

ABSTRACT
Adenosine deaminases bind double stranded RNA and convert adenosine to inosine. Editing creates multiple isoforms of neurotransmitter receptors, such as with Gria2. Adar2 KO mice die of seizures shortly after birth, but if the Gria2 Q/R editing site is mutated to mimic the edited version then the animals are viable. We performed RNA-Seq on frontal cortices of Adar2-/- Gria2R/R mice and littermates. We found 56 editing sites with significantly diminished editing levels in Adar2 deficient animals with the majority in coding regions. Only two genes and 3 exons showed statistically significant differences in expression levels. This work illustrates that ADAR2 is important in site-specific changes of protein coding sequences but has relatively modest effects on gene expression and splicing in the adult mouse frontal cortex.
Keywords: ADAR2, RNA editing, RNA-Seq
Introduction
RNA editing is the post-transcriptional alteration of an RNA molecule so it differs from its genomic sequence. One well characterized RNA editing event is carried out by adenosine deaminases (ADARs), which converts adenosine to inosine. There are 3 known isoforms: ADAR1, ADAR2 and ADAR3. ADAR1 and ADAR2 are ubiquitously expressed, with expression levels being highest in the brain, while ADAR3 is exclusively expressed in the brain.1 Correlated with ADAR expression levels, adenosine to inosine editing occurs at higher proportions in the brain compared to other tissues.2 RNA editing can result in a change of the amino acid sequence, altered splicing, nuclear retention, or changes in the stability of the transcript.3–5
RNA editing has specific roles in the control of neurotransmission. Modifications in the amino acid sequence of AMPA-subtype Glutamate channels such as Gria2 lead to alterations in neuronal excitability.6 Due to this altered neuronal response to stimulation, Adar2−/− mice die of seizures shortly after birth. However, if the Gria2 Q/R editing site is mutated to mimic the edited base then these mice have the same survival rate as their littermate controls.7 There are also many other examples of adenosine to inosine editing in neural receptors, most of which show an increase in the edited base during development, reaching peak editing several weeks after birth.8 Therefore, control of RNA editing appears to be important in development of brain function.
The editing of the Q/R site in Gria2 depends on an intronic sequence base-pairing with the editing site to create the necessary double stranded structure, suggesting that editing is co-transcriptional, i.e., that editing occurs before intron removal.9 Editing of Gria2 has also been shown to affect the efficiency of its splicing, illustrating that, at least in some cases, editing and splicing may be coordinated.10 Supporting this contention, there are differences in splicing of Gria2 receptors in different subfields of the mouse hippocampus after suppression of neuronal activity with tetrodotoxin.11 Editing enzymes themselves are also highly edited. For example the editing of Adar2 generates a novel splice site and inclusion of a 47-nucleotide cassette, which shifts the open reading frame to an internal translation initiation site producing a truncated protein.4 Although the majority of Adar2 mRNA contains this insert, only a minimal amount of the overall protein product does, indicating a potential negative auto-regulation of expression.4 Mice with a mutation at the editing site complementary sequence of Adar2 show no insertion of the 47-nucleotide cassette and an increase in ADAR2 protein levels in multiple tissues.12 ADAR1 and ADAR2 have also been found to associate with spliceosomal components in large nuclear ribonucleoprotein particles, further demonstrating their potential involvement in splicing.13 There are also correlations between editing and splicing in human tissues.2 One previous study has experimentally addressed the role of ADAR1 in splicing at the genome-wide level in cells using an siRNA plasmid to knockdown ADAR1,14 but there are no equivalent studies for ADAR2 in the brain.
Because ADAR enzymes do not edit every adenosine in the expressed genome, an important question is how ADARs are restricted to certain sites. Previous studies have demonstrated that ADAR2 has 5’ and 3’ preferences and that there are also favored triplet sequences.15,16 However, whether sequence is sufficient to determine ADAR2 specificity is unclear, as it has been observed that other double-stranded RNA binding proteins use structural rather than sequence information to define their specificity.17 Surveys of global editing in mouse brain also suggest that there is some sequence preference for edited sites,18 but there are not absolute restrictions on sequence. How much of this restriction is related to ADAR2 is unclear as in these studies the animals had all editing enzymes present.
To investigate specificity of ADAR2 for different RNA sequences, we used a previously described ADAR2 knockout mouse that has been engineered to be viable through adulthood by introducing a Gria2 mutation to mimic the edited base in the Q/R site.7 We used RNA sequencing (RNA-Seq) to examine RNA editing in a genome-wide manner in frontal cortices of Adar2−/− Gria2R/R mice compared to Adar2+/+ Gria2R/R littermate controls. We find 56 sites that show at least partial ADAR2 specificity for editing. Interestingly we find little evidence of gene expression or exon usage differences indicating ADAR2 does not play a major role in splicing and expression mechanisms and is therefore likely to be directed specifically to changes in protein coding sequences.
