

Abstract
RNA editing is a widespread mechanism that plays a crucial role in diversifying gene products. Its abundance and importance in regulating cellular processes were revealed using new sequencing technologies. The majority of these editing events, however, cannot be associated with regulatory mechanisms. We use tissue-specific high-throughput libraries of D. melanogaster to study RNA editing. We introduce an analysis pipeline that utilises large input data and explicitly captures ADAR's requirement for double-stranded regions. It combines probabilistic and deterministic filters and can identify RNA editing events with a low estimated false positive rate. Analyzing ten different tissue types, we predict 2879 editing sites and provide their detailed characterization. Our analysis pipeline accurately distinguishes genuine editing sites from SNPs and sequencing and mapping artifacts. Our editing sites are 3 times more likely to occur in exons with multiple splicing acceptor/donor sites than in exons with unique splice sites (p-value < 2.10−15). Furthermore, we identify 244 edited regions where RNA editing and alternative splicing are likely to influence each other. For 96 out of these 244 regions, we find evolutionary evidence for conserved RNA secondary-structures near splice sites suggesting a potential regulatory mechanism where RNA editing may alter splicing patterns via changes in local RNA structure.
Keywords: ADAR, alternative splicing, post-transcriptional regulation, RNA editing, RNA secondary structure
Introduction
The first release of the Homo sapiens genome1 made it quite clear that the number of protein coding genes (genes in the following) alone is only a poor measure for the complexity of the corresponding organism. Comparing, for example, the number of genes in the nematode genome of C. elegans (∼20,000) to those of the human genome (∼30,000) gives one an inkling that there must be substantial other cellular mechanisms at work beyond those of the Central Dogma in Biology2 to account for the discrepancy of 2 organisms' complexity. More recently, post-transcriptional mechanisms such as alternative splicing and RNA editing3-6 have been shown to significantly expand the number of functionally relevant gene products via differential regulation of transcripts of a single gene.
RNA editing is a widespread molecular mechanism in metazoa which modifies the primary transcripts of genomes.7 Nucleotide insertions were the first type of RNA editing, discovered in 1986 in trypanosomes.8 ADAR proteins (ADARs) are responsible for carrying out the most frequent type of RNA editing, A-to-I RNA editing, in mammals where an adenosine is converted to inosine in RNA transcripts.7 The notable abundance of A-to-I editing events in species such as human,6,9-13 Mus musculus (mouse),14-16 and Drosophila melanogaster (fly)17-19 (from thousands of sites in the fly genome to more than a million sites in human) demonstrates their significant potential to contribute to the regulation of other cellular mechanisms.
ADARs require double-stranded RNA regions to perform the deamination process.20 In primary transcripts, these regions are typically formed by local RNA secondary-structure features such as hair-pins. Once an appropriate double-stranded region is found, ADARs bind a base-paired adenosine and edit it without being very specific about the primary sequence surrounding the substrate.21 In other words, the requirement for a double-stranded structural context is much more important than the primary nucleotide composition in specifying a potential ADAR binding site.7 Somewhat surprisingly, this key feature has not yet been directly exploited in most RNA editing prediction programs.22,23 Many of the known double-stranded regions serving as ADAR binding sites are formed between exonic sequences and complementary intronic sequences24 (known as editing site complementary sequences). This supports the idea that editing usually precedes splicing.25 Also, for many editing sites levels of pre-mRNA editing and mRNA editing correlate well in D. melanogaster showing that RNA editing can happen co-transcriptionally.19 A well-studied example is the editing of RNA structures formed between inverted Alu repeats in human transcripts.26 Alu repeats constitute more than 10% of the human genome and can readily form double-stranded region and thus potential RNA editing sites by binding to their inverted copies in the same primary transcript. When one site is edited, other adenosine nucleotides in the same double-stranded region have a high chance of also being edited by the same ADAR protein; this may result in the conversion of several adenosines in a small region.16,27
Despite the considerable, recent efforts to discover functionalities of editing, there is still much to be discovered and understood regarding the molecular mechanisms and functional roles of RNA editing. Most cellular mechanisms interpret inosine as guanosine, including splicing and translation. Some cellular factors (e.g. Tudor staphylococcal nuclease involved in RNA interference), however, can distinguish inosine from guanosine.28
There is already some evidence showing that ADARs play a role in changing protein properties,29 modifying RNA secondary structures,20 changing splicing efficiencies,30 regulating gene expression,6 and recovering aberrant mutations.13 Although the modification of a single nucleotide within a transcript can have many potential consequences (similar to those just mentioned), the number of reported cases for each of the mechanisms does not provide a convincing explanation for the thousands of RNA editing events predicted in human, mouse and fly. Hence, there remains much to be understood.
