channel formed in the membrane by HN for BoNT/A and BoNT/E during translocation [45]. Reconciliation of this model with our observations would imply that the channel made by HN would be closed by the passage of LC between the lipids of the bilayer and one of the faces of HN embedded in the membrane.
Supporting Information
A, LC-HN, HN and LC in reducing and non-reducing conditions (+ or—β-mercaptoethanol, (β-ME)). B; LC-HN was treated with trypsin before mixing with loading buffer + or—β-ME and analyzed on the SDS-PAGE. On each gel, Lane 1 is a ladder of protein molecular weight markers shown in kDa. The results show that the proteolytic cleavage site between LC and HN is uncleaved in the recombinant protein before trypsin treatment and cleaved after. The disulfide bridge linking both domains is intact as well and reduced by β-ME.
(TIF)
These representations were calculated using Primus. They correspond to the logarithm of the SAXS intensity (log I) as a function of the square of the diffusion vector amplitude (q2), plotted up to a q*Rg limit of 1.3. The slope of these representations represents the radius of gyration (Rg) of the molecule.
(TIF)
Self fluorescence quenching of SRB entrapped inside the LUV define 0% fluorescence. The LUV are incubated at varying pH for over 150 s. The absence of fluorescence increase indicates the absence of SRB leakage from the LUV (in the absence of protein addition). After 150 s, the addition of Triton X-100 solubilizes the membrane of all the LUV and releases the entrapped SRB, leading to dequenching and maximum fluorescence (100%).
(TIF)
Kinetics of LUV leakage of LC, HN and LC-HN at pH 7 (black lines), pH 5 (green), and pH 4.5 (orange) in presence of neutral LUV (left) or negatively charged LUV with 200 mM NaCl (right). The arrows indicate the addition of protein and triton X-100 that gives the maximum of dye release.
(TIF)
ΔG0 is the free energy in the absence of chaotropic agent, D is the denaturant concentration and m is the coefficient of dependence of free energy on denaturant concentration. m is related to the variation of the solvent-accessible surface area between the folded and unfolded states.
(DOCX)
Acknowledgments
We thank Nicolas Durafourc and Maïté Paternostre for help with LUV preparation. For help with protein production and purification we thank Cassandra Porebski. For help with AUC experiments we thank Bertrand Raynal. AA was supported by DGA and CEA. This work was funded by the joint ministerial program of R&D against CBRNE threats. SIMOPRO is a member of the Laboratory of Excellence LERMIT supported by a grant from the Agence Nationale de la Recherche (ANR-10-LABX-33). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by the joint ministerial program of R&D against CBRNE threats. SIMOPRO is a member of the Laboratory of Excellence LERMIT supported by a grant from the Agence Nationale de la Recherche (ANR-10-LABX-33). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Poulain B, Popoff MR, Molgo J. How do the Botulinum Neurotoxins block neurotransmitter release: from botulism to the molecular mechanism of action. The Botulinum J. 2008; Vol.1: 14–87. [Google Scholar]
- 2.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, et al. Botulinum toxin as a biological weapon: medical and public health management. JAMA. 2001; 285: 1059–1070. [DOI] [PubMed] [Google Scholar]
- 3.Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000; 80: 717–766. [DOI] [PubMed] [Google Scholar]
- 4.Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat Struct Biol. 1998; 5: 898–902. [DOI] [PubMed] [Google Scholar]
- 5.Rossetto O, Montecucco C. Presynaptic neurotoxins with enzymatic activities. Handb Exp Pharmacol. 2008; 184: 129–170. [DOI] [PubMed] [Google Scholar]
- 6.Kumaran D, Eswaramoorthy S, Furey W, Navaza J, Sax M, Swaminathan S. Domain organization in Clostridium botulinum neurotoxin type E is unique: its implication in faster translocation. J Mol Biol. 2009; 386: 233–245. 10.1016/j.jmb.2008.12.027 [DOI] [PubMed] [Google Scholar]
- 7.Swaminathan S, Eswaramoorthy S. Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat Struct Biol. 2000; 7: 693–699. [DOI] [PubMed] [Google Scholar]
- 8.Montecucco C. How do tetanus and botulinum toxins bind to neuronal membranes? Trends in Biochemical Sciences. 1986; 11: 314–317. [Google Scholar]
- 9.Rummel A, Eichner T, Weil T, Karnath T, Gutcaits A, Mahrhold S, et al. Identification of the protein receptor binding site of botulinum neurotoxins B and G proves the double-receptor concept. Proc Natl Acad Sci U S A. 2007; 104: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rummel A. Double receptor anchorage of botulinum neurotoxins accounts for their exquisite neurospecificity. Curr Top Microbiol Immunol. 2013; 364: 61–90. 10.1007/978-3-642-33570-9_4 [DOI] [PubMed] [Google Scholar]