Results
We used RNA-Seq to estimate the effects of ADAR2 deficiency on the brain transcriptome, as this technique provides direct sequence information. We compared the frontal cortices of 6 adult male Adar2-/-/Gria2R/R and 6 littermate adult male control (Adar2+/+/Gria2R/R) mice. The generation of these mice has been previously described.7 In this line, a PGK-neo cassette insertion removes exon 4 that contains the catalytic deaminase domain (Fig. 1a). We generated between 31 and 81 million reads per sample, of which 90-93% could be mapped to the mm9 mouse reference genome (Table 1). As expected, there were far fewer reads for ADAR2 mice over the Adar2 gene, particularly exon 4 where the PGK-neo cassette was inserted (Fig 1b; see below for further discussion of splicing in this region).
Figure 1.

Genomic deletion in the Adar2 locus and measured effects on Adar2 expression. (a) Schematic figure of the mutant allele where a PGK-neo cassette is inserted into the mouse Adar2 genomic locus, as described previously.7 Individual numbered exons are in gray, and the +24nt in the partially retained intron 5’ to exon 2 (black) is shown below the figure. (b) RNA-Seq coverage from one representative Adar2+/+ (upper plot) and Adar2−/− (lower plot) animal. Read coverage per base is shown on the y-axes; note that due to disruption by the PGK-neo cassette there is lower overall coverage for Adar2 in the knockout animals and that exon 4 is below limits of detection. Similar results were obtained for all animals in each genotype group.
Table 1.
Quality control parameters for RNA-Seq reads and alignments.
| Adar2 −/− Gria2 R/R | ||||||
|---|---|---|---|---|---|---|
| Animal | 1 | 2 | 3 | 8 | 9 | 12 |
| Total reads | 41,352,506 | 89,460,826 | 53,026,848 | 52,307,792 | 75,605,052 | 40,633,704 |
| Unmapped | 2,761,592 | 7,770,117 | 4,306,666 | 4,981,857 | 6,671,062 | 26,33,712 |
| Total aligned | 38,590,914 | 81,690,709 | 48,720,182 | 47,325,935 | 68,933,990 | 37,999,992 |
| % Aligned | 93.32 | 91.31 | 91.87 | 90.47 | 91.17 | 93.51 |
| Adar2 +/+ Gria2 R/R | ||||||
| Animal | 4 | 5 | 6 | 7 | 10 | 11 |
| Total reads | 82,903,968 | 33,946,702 | 58,223,596 | 55,771,426 | 78,367,494 | 65,266,890 |
| Unmapped | 6261916 | 2,471,010 | 4,965,827 | 4,371,904 | 7,055,969 | 4,761,268 |
| Total aligned | 76,642,052 | 31,475,692 | 53,257,769 | 51,399,522 | 71,311,525 | 60,505,622 |
| % Aligned | 92.44 | 92.72 | 91.47 | 92.16 | 90.99 | 92.70 |
To discover ADAR2 specific editing sites, we first called variants that differed in sequence from the reference mouse genome. To limit any potential sequencing errors, we considered only A-to-I sequence differences within exons and untranslated regions that were shared by all animals within each group at a given base in the genome. After merging of the 2 groups, to allow for the possibility that a given site might be edited only in one group of animals, we then removed possible single nucleotide polymorphisms and ambiguously mapped reads using a BLAT-filter (see Materials and Methods). Each of these filters reduced the number of apparent editing sites such that using the most highly restrictive filtering approach, 143 sites could be mapped to the same base in the genome, each of which was edited in all of the control animals (Table 2). We then separated these sites into UTR (3’and 5’, with an indication of being within a repeat region), coding (synonymous and non-synonymous), and miRNAs (Fig. 2a,Table S1). Approximately 70% of the editing sites were within repeat regions, which is similar to the rates reported in other recent studies.19
Table 2.
Number of potential editing sites detected in the mouse brain using increasingly stringent filtering criteria.
| Adar2 +/+ Gria2R/R | Adar2 −/− Gria2 R/R | |
|---|---|---|
| Variants detected | 148302 | 147515 |
| Shared in all animals in group | 4589 | 6882 |
| Restricted to exon/UTR | 952 | 1695 |
| AtoI/TtoC | 282 | 389 |
| Merged (union) from both groups | 493 | |
| Filtered for known SNPs | 236 | |
| Unambiguously mapped (BLAT) | 143 |
Figure 2.

