

ABSTRACT
Most mammalian protein-coding gene promoters are divergent, yielding promoter upstream transcripts (PROMPTs) in the reverse direction from their conventionally produced mRNAs. PROMPTs are rapidly degraded by the RNA exosome rendering a general function of these molecules elusive. Yet, levels of certain PROMPTs are altered in stress conditions, like the DNA damage response (DDR), suggesting a possible regulatory role for at least a subset of these molecules. Here we manipulate PROMPT levels by either exosome depletion or UV treatment and analyze possible effects on their neighboring genes. For the CTSZ and DAP genes we find that TFIIB and TBP promoter binding decrease when PROMPTs accumulate. Moreover, DNA methylation increases concomitant with the recruitment of the DNA methyltransferase DNMT3B. Thus, although a correlation between increased PROMPT levels and decreased gene activity is generally absent, some promoters may have co-opted their divergent transcript production for regulatory purposes.
KEYWORDS: Divergent transcription, DNA damage response, DNA methylation, mRNA, PROMPT, RNA exosome, RNASeq
Introduction
Eukaryotic genomes are pervasively transcribed, giving rise to a large number of long non coding (lnc) RNAs.1-4 Many of these newly discovered transcripts have short half-lives and are only detectable when RNA degradation factors are compromised.5-7 In one example, depletion of the human ribonucleolytic RNA exosome provokes the accumulation of so-called ‘PROMoter uPstream Transcripts’ (PROMPTs), that are transcribed in reverse orientation to most active protein coding genes.5,8-10 Although some lncRNAs expressed from protein-coding gene promoters have been suggested to functionally impact the activity of the neighboring gene,11-13 a general role (if any) of such promoter-associated RNAs remains elusive.
A functional role of otherwise unstable transcripts might be revealed at physiological conditions altering RNA turnover rates. Indeed, it was recently suggested that the DNA damage response (DDR) entails the phosphorylation of the RBM7 protein, which is an integral component of the nuclear exosome targeting (NEXT) complex connecting at least a subset of PROMPTs to decay by the exosome.14-17 Such phosphorylation of RBM7 appears to alter the affinity of the protein for RNA, resulting in the accumulation of selected PROMPTs during the DDR due to their inefficient targeting by the NEXT complex.
In this study, we use both RNA exosome depletion and UV treatment to manipulate PROMPT levels. Our general observation is that increased abundance of PROMPTs does not correlate with altered expression of the neighboring gene. Yet, the CTSZ and DAP genes are found to exhibit a concurrent downregulated transcription initiation activity, suggesting that continued analysis of these genes may reveal new principles for lncRNA function.
Results and discussion
CTSZ and DAP gene transcription is reduced upon exosome inactivation
To study the relationship between PROMPT expression and transcription levels of neighboring protein-coding loci in some detail, we focused on the CTSZ and DAP genes. These were chosen because a low throughput screen performed in our laboratory indicated changes in their promoter methylation upon RRP40 depletion (data not shown) and preliminary analysis indicated a negative correlation between their PROMPT and mRNA levels. We depleted a core subunit of the exosome, RRP40 (Fig. 1A), which was previously shown to reduce exosome function5 and increase ‘Cap Analysis of Gene Expression’ (CAGE) and RNAseq signals from the respective proCTSZ (Fig. 1B, top panel) and proDAP (Fig. 1B, bottom panel) PROMPT regions.8,18 Upon RRP40 depletion and the expected PROMPT stabilization (Fig. 1C, left panel), a concomitantly mild, but significant, decrease in CTSZ and DAP mRNA levels was observed (Fig. 1C, right panel). To investigate whether this was based on lowered transcription initiation of the 2 genes, we analyzed their promoter occupancies by chromatin immunoprecipitation (ChIP)-PCR of the general transcription factors (GTFs) TFIIB and TBP. Since both promoter regions are CG-rich, we designed amplicons upstream of, but as close as possible to, the respective gene TSSs (Fig. 1B). These amplicons capture promoter-bound pre-initiation complexes (PICs) deposited within a distance of the amplicons matching the range of resolution defined by the sonicated DNA (approx. 200–500bp, data not shown). For both promoters, exosome depletion led to reduced binding of TFIIB and TBP (Fig. 1D). In support of this result, RNA polymerase II (RNAPII) occupancy at the intron 1 region of CTSZ and at the 3’ UTR region of DAP was also decreased upon RRP40 depletion (Fig. 1D).