- 11.Binz T, Rummel A. Cell entry strategy of clostridial neurotoxins. Journal of Neurochemistry. 2009; 109: 1584–1595. 10.1111/j.1471-4159.2009.06093.x [DOI] [PubMed] [Google Scholar]
- 12.Harper CB, Martin S, Nguyen TH, Daniels SJ, Lavidis NA, Popoff MR, et al. Dynamin inhibition blocks botulinum neurotoxin type A endocytosis in neurons and delays botulism. J Biol Chem. 2011; 286: 35966–35976. 10.1074/jbc.M111.283879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoch DH, Romero-Mira M, Ehrlich BE, Finkelstein A, DasGupta BR, Simpson LL. Channels formed by botulinum, tetanus, and diphtheria toxins in planar lipid bilayers: relevance to translocation of proteins across membranes. Proc Natl Acad Sci U S A. 1985; 82: 1692–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montal M. Translocation of botulinum neurotoxin light chain protease by the heavy chain protein-conducting channel. Toxicon. 2009; 54: 565–569. 10.1016/j.toxicon.2008.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montecucco C, Schiavo G, Dasgupta BR. Effect of pH on the interaction of botulinum neurotoxins A, B and E with liposomes. Biochem J. 1989; 259: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humeau Y, Doussau F, Grant NJ, Poulain B. How botulinum and tetanus neurotoxins block neurotransmitter release. Biochimie. 2000; 82: 427–446. [DOI] [PubMed] [Google Scholar]
- 17.Geny B, Popoff MR. Bacterial protein toxins and lipids: pore formation or toxin entry into cells. Biol Cell. 2006; 98: 667–678. [DOI] [PubMed] [Google Scholar]
- 18.Jiang J, Pentelute BL, Collier RJ, Zhou ZH. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature. 2015; 521: 545–549. 10.1038/nature14247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chassaing A, Pichard S, Araye-Guet A, Barbier J, Forge V, Gillet D. Solution and membrane-bound chaperone activity of the diphtheria toxin translocation domain towards the catalytic domain. FEBS J. 2011; 278: 4516–4525. 10.1111/j.1742-4658.2011.08053.x [DOI] [PubMed] [Google Scholar]
- 20.Fischer A, Montal M. Single molecule detection of intermediates during botulinum neurotoxin translocation across membranes. Proc Natl Acad Sci U S A. 2007; 104: 10447–10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koriazova LK, Montal M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat Struct Biol. 2003; 10: 13–18. [DOI] [PubMed] [Google Scholar]
- 22.Montecucco C, Schiavo G, Gao Z, Bauerlein E, Boquet P, DasGupta BR. Interaction of botulinum and tetanus toxins with the lipid bilayer surface. Biochem J. 1988; 251: 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galloux M, Vitrac H, Montagner C, Raffestin S, Popoff MR, Chenal A, et al. Membrane Interaction of botulinum neurotoxin A translocation (T) domain. The belt region is a regulatory loop for membrane interaction. J Biol Chem. 2008; 283: 27668–27676. 10.1074/jbc.M802557200 [DOI] [PubMed] [Google Scholar]
- 24.Baldwin MR, Bradshaw M, Johnson EA, Barbieri JT. The C-terminus of botulinum neurotoxin type A light chain contributes to solubility, catalysis, and stability. Protein Expr Purif. 2004; 37: 187–195. [DOI] [PubMed] [Google Scholar]
- 25.Stura EA, Le Roux L, Guitot K, Garcia S, Bregant S, Beau F, et al. Structural framework for covalent inhibition of Clostridium botulinum neurotoxin A by targeting Cys165. J Biol Chem. 2012; 287: 33607–33614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu Z, Chen S, Baldwin MR, Boldt GE, Crawford A, Janda KD, et al. Light chain of botulinum neurotoxin serotype A: structural resolution of a catalytic intermediate. Biochemistry. 2006; 45: 8903–8911. [DOI] [PubMed] [Google Scholar]
- 27.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000; 78: 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chenal A, Savarin P, Nizard P, Guillain F, Gillet D, Forge V. Membrane protein insertion regulated by bringing electrostatic and hydrophobic interactions into play. A case study with the translocation domain of diphtheria toxin. J Biol Chem. 2002; 277: 43425–43432. [DOI] [PubMed] [Google Scholar]
- 29.Montagner C, Perier A, Pichard S, Vernier G, Menez A, Gillet D, et al. Behavior of the N-terminal helices of the diphtheria toxin T domain during the successive steps of membrane interaction. Biochemistry. 2007; 46: 1878–1887. [DOI] [PubMed] [Google Scholar]
- 30.Nozaki Y. The preparation of guanidine hydrochloride. Methods Enzymol. 1972; 26: 43–50. [DOI] [PubMed] [Google Scholar]
- 31.Chenal A, Karst JC, Sotomayor Perez AC, Wozniak AK, Baron B, England P, et al. Calcium-induced folding and stabilization of the intrinsically disordered RTX domain of the CyaA toxin. Biophys J. 2010; 99: 3744–3753. 10.1016/j.bpj.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai S, Kukreja R, Shoesmith S, Chang TW, Singh BR. Botulinum neurotoxin light chain refolds at endosomal pH for its translocation. Protein J. 2006; 25: 455–462. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Singh BR. Spectroscopic analysis of pH-induced changes in the molecular features of type A botulinum neurotoxin light chain. Biochemistry. 2000; 39: 6466–6474. [DOI] [PubMed] [Google Scholar]