ADAR2 influences editing of a subset of coding RNAs. (a) Adenosine to Inosine editing. Average percent edited in Control (Adar2+/+Gria2R/R) mouse frontal cortex (n = 6) on the x-axis and average percent editing in the Adar2−/−Gria2R/R mouse frontal cortex (n = 6) on the y-axis. Colors indicate the type of edit sites; green are coding edits; red are miRNA; yellow are UTR and purple are repeat sequences. (b) Technical validation of RNA editing. Two sites in the genes Gria3 and Gabra3 were chosen for validation. We sequenced both cDNA and genomic DNA clones for each site. For each site, an example of edited and non-edited cDNA clones are shown in the top 2 chromatograms and the genomic sequence is shown in the bottom chromatogram. (c,d) Counts of edited and non-edited cDNA clones for Gria3 (c) and Gabra3 (d) (filled bars) showed similar averages to those found in RNA-Seq data (open bars). Error bars indicate SEM.
Using a Fisher's exact test for proportions, comparing edited to unedited base counts in control versus Adar2-/-/Gria2R/R, 69 of the 143 identified sites were differentially edited in a statistically significant manner (p < 0.05 after false discovery rate (FDR) adjustment; Fig 2a). Fifty-six of these sites showed decreased editing in the ADAR2 knockout group. Some sites, such as those found in Cacna1d, were not detectably edited in Adar2-/-/Gria2R/R mice. Differential sites were found within both coding and non-coding regions; although there was a higher proportion in coding regions (p = 0.001; Fisher's exact comparing the number of coding to non-coding sites in differential vs. nondifferential editing when comparing control versus Adar2−/−/Gria2R/R).
We chose 2 differentially edited sites, one in Gria3 the other in Gabra3, for technical validation using cDNA cloning and sequencing. To confirm these sites were not polymorphisms we also sequenced the genomic DNA and only adenosine was found at both sites (Fig. 2b). For Gabra3, we found guanosine in 96% of sequences in the control animals and only 19% in the ADAR2 knockout mice using RNA-Seq. Similarly, for cDNA cloning and sequencing, 90% of sequences were guanosine in control animals and 17% were edited in the Adar2-/-/Gria2R/R mice. The Gria3 site was edited in 94% of sequences in the control group and edited at 81% in ADAR2 knockout mice in the RNA-Seq dataset. We validated these difference using cloning and sequencing, with 95% editing in the control animals and 86% editing in the Adar2-/-/Gria2R/R mice (Figs. 2c,d).
We also specifically investigated whether the 47-nucleotide cassette, reported to be dependent on ADAR-editing, was modified, as these sites were not in the final list of edit sites due to annotation as an intronic region. Interestingly, we find that the cassette is still present with evidence of editing in all ADAR2 knockout animals for the +24 site (Table 3), although the actual proportion of edited sites in each animal was highly variable.
Table 3.
Editing percentage for the +24 site found within the 47-casette insert in Adar2 in individual animals.
| Adar2 −/− Gria2 R/R | Adar2 +/+ Gria2 R/R |
|---|---|
| 43 | 76 |
| 50 | 79 |
| 71 | 69 |
| 86 | 88 |
| 82 | 80 |
| 35 | 82 |
To explore potential sequence specificity of mouse ADAR2, we calculated the frequency of bases either side of the 56 editing sites with significantly decreased editing in our restricted data set. Looking immediately 5′ to the edited base, U had the most representation comprising 41.1% of sequences. The next common base was A at 32.1% than C with 21.4% and lastly the least represented base was G with only 5.4% of sequences. Investigating immediate 3′ base preference we found G comprised the majority of sequences at 62.5%. Bases A, C, and U were fairly evenly represented at 14.3%, 12.5%, and 10.7% respectively. Preference for triplet sequences was also calculated with UAG, AAG, and CAG being the most common at 23.2%, 19.6% and 14.3% respectively.
The highly restrictive nature of filtering used to generate the list of A to I sites that could be mapped unambiguously to the same genomic region in all animals, thereby allowing for strict comparisons between genotypes, resulted in a much smaller set of candidate sites than has previously been reported elsewhere.18 We therefore examined the effect of using less stringent filtering approaches. Specifically, we removed the filter requiring that the edits were in either cds or UTR. However, excluding this filter resulted in a dataset with a larger set of 551 sites that had similar properties to the more restrictive data set outlined above (Fis. S1). Specifically, we found that; more editing sites were diminished in the Adar2 knockout mice compared to controls with variable proportions (Fig. S1A and 1B,b); more sites were in SINE repeats than other types of repeats or non-repeat elements (Fig. S1C and 1D); and that the sequence preferences around the edited site were similar with either more or less restrictive filters (Fig. S1E and 1F). These data suggest that the effects of Adar2 deficiency are retained irrespective of at least some choices in data analysis and are therefore likely to be robust.