Figure 1.

Transcription of the CTSZ and DAP genes decreases upon exosome depletion. (A) Western blotting verification of RRP40 levels in control cells treated with EGFP siRNAs and in cells treated with siRNAs against RRP40. Tubulin was used as a loading control. (B) Genome browser screenshots of PROMPT regions of the CTSZ (top) and DAP (bottom) genes, displaying mapped CAGE and RNAseq sequence reads from both EGFP (control) and RRP40-depleted HeLa cells.8,18 Note that Y-axes scales are comparable for the control and RRP40 knockdown samples. Only plus strand information is shown (data from the minus strand containing mRNAs are not displayed). Relative position of amplicons used for RT-qPCR, ChIP and HpaII-methylation (HpaII) assays are indicated at the bottom of each panel. (C) PROMPT (left panel) and mRNA (right panel) levels measured by RT-qPCR in control (dark gray) and knockdown (light gray) cells from (A). RNA levels were normalized to GAPDH RNA levels from the same sample and plotted relative to levels of the EGFP control. Error bars represent the standard deviation from 6 biological replicates. (D) Occupancy of TFIIB and TBP at the DAP and CTSZ promoters and of RNAPII at the CTSZ intron 1 and the DAP 3’UTR as measured by ChIP-PCR in control (dark gray) and RRP40-depleted (light gray) cells. IP efficiencies were normalized to levels at the GAPDH gene and plotted relative to levels of the EGFP control. Error bars represent the standard deviation from 3 biological replicates in the case of TFIIB and TBP and the standard deviation from 3 technical repeats of a representative experiment in the case of RNAPII. For all relevant figures, P-values were calculated using an unpaired 2 tailed Student's t-test comparing the RRP40 depleted cells to the corresponding EGFP control. (*) P-value < 0.05; (**) P-value < 0.01.
The observed reduction in gene transcription could be caused by PROMPTs accumulating at their sites of production from the same promoter. If so, the respective RNA would be expected to be chromatin-associated. Indeed, 27% of proCTSZ transcripts accumulated in the chromatin fraction of HeLa cells subjected to RRP40 depletion (Fig. 2A, left panel). In contrast, only 16% of CTSZ mRNA was chromatin-associated (Fig. 2A, right panel). This difference is likely an underestimate because of an expected larger fraction of nascent mRNA than PROMPT in the chromatin fraction, given the length difference between these transcripts. The observed increase in chromatin-associated mRNA upon exosome-depletion was surprising but might be due to an accumulation of aberrant molecules that would normally be degraded (Fig. 2A, right panel).
Figure 2.

Exosome depletion triggers increased PROMPT-association with chromatin and methylation of the CTSZ promoter. (A) CTSZ PROMPT (left panel) and mRNA (right panel) levels within fractions (cytoplasm, nucleoplasm and chromatin) of control and RRP40-depleted HeLa cells and as measured by RT-qPCR. RNA levels are displayed as 2−Ct, with Ct (cycle treshold) values obtained from RT-qPCR reactions performed on RNA from an equivalent number of cells. (B) HpaII resistance ratio measured by qPCR by dividing the amount of amplified product in samples digested with HpaII with their non-digested controls. Results were normalized to qPCR levels from a DNA region without an HpaII site. (C) ChIP efficiencies of DNMTs at the CTSZ PROMPT region (1026bp upstream the CTSZ gene TSS) as measured by ChIP-qPCR with antibodies to the 3 human DNMTs. (D) DNMT3B ChIP efficiencies at different positions of the CTSZ PROMPT region measured as in (C). Distances from the CTSZ TSS to the midpoint of the utilized amplicon are indicated below the graph. For all displayed experiments in the figure, control cells are depicted in dark gray and RRP40-deplete cells in light gray. Error bars show standard deviations from 3 technical replicates of a representative dataset.