Although studies suggested some primary sequence features and also proteins that affect ADAR activity in specific target regions, the general regulation of RNA editing is unclear. Inverted copy sequences in proximity of a region increase the editing probability of that region, probably by having the potential to form the double-stranded region required for ADAR binding; in support of this, Alu repeats were observed to constitute the majority of ADAR targets in human transcripts.9,12 Moreover, some short primary sequence preferences have been observed for ADAR proteins in human,13,31 mouse23 and fly.32 On the other hand, few RNA-binding proteins have so far been shown to suppress the editing levels of specific targets.33 The SFRS9 gene, which encodes a splicing factor, represses the editing of the cyFIP2 gene. This could be the result of competition between the 2 proteins for common substrates or due to the protein-protein interaction between ADAR2 and SFRS9.33 The level of ADAR expression is another regulatory factor, despite it not usually correlating well with the level of RNA editing.34
Recent studies suggest that alternative splicing and RNA editing mechanisms have the potential to influence each other.25,35 Obviously, RNA editing can directly modify splicing patterns by editing primary sequence motifs required such as splice sites, splicing enhancers or silencers.25,36 Other studies in human and fly suggest that many of the editing sites occur in transcripts encoding RNA-binding proteins that play roles in alternative splicing. This may alter the expression, efficiency or binding properties of these proteins which may in turn affect the splicing of many genes.32,35 On the other hand, different ADAR isoforms have different editing efficiencies,37 so the splicing machinery also has the potential to influence RNA editing. It thus seems obvious to hypothesize that there are feedback loops between RNA editing and alternative splicing waiting to be discovered.
The recent advent of new technologies for high-throughput sequencing has brought new opportunities to studying RNA editing. In the past few years, thousands of editing sites have been discovered by calling A-to-G differences between the reference genome and the transcriptome reads in human,6,12,13 mouse,16 and fly.17,19 Recent RNA-Seq technologies are capable of generating hundreds of millions of reads with a much lower cost compared to traditional Sanger sequencing. Moreover, the large number of reads aligned to a single genomic location makes the prediction of the RNA editing sites with a low level of editing not only feasible, but also more reliable. One downside of high-throughput sequencing as opposed to Sanger sequencing is certainly the shorter read length as well as the higher sequencing error rate (not to mention its position dependence). One key challenge when analyzing RNA-seq is thus to discriminate true editing events from different kinds of artifacts.6,12,13 More specifically, short reads with several sequencing errors can easily be misaligned to the genome (or not aligned at all), especially when dealing with repeat-rich sequences such as part of the human genome.38 The high mutation rates of some species can make it difficult to align transcriptome reads to a reference genome unless genomic reads from the same individual are also available. Also, if the transcriptome data are not generated in a strand-specific manner, incompletely annotated parts of overlapping genes can cause misleading mismatch calls.16
As a result of these and many other potential challenges, RNA-seq data require sophisticated and statistical data analysis methods for reliably detecting RNA editing events. Fortunately, the large number of experimentally confirmed A-to-I RNA editing events in Drosophila melanogaster and the considerable amount of publicly available data make the fly a promising model organism to study ADAR mechanisms. In recent years, there has been an increasing amount of studies on the importance and abundance of RNA editing in this organism.17,19,37 D. melanogaster has one ADAR gene (dADAR), and among vertebrate ADARs, ADAR2 is the most similar gene to dADAR.39 In fly, dADAR is highly expressed in the central nervous system, and similar to vertebrates, its expression shows tight temporal regulation.37 Thousands of RNA editing sites have been previously reported.17,19
Here, we use tissue-specific high-throughput data sets of D. melanogaster from the modENCODE project 40 to identify RNA editing events in multiple tissues. To achieve this, we introduce a new computational analysis pipeline to accurately identify editing events and to distinguish genuine editing events from sequencing and mapping artifacts. In our analysis of the resulting, predicted cases of RNA editing, we search for cases of differential exon usage between pairs of different tissues to identify regions where RNA editing and alternative splicing may influence each other. Finally, in order to discover potential molecular mechanisms underlying this interplay, we identify many cases of evolutionarily conserved RNA secondary-structures that have the potential to regulate alternative splicing via RNA editing.