As previous studies have suggested that ADARs can affect expression and splicing, we investigated this possibility in a genome-wide manner. Differential expression revealed that Adar2, Flnb, and Cdh13 had a statistically significant difference comparing ADAR2 knockouts to controls (Figs. 3a, b). However, these were relatively modest differences in expression, with only Adar2 having more than a 2-fold difference in expression. Measuring differential exon usage, exon 4 of ADAR2 had the largest statically significant change, with an average of over 2 hundred counts for their littermate controls compared to close to zero counts for Adar2−/−/Gria2R/R animals. Exons 5, 6, 7 were present at around 10 % compared to their littermate controls, likely due to exon skipping of the PGK-neo cassette present in the knock out animals. Along with multiple exons of ADAR2, exons found in Hcfc2, Zfp941, and Vps52 were differential expressed but only exon 4 of ADAR2 reached over 2-fold difference between genotype. There is some evidence that ADAR2 may play a role in Gria2 splicing10 and hence we specifically examined Gria2 exon usage. However, we found no difference between Adar2-/-/Gria2R/R and their littermate controls (Fig. 4). Overall, these data suggest that the net effect of ADAR2 deficiency in the mouse frontal cortex on splicing is relatively modest.
Figure 3.

ADAR2 deficiency has only modest effects on mRNA expression levels or exon utilization. (a) Plot of fold difference of log2 normalized expression in Control (Adar+/+Gria2R/R) frontal cortex (n = 6) versus Adar2−/− Gria2R/R frontal cortex (n = 6) on the x-axis against –log10 adjusted p-value on the y-axis for all detected genes. Each gene is colored based on the log10 base mean expression where more highly expressed genes are in red. Adar2, Flnb, and Cdh13 are the only genes that were significantly differentially expressed (adjusted p < 0.05). (b) Differential alternative exon events. Plot of differential exon expression with fold difference of log2 normalized expression in Control (Adar2+/+Gria2R/R) frontal cortex (n = 6) vs. Adar2−/−Gria2R/R frontal cortex (n=6) on the x-axis and –log10 adjusted p-value on the y-axis for all detected exons. Blue colors show individual exons of Adar2, which account for all but 3 of the significantly different exons found in the genes Hcfc2, Zfp941, and Vps52 (p adjusted < 0.05).
Figure 4.
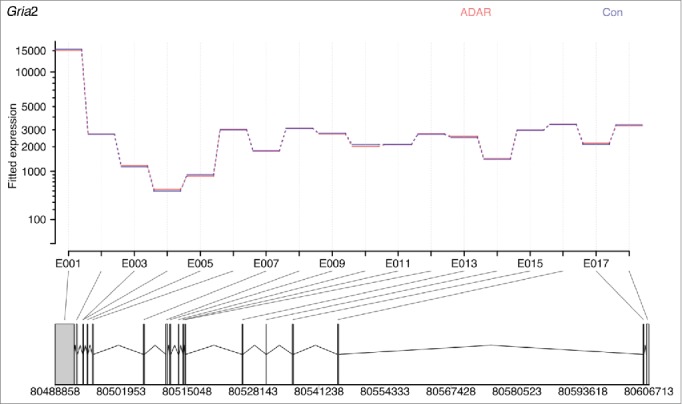
ADAR2 deficiency does not affect Gria2 exon expression levels. Image was generated inputting read counts for each exon into DEXSeq. The data is normalized for overall number of counts for Gria2, with blue showing the average number of normalized counts for all Adar2−/−Gria2R/R animals, and red the average number of normalized counts for all littermate control (Adar2+/+Gria2R/R) animals. The lines are completely overlapping showing that expression of each exon is the same in both groups, and hence there is no differences in splicing despite a decrease in editing of the R/G site of Gria2.
Discussion
In the current study, we used RNA-Seq to measure changes in expression, splicing and editing in Adar2-/-/Gria2R/R mice and littermate controls to elucidate the potential role of ADAR2 in these mechanisms. With this dataset we were able to look for editing changes in an unbiased genome-wide manner to elucidate ADAR2 specificity, providing a global view of the role of one of the ADAR isoenzymes in RNA editing.