It was previously described that a ncRNA upstream of the SPHK1 gene affected the methylation of CpG dinucleotides within the SPHK1 promoter region.19 Moreover, in RRP40 depletion-conditions, the same ncRNA is stabilized and SPHK1 promoter methylation levels increase.5 Since DNA methylation of promoters can be associated with gene silencing (reviewed in20,21), we analyzed the methylation levels of the PROMPT-expressing regions of the CTSZ and DAP promoters taking advantage of the methylation sensitive enzyme HpaII, that is unable to digest methylated DNA (see Methods). To this end, PCR fragments encompassing the HpaII target sequence CCGG were required, thus, amplicons were designed to reconcile the general difficulty of promoter amplification and the desire to monitor regions close to the PROMPT amplicons (Fig. 1B). Using this approach, both promoters displayed an increase in methylation upon RRP40 depletion (Fig. 2B). DNA methylation can be deposited by any of the 3 DNA methyltransferases: DNMT1, DNMT3A and DNMT3B (reviewed in.20,22) To examine whether one, or more, of these were recruited to the CTSZ promoter region, we conducted ChIP analyses with antibodies against the 3 enzymes. An increased DNMT3B ChIP signal was observed in the CTSZ PROMPT region upon RRP40 depletion (Fig. 2C), which was not simply due to altered DNMT levels in the knockdown cells (Fig. S1). A more precise delineation of the region onto which DNMT3B was recruited revealed a specific peak around 1026 bp upstream of the CTSZ gene TSS (Fig. 2D), overlapping the CTSZ PROMPT region (Fig. 1B) and where we also detected a difference in methylation by the HpaII digestion assay (Fig. 2B). Taken together these analyses demonstrate that exosome depletion can lead to transcription downregulation and altered promoter DNA methylation.
RRP40 depletion does not generally decrease protein coding gene expression
Most, if not all, protein coding gene promoters produce PROMPTs. Thus, if the phenotype seen at the DAP and CTSZ genes was widespread, gene expression should be generally altered upon exosome inactivation. However, this is not the case: only a small fraction of RefSeq annotated protein coding genes were differentially expressed as measured by RNASeq (Andersen et al. NSMB 2013) in RRP40-depleted vs. control samples (Fig. 3). Analysis of mRNA reads (see Methods) resulted in few down- and up-regulated transcripts (288 vs. 194 from a total of 12403 inspected genes, padj < 0.1) (Fig. 3). CTSZ mRNA was found to be significantly downregulated in this analysis (log2 fold change = −0.74, with padj = 0.025). DAP mRNA levels were only slightly affected, perhaps reflecting that the relatively modest effect we observe by qPCR goes undetected in this RNASeq experiment. We conclude that RRP40 depletion does not generally lead to lowered gene expression, although a small subset of genes appear affected.
Figure 3.
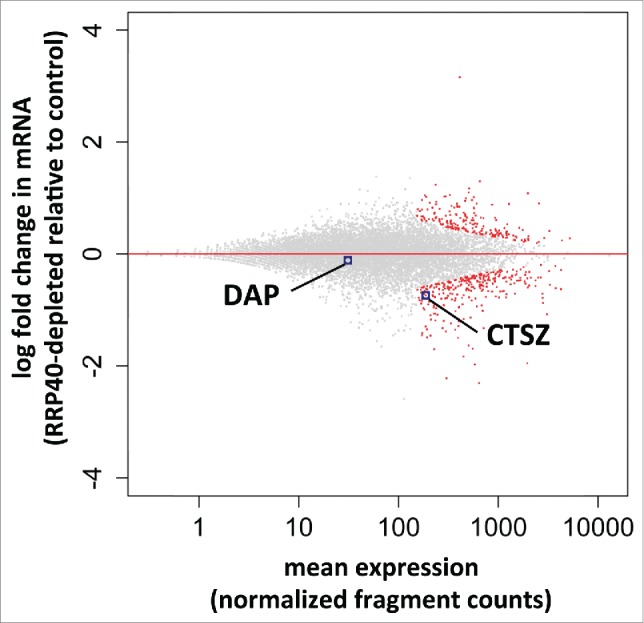
Global mRNA analysis of RRP40 depleted cells. RNASeq-based expression changes of RRP40 knockdown vs. EGFP control cells. Y-axes display log2-fold changes of individual transcript levels while x-axes show the mean normalized expression (fragment counts normalized by size factors). Data were computed by the DESeq2 software.29 Individual RefSeq protein coding genes are shown in black and significantly differentially expressed genes (padj = FDR < 0.1) are colored red. Dots corresponding to DAP and CTSZ mRNAs are highlighted.