- 34.Lai B, Agarwal R, Nelson LD, Swaminathan S, London E. Low pH-induced pore formation by the T domain of botulinum toxin type A is dependent upon NaCl concentration. J Membr Biol. 2010; 236: 191–201. 10.1007/s00232-010-9292-z [DOI] [PubMed] [Google Scholar]
- 35.Eswaramoorthy S, Kumaran D, Keller J, Swaminathan S. Role of metals in the biological activity of Clostridium botulinum neurotoxins. Biochemistry. 2004; 43: 2209–2216. [DOI] [PubMed] [Google Scholar]
- 36.Fu FN, Busath DD, Singh BR. Spectroscopic analysis of low pH and lipid-induced structural changes in type A botulinum neurotoxin relevant to membrane channel formation and translocation. Biophys Chem. 2002; 99: 17–29. [DOI] [PubMed] [Google Scholar]
- 37.Falguieres T, Johannes L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol Cell. 2006; 98: 125–134. [DOI] [PubMed] [Google Scholar]
- 38.Spooner RA, Lord JM. How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr Top Microbiol Immunol. 2012; 357: 19–40. 10.1007/82_2011_154 [DOI] [PubMed] [Google Scholar]
- 39.Feld GK, Brown MJ, Krantz BA. Ratcheting up protein translocation with anthrax toxin. Protein Sci. 2012; 21: 606–624. 10.1002/pro.2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid MF, Robinson JP, DasGupta BR. Direct visualization of botulinum neurotoxin-induced channels in phospholipid vesicles. Nature. 1993; 364: 827–830. [DOI] [PubMed] [Google Scholar]
- 41.Sun S, Tepp WH, Johnson EA, Chapman ER. Botulinum neurotoxins B and E translocate at different rates and exhibit divergent responses to GT1b and low pH. Biochemistry. 2012; 51: 5655–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Malenbaum SE, Kachel K, Zhan H, Collier RJ, London E. Identification of shallow and deep membrane-penetrating forms of diphtheria toxin T domain that are regulated by protein concentration and bilayer width. J Biol Chem. 1997; 272: 25091–25098. [DOI] [PubMed] [Google Scholar]
- 43.Hammond K, Caputo GA, London E. Interaction of the membrane-inserted diphtheria toxin T domain with peptides and its possible implications for chaperone-like T domain behavior. Biochemistry. 2002; 41: 3243–3253. [DOI] [PubMed] [Google Scholar]
- 44.Ren J, Kachel K, Kim H, Malenbaum SE, Collier RJ, London E. Interaction of diphtheria toxin T domain with molten globule-like proteins and its implications for translocation. Science. 1999; 284: 955–957. [DOI] [PubMed] [Google Scholar]
- 45.Montal M. Botulinum neurotoxin: a marvel of protein design. Annu Rev Biochem. 2010; 79: 591–617. 10.1146/annurev.biochem.051908.125345 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, LC-HN, HN and LC in reducing and non-reducing conditions (+ or—β-mercaptoethanol, (β-ME)). B; LC-HN was treated with trypsin before mixing with loading buffer + or—β-ME and analyzed on the SDS-PAGE. On each gel, Lane 1 is a ladder of protein molecular weight markers shown in kDa. The results show that the proteolytic cleavage site between LC and HN is uncleaved in the recombinant protein before trypsin treatment and cleaved after. The disulfide bridge linking both domains is intact as well and reduced by β-ME.
(TIF)
These representations were calculated using Primus. They correspond to the logarithm of the SAXS intensity (log I) as a function of the square of the diffusion vector amplitude (q2), plotted up to a q*Rg limit of 1.3. The slope of these representations represents the radius of gyration (Rg) of the molecule.
(TIF)
Self fluorescence quenching of SRB entrapped inside the LUV define 0% fluorescence. The LUV are incubated at varying pH for over 150 s. The absence of fluorescence increase indicates the absence of SRB leakage from the LUV (in the absence of protein addition). After 150 s, the addition of Triton X-100 solubilizes the membrane of all the LUV and releases the entrapped SRB, leading to dequenching and maximum fluorescence (100%).
(TIF)
Kinetics of LUV leakage of LC, HN and LC-HN at pH 7 (black lines), pH 5 (green), and pH 4.5 (orange) in presence of neutral LUV (left) or negatively charged LUV with 200 mM NaCl (right). The arrows indicate the addition of protein and triton X-100 that gives the maximum of dye release.
(TIF)
ΔG0 is the free energy in the absence of chaotropic agent, D is the denaturant concentration and m is the coefficient of dependence of free energy on denaturant concentration. m is related to the variation of the solvent-accessible surface area between the folded and unfolded states.
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.