We found evidence for many edit sites in control animals in both coding and non-coding regions. Most of these sites have been previously identified in our work20 or by Wahlstedt et al.,8 suggesting they are both technically and biologically robust. The number of edited sites is lower than some previous estimates, likely because we used very stringent filtering to allow for more robust comparisons between groups. For example Danecek et al reported approximately 4,800 editing sites in whole brain samples of different mouse strains but half of these were edited to less than 20%.18 We required that the exact editing sites were present in all of the wild type animals. Furthermore, as editing levels can be strain- and brain-region specific, our approach might yield different results in other animals. Nonetheless, this approach clearly provides a reliable, if likely conservative, estimate of the editing percentage for at least some sites as supported by our ability to validate proportions of editing by cloning and sequencing.
Comparing Adar2−/−/Gria2R/R animals to their littermate controls we found editing differences in 69 out of 143 sites. These data confirm that ADAR2 affects editing of multiple coding sequences. Very few of these sites were completely unedited in the knockout animals. The most likely explanation for the residual editing found in most sites is that they are edited by multiple ADARs. There is support for this hypothesis as, for example, the secondary editing site (R/G) in Gria2 can be efficiently edited by recombinant ADAR1 or 2.21 Our data shows that this specific site retains some editing in the Adar2-/-/Gria2R/R animals. The characterization of the Adar2-/- Gria2R/R mice7 by Higuchi et al suggested that there is residual Adar2 expression due to transcripts that skip the PGK-neo cassette but that the enzyme would be inactive as the neo-cassette replaces the part of exon 4, thought to encode an essential adenosine deaminase motif. In the Adar2 gene itself we also find evidence of editing activity in the Adar2-/-/Gria2R/R animals, since all samples showed some level of inclusion of the 47-nucleotide cassette, which is determined, by editing of the -1 position of exon 2 of the Adar2 gene. As the ADAR2 enzyme is not present to edit this site in these animals, a possible explanation for cassette retention is that ADAR1 can edit ADAR2, as proposed above for other A to I editing sites. Previous data suggests that the -1 editing site is necessary for inclusion of the 47-nucleotide cassette in ADAR2, but did not address if ADAR2 is necessary for that editing event, notwithstanding that ADAR2 can edit itself in vitro. Our data would support the contention that ADAR1 can, at least in the context of chronic ADAR2 deficiency, contribute to editing at the same site, as we have suggested in general and would be consistent with the maintenance of some editing when ADAR2 is knocked out in younger animals. The editing of ADAR2 is therefore likely to still be an auto-editing site, but not purely restricted to ADAR2 vs. other ADAR enzymes.
Further illustrating potential interaction between ADAR isoforms, we found 13 sites that have increased editing in Adar2-/-/Gria2R/R animals, which may indicate that there is competition between ADAR isoenzymes when there are 2 sites within close proximity. For instance, within the gene Ebna1bp2 a site at position 118298748 editing doubles in Adar2-/-/Gria2R/R animals as compared to controls, while a site only 17 bp away is only half as edited in Adar2-/-/Gria2R/R animals. It may be that if ADAR2 is not present to edit a given site, as is the case with the Adar2-/-/Gria2R/R animals, then ADAR1 might have better availability to its nearby site. Such a scenario may occur with the D site of 5-HT(2C) receptor. Sites A and B show ADAR1 specificity and site D, less than 15 base pairs away, is ADAR2 specific. Knocking out ADAR1 results in a complete loss of both A and B site editing, but an increase in editing of site D when compared to controls.22 It is unlikely that increases in editing at sites after ADAR2 deletion in our study were due to changes in ADAR1 levels as the base mean expression was 4361 in the Adar2-/-/Gria2R/R compared to 4304 in their littermate controls. More likely is that ADAR1 and ADAR2 compete for a subset of sites where neither enzyme is completely specific, leading to enhanced access by ADAR1 when ADAR2 is absent.
One aim of this study was to use ADAR2 specific editing sites to understand substrate specificity. Because there may be some differences in specificity between species as human ADARs mainly target ALU repeats while mouse ADARs target SINE, LINE, and LTR repeats,23 we inferred specificity from ADAR2 knockout mice. Lehmann and Bass suggested that human ADAR2 has a preference both 5’ and 3’ of the edited adenosine, as A≈U>C=G for the 5’ and U=G>C=A for the 3’. The same study also found triplet preference for UAU, AAG, UAG and AAU.16 In mouse tissue Danecek et al., reported a slightly different preference for editing in the mouse brain where all ADARs are expressed, showing edited preferences for U>A∼C>>G at the 5’ base of the edited site and G>A∼C∼U at the 3’ base.18 Using all statistically differential edited sites in the mouse frontal cortex we looked at sequence similarity within 20 base pairs on either side of the edited site and found a preference for U or A immediately 5’ of the edited site, accounting for about 70% of the sequences, consistent with the human ADAR2 sequence identified previously.16 Immediately 3’ to the edited site, we confirmed a preference for G which again is consistent with the equivalency of G and U in human ADAR216 but also with the overall preference for editing of sites in mice.18 There was also a triplet sequence preference for UAG, again generally consistent with what has been reported for editing in the mouse brain.18 Overall, these data suggest that ADAR2 has some sequence specificity but, allied with the inference that ADAR1 may also edit overlapping sites, ADAR2 is not absolutely specific for editing of all expressed mRNA species in the mouse brain.