Results
Our pipeline accurately distinguishes genuine editing sites from SNPs, and sequencing and mapping artifacts
To study RNA editing, we selected tissue-specific RNA-seq libraries of Drosophila melanogaster from the modENCODE project,40,41 classified into 10 tissues (Fig. S1). For detailed information on our data set see Materials and Methods section.
The set of sites predicted by our pipeline is highly enriched in A-to-G conversions. Figure 1A shows the number of unique RNA editing sites identified for each of the 12 possible types of DNA/RNA differences after applying our pipeline to the combined data set comprising all 29 libraries. We find 3680 unique conversion sites in multiple tissues of D. melanogaster of which 2879 (78.2%) correspond to A-to-G conversions (Table S1). Assuming similar A-to-G and G-to-A mutation rates as well as similar rates of sequencing and mapping errors for these 2 types of transitions, we can estimate the false positive error rate of our predictions. Of the 3680 sites in our set, 112 of them are G-to-A conversions. By assuming that up to 112 of these A-to-G detected sites are false positive predictions, we estimate the false positive rate to be at most 3.9% (112/2879). Estimates of false positive rates for each tissue separately are listed in Figure S2.
Figure 1.
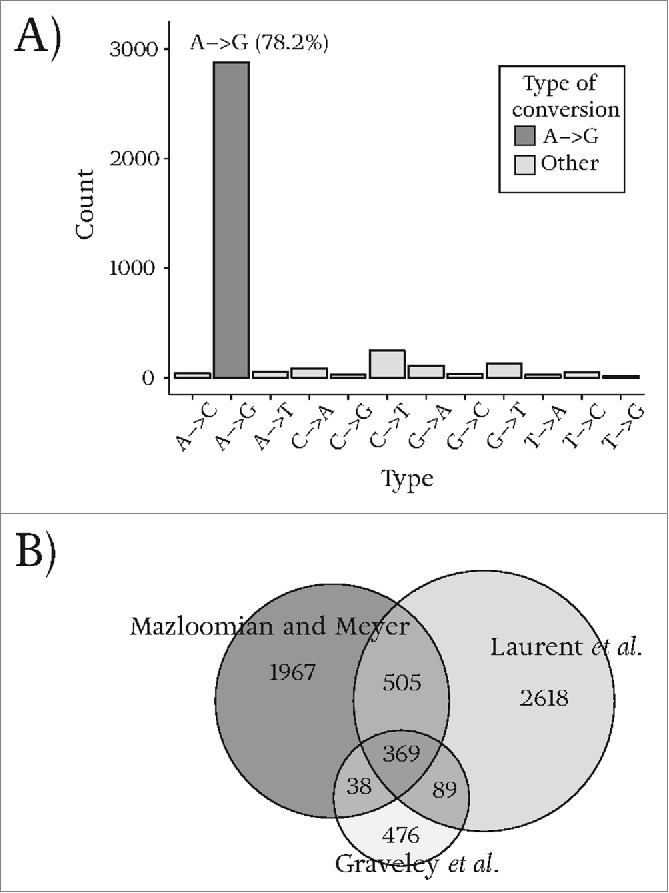
Types of identified conversions and the overlap of A-to-G conversions with other high-throughput studies. (A) Number of different types of conversions identified by our analysis pipeline. Most of the identified sites correspond to A-to-G conversion. (B) Venn diagram showing the overlap between our study and 2 other high-throughput studies by Graveley et al.17 and Laurent et al.32
Figure 1B shows the extent of overlap of the 2879 RNA editing sites identified by us and those of 2 other genome wide studies in D. melanogaster by Graveley et al.17 and Laurent et al.32 In contrast to our study, Graveley et al. analyze RNA-seq data sets of the modENCODE project from different developmental stages, i.e. their read samples do not overlap our tissue-specific data sets. In another high-throughput genome wide study of RNA editing in D. melanogaster, Laurent et al. employ single molecule sequencing for data generation. As Figure 1B shows, the overlap of sites predicted by this and both previous studies is not very high (369/2879(13%)), yet the overlap between our sites and each study separately, especially Laurent et al., is considerable (874/2879 (30%) Laurent et al., 407/2879 (14%) Graveley et al.), implying that a reassuring third (912/2879 (32%)) of our RNA editing sites have been detected by either of these earlier studies, while still adding a large number (1967) of new potential RNA editing to the existing D. melanogaster annotation.