UV irradiation can induce PROMPT stabilization and transcription reduction
We next considered ‘physiological’ situations where PROMPT levels are altered. The exosome co-factor complex, NEXT, connects the exosome to the DNA damage response as the NEXT component RBM7 becomes phosphorylated upon UV irradiation with a concomitant accumulation of PROMPTs.14 To investigate whether such a situation would recapitulate the transcription effects observed upon RRP40 depletion, we treated HeLa cells with 20 J/m2 of UVC light and harvested them at different time points after irradiation. That the cells responded appropriately to the treatment was confirmed by increased levels of GADD45A RNA (Fig. 4A), which is reportedly induced in response to DNA damage.23,24 Upon UV treatment both CTSZ and DAP PROMPT levels were increased (Fig. 4B), whereas the neighboring CTSZ and DAP mRNAs were downregulated (Fig. 4C). Consistently, TFIIB occupancy (Fig. 4D) and methylation (Fig. 4E) of the CTSZ and DAP promoter regions were decreased and increased, respectively. As in the case of RRP40 depletion, UV treatment also induced DNMT3B recruitment to the CTSZ PROMPT region (Fig. 4F), which was RNA-dependent as demonstrated by RNase-treatment of samples prior to the ChIP. Thus, UV irradiation induces PROMPT accumulation and a concomitant gene transcription decrease, with shared characteristics to the transcription effects observed upon exosome depletion.
Figure 4.

PROMPT accumulation and gene transcription repression upon UV irradiation. (A) GADD45A mRNA levels as measured by RT-qPCR along the indicated time course after UV irradiation of HeLa cells. (B) CTSZ (dark gray) and DAP (light gray) PROMPT levels measured and plotted as in (A). (C) CTSZ (dark gray) and DAP (light gray) mRNA levels measured and plotted as in (B). For A), B) and C) a representative experiment is shown out of 3 independent biological replicates. Graphs show RNA levels relative to the untreated sample. Error bars show standard deviations from 3 technical replicates. (D) TFIIB IP efficiencies along the UV time course from (A) as measured by ChIP-qPCR of the CTSZ (dark gray) and DAP (light gray) promoter regions. Error bars show standard deviations from 3 technical replicates of a representative experiment of 3 biological replicates. (E) Methylation levels at the CTSZ (dark gray) and DAP (light gray) promoter regions as measured by HpaII resistance along the UV time course from (A). Values were plotted relative to levels of the untreated sample. Error bars show standard deviations from 3 biological replicates. P-values were calculated with an unpaired 2-tailed Student's t-test comparing each time point to the corresponding untreated sample. (*) P-value < 0.05; (**) P-value < 0.01. (F) DNMT3B ChIP efficiencies measured by the ‘-1026’ amplicon from Figure 2D along the indicated UV time course. IP efficiences of samples treated (light gray) or not (dark gray) with RNase A before the IP are shown. Error bars show standard deviations from 3 technical replicates of a representative experiment.
A local function of some PROMPTs?
Decreased transcription of the CTSZ and DAP genes occurs concomitant with their increased PROMPT levels. It is therefore possible that in these cases promoter-associated ncRNAs are involved in gene silencing. A recurrent mechanism of action for such ncRNAs is their ability to recruit effector proteins to chromatin (reviewed in25 and26). For example, an RNA:DNA triplex mechanism was suggested to link the rDNA promoter with DNMT3B in an ncRNA dependent manner.11 We have observed that DNMT3B recruitment to the CTSZ promoter upon UV irradiation of cells is RNA dependent (Fig. 4F). Thus, CTSZ PROMPTs may operate by a similar mechanism. However, since DNA methylation is rarely the initiating event in gene silencing20,21 such an activity would probably rather serve to maintain a silenced state subsequent to other repressive events. Whether promoter-associated RNAs can also directly impact assembly of GTFs at some gene promoters is an interesting possibility. Alternatively, enhancer RNAs (eRNAs), which are also exosome-7,27 and NEXT complex-27 sensitive, may play a role in modulating CTSZ and DAP transcription from more distant genomic positions.
The transcription effects we observe for CTSZ and DAP do not extend genome wide. In addition, for the genes analyzed by Blasius et al., UV treatment triggers increased PROMPT levels with no selective effect on neighboring promoters.14 While this implies that PROMPTs are not widespread effectors of protein-coding gene transcription, our analysis indicates that some may take advantage of the divergent behavior of their promoters as part of a regulatory mechanism. Further studies are required to delineate any causal relations in these cases.