ADAR2 has been suggested to play a role in Gria2 splicing and Adar2 gene expression regulatory machinery. Furthermore, ADAR1 knockdown in cells may result in global changes in splicing.14 We therefore looked at expression and exon usage in a global manner in our data set to see if ADAR2 would have the same global effect. We found relatively few examples of differences in expression and exon usage in the frontal cortex Adar2-/-/Gria2R/R mice in comparison to littermate controls. For the specific splicing of Gria2, exon expression patterns were the same in Adar2-/-/Gria2R/R and their littermate controls. This would be consistent with previous work of Bratt et al. who found that although editing of the R/G site negatively affected splicing in vitro the same did not hold true in vivo.24
The potential correlation between editing and splicing is further complicated by the fact that both the R/G site and flip inclusion are regulated during development.20 Instead, we propose that at least in mouse frontal cortex ADAR2 has a predominant role in control of protein diversity, supported by the observation that there is an excess of changes in editing sites that are in coding regions. We speculate that ADAR2 may have evolved a specific function that is distinct from the effect of ADAR1 on editing RNA species in response to infection.25 Such a prediction could be tested in future experiments using RNA-Seq from ADAR1 deficient animals, possibly after challenge with viral infection. In those experiments where a relationship between editing and splicing has been noted for ADAR1 the precise mechanism(s) involved are not fully identified, and it has been proposed that ADAR1 might affect splicing through indirect, trans-acting mechanisms rather than acting on splicing in cis.14 Therefore, it is possible that the lack of effects of ADAR2 deficiency are because ADAR enzymes affect splicing in an indirect manner, notwithstanding that there are important correlations between the 2 ways in which protein diversity is generated.
It is also important to acknowledge that although A to I editing occurs at highest levels in the brain compared to other tissues, it is possible that there are stronger effects in other brain regions than the frontal cortex. We have not examined either the striatum2 or the CA1 and CA3 hippocampal subfields,11 where there may be a stronger relationship between splicing and editing. Estimation of some RNA editing sites may be dependent on both the tissue sampled and the approach used. For example, the +24 site of ADAR2 is reported to be edited to ∼45% in whole brain samples using estimates based on peak height in sequencing.7 We found the same site edited to ∼80% in the samples from frontal cortex used here, which is consistent with a more recent RNA-Seq based study in wild type mice.8 In our hands, the level of editing at this site is highly variable between animals but not lost completely in the ADAR2-/- group which contrasts to the whole brain results from Higuchi et al that found ∼10% residual editing in these animals. We speculate that the retention of some editing at this site might also relate to the ability of ADAR1 to partially compensate for some ADAR2 sites. A final caveat is that, in order to test experimentally the effects of ADAR2 deficiency in vivo, we used a mouse model whereas there is some evidence that primate-specific genes are edited by ADAR2, which we have not addressed here.
Overall this dataset provides a set of ADAR2 specific editing sites in an unbiased genome-wide manner in the mouse brain. We propose that some of the observed specificity relates to a derived function of ADAR2 on editing of coding sequences.
Methods
RNA extraction and sequencing
Total RNA was extracted from the frontal cortex of adult male mice using a glass dounce tissue homogenizer and trizol. An Agilent 2100 Bioanalyzer RNA Nano Chip was utilized to measure the RNA quality; samples had a mean RIN of 9.6 (+/-0.3). Library preparation was performed on 1 μg total RNA. cDNA libraries were synthesized using the TruSeq stranded total RNA sample preparation kit (Illumina cat. no. RS-122-2303) with ribo-Zero gold rRNA removal beads as per the manufacturer's protocol (http://supportres.illumina.com/documents/documentation/chemistry_documentation/samplepreps_truseq/truseqstrandedtotalrna/truseq_stranded_totalrna_sampleprep_guide_15031048_d.pdf). Six libraries were pooled and 4.5 pM of each library ran on a single flowcell lane of an Illumina Hi-Seq sequencer generating 100bp paired end reads.