Apart from the obvious differences in the sampled cells, effects such as random sampling in high-throughput studies, sequencing errors and other challenges in distinguishing editing sites from artifacts42 can account for the observed differences in detected sites. One of the key features of ADAR-derived RNA editing is that even in the same cell, the editing of 2 transcripts of the same gene does not necessarily involve identical RNA editing sites, but only the same double-stranded region which seems to be necessary and sufficient requirement for RNA editing to have the desired functional effect. Furthermore, differences in the proposed pipelines of these studies (different sets of thresholds, tests and captured features) and differences in the type of input data (strandedness, single end reads as opposed to paired end reads only) could at least partially account for some of the observed variations in results.
Among all output sites of our pipeline, 45% (1288/2879) have been predicted by at least one of 4 existing RNA-seq studies of RNA editing in D. melanogaster,17-19,32 and at least 14% (400/2879) have been experimentally validated. In summary, the number of previously validated and identified sites combined with the low estimated false positive error rate indicates that most of our reported sites are likely to be genuine RNA editing sites.
Characterization of identified RNA editing sites
As would be expected when analyzing libraries of RNA-seq data (i.e., reads derived from mRNAs), most (40%, 1149/2879) our RNA editing sites occur in coding regions. Figure 2A represents the distribution of RNA editing sites (which obviously derive from the transcriptome) onto different types of genomic regions. The abundance of RNA editing events in coding regions when analyzing pre-mRNAs of the fly genome has been reported earlier.19 Editing in coding regions can cause non-synonymous changes. These may alter the sequence of a protein (and possibly its length) and also change the protein's structure and function.
Figure 2.

Characterization of the identified editing sites. (A) Number and percentage of identified sites in different genomic regions. Coding regions contain more sites than other regions. (B) Number of all 12 types of conversions for 4 tissues of our study: Central Nervous System (CNS), Digestive System, Head, and Imaginal Discs. Head and CNS contribute most to the list of our predictions. (C) Percentage of overlapping sites between pairs of tissues as encoded by color shading. To compute the overlap ratio, the number of common sites between pairs of tissues is divided by the smaller number of detected sites between corresponding tissues.
The next class of genomic regions with a large number of identified sites are 3′ untranslated regions (3′ UTRs). Editing of 3′ UTRs may alter gene expression by changing nucleotides in target sequences, e.g., of miRNAs. On the other hand, binding of ADAR to a target region can also prevent miRNAs and other molecules from binding.6 Indeed, we find that 165 of our editing sites overlap known miRNA target regions. Another mechanism for altering gene expression patterns is to directly edit the miRNA molecules themselves or by interfering with miRNA processing.38,43,44 We find 6 editing sites in 4 miRNA molecules: mir-4971 (1 site), mir-2a-2 (2 sites), mir-4961 (2 sites), and mir-4956 (1 site). These miRNA editing sites have the potential to influence miRNA processing and targeting.
Although our data derives from spliced transcripts, i.e. mRNAs (polyA enriched), we find 580 editing sites (20%) in genomic regions that are annotated as being intronic. The prevalence of editing in retained introns has already been reported.19 Editing in introns can happen when the editing site falls into an editing site complementary sequence (ECS) which forms a double-stranded region with a region in an adjacent exon.24 RNA editing may then lead to changes in the local RNA secondary structure which may result in the exon being retained.45 Via this molecular mechanism, RNA editing thus has the potential to alter splicing patterns by changing local RNA secondary-structure.