Methods
Cells and siRNA-mediated depletion of RRP40
HeLa cells originating from the S2 strain were cultured and transfected with enhanced green fluorescent protein (EGFP; control) or hRRP40 (EXOSC3) siRNAs as described previously.8 Briefly, cells cultured in DMEM supplemented with 10% FBS were transfected using Lipofectamine 2000 (Life Technologies) at a final siRNA concentration of 20 nM. Transfections were performed 1 and 3 d after cell seeding and the cells were harvested 5 d after seeding. The following siRNA sequences were used: EGFP 5′-GACGUAAACGGCCACAAGU[dT][dT]-3′, EGFP_as 5′-ACUUGUGGCCGUUUACGUC[dT][dT]-3′, hRRP40 5′-CACGCACAGUACUAGGUCA [dT][dT]-3′ and hRRP40_as 5′-UGACCUAGUACUGUGCGUG [dT][dT]-3′.
Western blotting analysis
Cells washed twice in PBS, harvested and centrifuged for 5 minutes at 1500rpm were lysed in RSB100 containing 0.5% Triton X-100 (10mM Tris pH 7.4, 100mM NaCl, 2.5mM MgCl2, 0.5% Triton X-100). SDS-PAGE and western blotting analysis were carried out according to standard procedures using the following antibodies: anti-RRP40 (Proteintech, 15062–1-AP) at 1:4000; anti-α-tubulin (Rockland, 600–401–880) at 1:2500; anti-DNMT1 (Abcam, ab87656) at 1:500; anti-DNMT3A (Abcam, ab2850) at 1:500; anti-DNMT3B (Abcam, ab2851) at 1:1000. Secondary HRP goat-anti-rabbit antibody (Dako, P0448) was used at 1:5000.
RNA isolation and RT-qPCR analysis
RNA was isolated using TRIzol (Ambion) and treated with TURBO DNAse (Ambion) according to the manufacturer's instructions. cDNA was then prepared with the SuperScript II kit (Invitrogen), using 1μM oligo dT18 and 5ng/μl random hexamers. Only random hexamers were used for the UV experiments. To evaluate genomic DNA contamination, a negative control was prepared in parallel by treating the same amount of RNA in the same way but without adding the reverse transcriptase enzyme. qPCR was performed with Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) in a MX3000P (Agilent technologies) machine.
ChIP analysis
ChIP was performed as described previously.18 Briefly, HeLa cells were crosslinked during 10 minutes at room temperature (RT) with 1% formaldehyde added to the culture medium. Cross-linking was stopped by addition of 125mM glycine at RT. After washing, cells were successively incubated in cell lysis buffer (200mM Tris pH 8, 85mM KCl, 0,5% NP40) and nuclei lysis buffer (50mM TrisHCl pH 8, 10mM EDTA, 1% SDS). In the case of the RNase A treated samples (Fig. 4F), 50μg/ml RNase A was added to the lysis buffers. The obtained chromatin was sheared to a 250–500 bp length using a COVARIS® sonicator (Covaris Inc., Massachusetts, USA). Chromatin was incubated overnight at 4°C with appropriate antibodies (and corresponding negative control without antibody). 1% of the total chromatin volume was saved as input material for the analysis of IP efficiency. Antibody-bound chromatin was recovered with protein A sepharose beads (GE-Healthcare). After reverse crosslinking of protein-DNA complexes, purified DNA was used for qPCR. Antibodies used: 5μg anti-TFIIB (Santa Cruz Biotechnology, sc-225 X), 5μg anti-TFIID (Santa Cruz Biotechnology, sc-204 X), 5μg anti-DNMT1 (Abcam, ab87656), 5μg anti-DNMT3A (Abcam, ab2850) and 5μg anti-DNMT3B (Abcam, ab2851).
Subcellular cell fractionation
Confluent cells (50–80%) were harvested and re-suspended in RSB100 buffer (10mM Tris, 100mM NaCl, 2.5mM MgCl2) containing 40μg/ml digitonin. After 5 minutes of incubation on ice, the cell suspension was centrifuged at 2000xg for 8 minutes at 4°C. The supernatant (cytoplasmic fraction) was collected and kept on ice until further use. The pellet was completely resuspended in RSB100 buffer containing 0.5% Triton X-100 and incubated on ice for 5 minutes. After centrifugation, the nuclear cell pellet was resuspended in RSB100/Triton X-100 and sonicated for 15 seconds. The sonicated nuclear suspension was centrifuged at 4000xg for 5 minutes at 4°C. The supernatant (nucleoplasmic fraction) was kept on ice and the pellet (chromatin fraction) was resuspended in 500 μl TES buffer (10mM Tris/HCl pH 7.5, 10mM EDTA pH 8, 0.5% SDS). Immediately after, 500μl of phenol pH 6.6 were added to the chromatin suspension and the sample was incubated at 65°C for one hour. RNA from the different fractions was extracted by phenol/chloroform and eluted in 30μl RNAse-free water.