Statistical analysis of gene expression and alternative exon utilization
Fastq files were aligned to the mouse reference genome (mm9) using Tophat (v2.0.6) and Bowtie (2.02.0) applying the ensembl gtf option with the Mus_musculus.NCBIM37.61 gtf to build bowtie indexes. Overall quality and total read counts are listed in Table 1. Reads were annotated and quantified to a given gene or exon using the Python module HT-SEQ. For gene counts the same ensembl gtf mentioned above was used to provide reference boundaries. For exon counts this gtf was flattened using the python script found with HT-SEQ to create the appropriate counting bins needed for downstream analysis. We used either the R/Bioconductor package DESeq for genes or DEXSeq for exons to normalize for library size and to perform a variance-stabilizing transformation. Poisson distributions of normalized counts for each transcript were then compared across Adar2-/-/Gria2R/R and control groups using a negative binomial test. Multiple testing was corrected for using the Benjamini-Hochberg procedure. For gene expression analysis genes with base mean expression of less than 15 were filtered out for low coverage. For exon analysis exons with base mean expression of less than 20 were filtered out for low coverage.
Editing
Bams were first filtered for only singly mapped reads. Variants were then called for each sample using SAMtools mpileup (samtools-0.1.19.0) with mm9 as a reference. We extracted variants within a gene boundary, rather than introns, and with A to G in the positive strand or T to C changes in the negative strand, as this is the known edited base for Adar. We created a merged table to include only variants shared by all samples within either the Adar2-/-/Gria2R/R or control group then filtered out any SNPs based on the snp128 build. Finally, we submitted sequences containing the candidate edit sites to UCSC BLAT to distinguish reads with single mismatches to the genomic sequence due to RNA editing from those due to inappropriate mapping to another part of the genome. We accepted candidate edits if the best scored alignment of the read included the site of the edited base from the original alignment; if there were multiple genomic alignments of equal or higher score, then the candidate edit site was discarded. The number of candidate sites remaining at each step of the filtering procedure is shown in Table 2.
To determine sequence preference we used the 56 sites found to have a decrease in editing when comparing Adar2-/-/Gria2R/R to littermate controls and summed the immediate base both 3′ and 5′ from the edited base for A, G, C, and T. This value was then divided by the total number of sites to calculate the percentage preference and a position weighted matrix was used as an input into the R package seqLogo and displayed without scaling for information content. Triplet preference was calculated in a similar manner.
For supplementary analyses, we used the same approaches for differential editing, classification into repeat and non-repeat sequences and sequence preference as above but without the filter for gene boundaries, thus including intergenic and intronic reads. This increased the number of A to I sites from 143 to 554 and the number of differential sites from 69 to 103.
RNA editing validation by RT-PCR and sequencing
cDNA was made from RNA extracted with trizol using the Superscript III First-Strand Synthesis System from Invitrogen. We designed primer pairs that were approximately 150 base pairs away from the editing site and spanned an exon junction to avoid genomic DNA amplification. We also designed a genomic DNA primer to span the same site that amplified a product of similar size when used with one of the cDNA primers. Primer sequences are available upon request. Amplification of cDNA and genomic DNA was then performed with Fast Start Master Mix (Roche). Products were separated on a 1.5% agarose gel, cut out and extracted DNA using a QIAquick gel extraction kit (Qiagen). We cloned DNA into the PCR8-TA cloning vector (Invitrogen) in One Shot Mach1-T1 Chemically competent cells and sequenced 24 colonies from 3 biological replicates for each edit site and 12 colonies from genomic DNA using a 3730 capillary sequencer.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (Project number AG000947) and by the Swedish Research Council (Project 2012-2353) and Swedish brain power. This study used the high-performance computational capabilities of the Biowulf Linux cluster (http://biowulf.nih.gov). We are also very grateful to Dr. Peter Seeburg and Dr. Miyoko Higuchi for their kind gift of the ADAR2 knockout and control mouse brains required for this study.