Genomic DNA isolation and HpaII treatment
Cells were lysed in a buffer containing 10mM Tris pH 8, 100mM NaCl, 10mM EDTA, 0.5%SDS and 1μg/μl proteinase K. The lysate was incubated for 3 hours at 42°C to allow for protein digestion. DNA was then extracted using the phenol/chloroform method. 2μl of DNA were incubated with Fast Digest HpaII restriction enzyme (Thermo Fischer Scientific). Digested DNA was diluted in 100μl, and 2μl were used in qPCR using primers spanning an HpaII restriction site. Methylation levels were calculated by dividing the amount of amplified product in digested samples by that in non-digested controls. Results were normalized to a DNA region without HpaII recognition sites within the ESWR1 gene (Table S1).
UV treatment
Media was removed from culture dishes and cells were irradiated with 20J/m2 of UVC immediately after media removal. Media was added back after the treatment and cells harvested at the indicated time points after irradiation according to the ChIP protocol or the RNA or DNA isolation methods (Fig. 4).
RNASeq data analysis
We used 3 previously described RNASeq libraries mapped to the human genome version 19 (hg19) in18: 2 from HeLa cells treated with EGFP siRNA and one depleted for RRP40. The data can be found in the Sequence Read Archive (SRA) with the following accession numbers: SRX365666, SRX365672 and SRX365673. Subsequent analyses were conducted by use of Python2.7 and R software and associated packages.28 The ‘refFlat’ (Feb. 2009 GRCh37/hg19) annotation file was downloaded from the UCSC Table Browser under the tabs ‘RefSeq genes’ and ‘Genes and Gene Predictions’. Only protein coding genes mapping to conventional chromosomes (chromosomes 1–22, X, Y and M) were used. Protein coding genes were chosen based on the presence of an annotated open reading frame with a length larger than 0 nucleotides. Each nucleotide represented in the filtered 'refFlat' annotation file was then indexed as being either exonic, intronic or both (because of alternative splicing events some nucleotides can be either exonic or intronic). Ambiguous regions of genes were masked, e.g. a region within a gene that also codes for an intron-hosted snoRNA is not included in the analysis. Read pairs from the mapped RNASeq data were then counted as mRNA if all nucleotides within the read pair were exonic according to the above mentioned index. This yielded a list of 12403 mRNAs with at least one read fragment in one of the 3 libraries. For differential expression analysis, mRNA fragment counts were analyzed by the DESeq2 software version 1.4.529 using the total number of uniquely mapped fragments for each sample as sizeFactors, other functions using default settings.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Manfred Schmid and Toomas Silla for comments on the manuscript. Claudia Scheffler is thanked for excellent technical assistance. Melanie Blasius and Steve Jackson are thanked for fruitful discussions. This work was supported by the Danish National Research Foundation (grant DNRF58), the Danish Cancer Society and the Novo Nordisk Foundation (T.H.J). M.L.L. is the recipient of a grant from the Danish Council for Independent Research (DFF- 1333–00059B).
References
- 1. Jensen TH, Jacquier A, Libri D. Dealing with pervasive transcription. Mol Cell 2013; 52:473-84; PMID:24267449; http://dx.doi.org/10.1016/j.molcel.2013.10.032 [DOI] [PubMed] [Google Scholar]
- 2. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell 2013; 154:26-46; PMID:23827673; http://dx.doi.org/10.1016/j.cell.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature 2012; 482:339-46; PMID:22337053; http://dx.doi.org/10.1038/nature10887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. . Landscape of transcription in human cells. Nature 2012; 489:101-8; PMID: 22955620; http://dx.doi.org/ 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science 2008; 322:1851-4; PMID:19056938; http://dx.doi.org/10.1126/science.1164096 [DOI] [PubMed] [Google Scholar]
- 6. Flynn RA, Almada AE, Zamudio JR, Sharp PA. Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc Natl Acad Sci USA 2011; 108:10460-5; PMID:21670248; http://dx.doi.org/10.1073/pnas.1106630108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andersson R, Refsing Andersen P, Valen E, Core LJ, Bornholdt J, Boyd M, Heick Jensen T, Sandelin A. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat Commun 2014b;5:5336; PMID: 25387874; http://dx.doi.org/10.1038/ncomms6336. [DOI] [PubMed] [Google Scholar]
- 8. Ntini E, Järvelin AI, Bornholdt J, Chen Y, Boyd M, Jørgensen M, Andersson R, Hoof I, Schein A, Andersen PR, et al. . Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol 2013; 20:923-8; PMID:23851456; http://dx.doi.org/10.1038/nsmb.2640 [DOI] [PubMed] [Google Scholar]
- 9. Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG, Almada AE, Lin C, Sharp PA, Giallourakis CC, et al. . Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci USA 2013; 110:2876-81; PMID:23382218; http://dx.doi.org/10.1073/pnas.1221904110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 2013; 499:360-3; PMID:23792564; http://dx.doi.org/10.1038/nature12349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmitz K-M, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 2010; 24:2264-9; PMID:20952535; http://dx.doi.org/10.1101/gad.590910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008; 454:126-30; PMID:18509338; http://dx.doi.org/10.1038/nature06992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K, Ward AJ, Raj A, Lee JT, Sharp PA, et al. . LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Mol Cell 2014; 54:777-90; PMID:24857549; http://dx.doi.org/10.1016/j.molcel.2014.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blasius M, Wagner SA, Choudhary C, Bartek J, Jackson SP. A quantitative 14-3-3 interaction screen connects the nuclear exosome targeting complex to the DNA damage response. Genes Dev 2014; 28:1977-82; PMID:25189701; http://dx.doi.org/10.1101/gad.246272.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell 2011; 43:624-37; PMID:21855801; http://dx.doi.org/10.1016/j.molcel.2011.06.028 [DOI] [PubMed] [Google Scholar]
- 16. Tiedje C, Lubas M, Tehrani M, Menon MB, Ronkina N, Rousseau S, Cohen P, Kotlyarov A, Gaestel M. p38MAPK/MK2-mediated phosphorylation of RBM7 regulates the human nuclear exosome targeting complex. RNA 2015; 21(2):262-78; PMID:25525152; http://dx.doi.org/ 10.1261/rna.048090.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lubas M, Andersen PR, Schein A, Dziembowski A, Kudla G, Jensen TH. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep 2015; 10:178-92; PMID:25578728; http://dx.doi.org/ 10.1016/j.celrep.2014.12.026 [DOI] [PubMed] [Google Scholar]
- 18. Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, et al. . The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol 2013; 20:1367-76; PMID:24270879; http://dx.doi.org/ 10.1038/nsmb.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imamura T, Yamamoto S, Ohgane J, Hattori N, Tanaka S, Shiota K. Non-coding RNA directed DNA demethylation of Sphk1 CpG island. Biochem Biophys Res Commun 2004; 322:593-600; PMID:15325271 [DOI] [PubMed] [Google Scholar]
- 20. Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13:484-92; PMID:22641018; http://dx.doi.org/ 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 21. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25:1010-22; PMID: 21576262; http://dx.doi.org/ 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010; 11:204-20; PMID:20142834; http://dx.doi.org/ 10.1038/nrg2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fornace AJ, Nebert DW, Hollander MC, Luethy JD, Papathanasiou M, Fargnoli J, Holbrook NJ. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol Cell Biol 1989; 9:4196-203; PMID:2573827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fornace AJ, Alamo I, Hollander MC. DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci USA 1988; 85:8800-4; PMID:3194391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keller C, Bühler M. Chromatin-associated ncRNA activities. Chromosome Res 2013; 21:627-41; PMID:24249576; http://dx.doi.org/ 10.1007/s10577-013-9390-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell 2011; 43:904-14; PMID:21925379; http://dx.doi.org/ 10.1016/j.molcel.2011.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. . An atlas of active enhancers across human cell types and tissues. Nature 2014a; 507:455-61; PMID:24670763; http://dx.doi.org/ 10.1038/nature12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Team RC. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012:2012 [Google Scholar]
- 29. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Gen Biol 2014; 15:550; PMID:25516281 [DOI] [PMC free article] [PubMed] [Google Scholar]