References
- 1. Hogg M, Paro S, Keegan LP, O’Connell MA. RNA editing by mammalian ADARs. Adv Genet 2011; 73:87-120; PMID:21310295; http://dx.doi.org/ 10.1016/B978-0-12-380860-8.00003-3 [DOI] [PubMed] [Google Scholar]
- 2. Wu D-D, Ye L-Q, Li Y, Sun Y-B, Shao Y, Chen C, Zhu Z, Zhong L, Wang L, Irwin DM, et al. Integrative analyses of RNA editing, alternative splicing, and expression of young genes in human brain transcriptome by deep RNA sequencing. J Mol Cell Biol 2015; 7:314-25; PMID:26186942; http://dx.doi.org/ 10.1093/jmcb/mjv043 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Z, Carmichael GG. The fate of dsRNA in the nucleus: a p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 2001; 106:465-75; PMID:11525732; http://dx.doi.org/ 10.1016/S0092-8674(01)00466-4 [DOI] [PubMed] [Google Scholar]
- 4. Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature 1999; 399:75-80; PMID:10331393; http://dx.doi.org/ 10.1038/19992 [DOI] [PubMed] [Google Scholar]
- 5. Serra MJ, Smolter PE, Westhof E. Pronounced instability of tandem IU base pairs in RNA. Nucleic Acids Res 2004; 32:1824-8; PMID:15037659; http://dx.doi.org/ 10.1093/nar/gkh501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenthal JJC, Seeburg PH. A-to-I RNA editing: effects on proteins key to neural excitability. Neuron 2012; 74:432-9; PMID:22578495; http://dx.doi.org/ 10.1016/j.neuron.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000; 406:78-81; PMID:10894545; http://dx.doi.org/ 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- 8. Wahlstedt H, Daniel C, Ensterö M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 2009; 19:978-86; PMID:19420382; http://dx.doi.org/ 10.1101/gr.089409.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 1993; 75:1361-70; PMID:8269514; http://dx.doi.org/ 10.1016/0092-8674(93)90622-W [DOI] [PubMed] [Google Scholar]
- 10. Schoft VK, Schopoff S, Jantsch MF. Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res 2007; 35:3723-32; PMID:17517775; http://dx.doi.org/ 10.1093/nar/gkm314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Penn AC, Balik A, Wozny C, Cais O, Greger IH. Activity-mediated AMPA receptor remodeling, driven by alternative splicing in the ligand-binding domain. Neuron 2012; 76:503-10; PMID:23141062; http://dx.doi.org/ 10.1016/j.neuron.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng Y, Sansam CL, Singh M, Emeson RB. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol Cell Biol 2006; 26:480-8; PMID:16382140; http://dx.doi.org/ 10.1128/MCB.26.2.480-488.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raitskin O, Cho DS, Sperling J, Nishikura K, Sperling R. RNA editing activity is associated with splicing factors in lnRNP particles: The nuclear pre-mRNA processing machinery. Proc Natl Acad Sci U S A 2001; 98:6571-6; PMID:11381114; http://dx.doi.org/ 10.1073/pnas.111153798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Solomon O, Oren S, Safran M, Deshet-Unger N, Akiva P, Jacob-Hirsch J, Cesarkas K, Kabesa R, Amariglio N, Unger R, et al. Global regulation of alternative splicing by adenosine deaminase acting on RNA (ADAR). RNA N Y N 2013; 19:591-604; http://dx.doi.org/ 10.1261/rna.038042.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuttan A, Bass BL. Mechanistic insights into editing-site specificity of ADARs. Proc Natl Acad Sci U S A 2012; 109:E3295-304; PMID:23129636; http://dx.doi.org/ 10.1073/pnas.1212548109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry (Mosc) 2000; 39:12875-84; http://dx.doi.org/ 10.1021/bi001383g [DOI] [PubMed] [Google Scholar]
- 17. Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J 1998; 17:7505-13; PMID:9857205; http://dx.doi.org/ 10.1093/emboj/17.24.7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Danecek P, Nellåker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol 2012; 13:26; PMID:22524474; http://dx.doi.org/ 10.1186/gb-2012-13-4-r26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim DDY, Kim TTY, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res 2004; 14:1719-25; PMID:15342557; http://dx.doi.org/ 10.1101/gr.2855504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, Rudenko IN, Galter D, Cookson MR. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat Neurosci 2013; 16:499-506; PMID:23416452; http://dx.doi.org/ 10.1038/nn.3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature 1996; 379:460-4; PMID:8559253; http://dx.doi.org/ 10.1038/379460a0 [DOI] [PubMed] [Google Scholar]
- 22. Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 2004; 279:4894-902; PMID:14615479; http://dx.doi.org/ 10.1074/jbc.M311347200 [DOI] [PubMed] [Google Scholar]
- 23. Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA N Y N 2006; 12:1802-9; http://dx.doi.org/ 10.1261/rna.165106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bratt E, Ohman M. Coordination of editing and splicing of glutamate receptor pre-mRNA. RNA N Y N 2003; 9:309-18; http://dx.doi.org/ 10.1261/rna.2750803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hartwig D, Schütte C, Warnecke J, Dorn I, Hennig H, Kirchner H, Schlenke P. The large form of ADAR 1 is responsible for enhanced hepatitis delta virus RNA editing in interferon-alpha-stimulated host cells. J Viral Hepat 2006; 13:150-7; PMID:16475990; http://dx.doi.org/ 10.1111/j.1365-2893.2005.00663.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.