Our remaining sites overlap intergenic regions, 5′ UTRs and exons of non-coding genes. Sites classified as intergenic may be due to an incomplete annotation of the D. melanogaster genome. The number of editing sites in the other 2 classes is small, but may have interesting biological consequences.
We took advantage of the large number of predicted RNA editing sites to investigate the primary sequence and structural binding preferences of ADAR. In agreement with earlier studies,32 we find that a guanosine directly adjacent in the 5′ position of an adenosine decreases the chance of the adenosine being edited, see Figure S3A. Analyzing the estimated base-pairing probabilities of small regions around the predicted RNA editing sites using RNAplfold,46 we find that the 2 nucleotides directly adjacent to the site are the most important to be base-paired in ADAR target regions (Fig. S3B).
Analyzing different tissue-specific data, we find that RNA editing happens in multiple tissues of D. melanogaster, predominantly in head. We highlight the number of DNA/RNA mismatches for 4 tissues in Figure 2B. The majority of detected editing sites occur exclusively in head and central nervous system. In other tissues, RNA editing is rare (Fig. S4). Reassuringly, we find that in heavily edited tissues most of the predicted sites are A-to-G conversions that can be attributed to ADAR activity; the false positive rate of our analysis is thus low, conversely, in other tissues the estimated error rate is higher (Fig. S2).
Editing patterns differ considerably between different types of tissues. Figure 2C illustrates the relative overlap between sets of predicted sites in the 10 studied tissues. Generally, different pairs of tissues do not share most of their editing events. One obvious candidate for regulating RNA editing is the expression of the ADAR gene itself. We find that ADAR expression is highest in head and central nervous system (CNS), but that the gene is also expressed in other tissues (Fig. S5). Over-expression of ADAR in head and CNS is in agreement with the number of detected sites in these tissues, however, a higher expression of ADAR in one tissue compared to the other, does not necessarily imply a greater level of RNA editing; thus, as suggested before,34 the level of ADAR expression alone cannot explain how RNA editing levels are regulated.
Our functional enrichment analysis using DAVID47 confirms that edited genes are involved in ion transport (Benjamini Hochberg (BH) adjusted p-value: 2.10-13), gated channel activity (BH adjusted p-value: 3.10-8) and cell-cell signaling (BH adjusted p-value: 8.10-8), the well known functions of ADAR targets.24,29 Additionally, functional annotation clustering using DAVID47 identifies a cluster of genes involved in locomotory behavior (BH adjusted p-value: 2.10-3) and similar genes which is in agreement with the phenotype associated with ADAR knock-down flies.48,49
Evidence for cross-regulation of RNA editing and alternative splicing and the potential underlying regulatory mechanism
As discussed in the introduction, there already exists some evidence for an inter-relation between alternative splicing and RNA editing mechanisms. Leveraging the large number of selected tissue-specific data sets used in our study, we decided to investigate the reciprocal effect between alternative splicing and RNA editing in much greater details and to discover potential underlying regulatory mechanisms. Alternative splicing and RNA editing both play key roles in diversifying gene products and in fine tuning gene expression on RNA level. It would thus be of great conceptual importance to identify potential mechanisms of their cross-regulation.
We find that a gene with a greater number of known isoforms has a higher chance of being edited. Figure 3A illustrates the positive correlation (Rpearson = 0.33, p-value 2.10-15) between the number of annotated isoforms and the number of predicted RNA editing sites in our study. One would expect longer genes to have a higher probability of being edited and to also have more splice variants (based on the larger number of exons). In order to test if the correlation observed in our data can be explained by gene length alone, we grouped genes according to their lengths and calculated the average number of known isoforms per group, once for the sub-group of edited and once for the complementary sub-group of un-edited genes (Fig. 3B). Although we find that longer genes tend to contain more editing sites, edited genes have a significantly greater number of known isoforms than un-edited genes.
Figure 3.
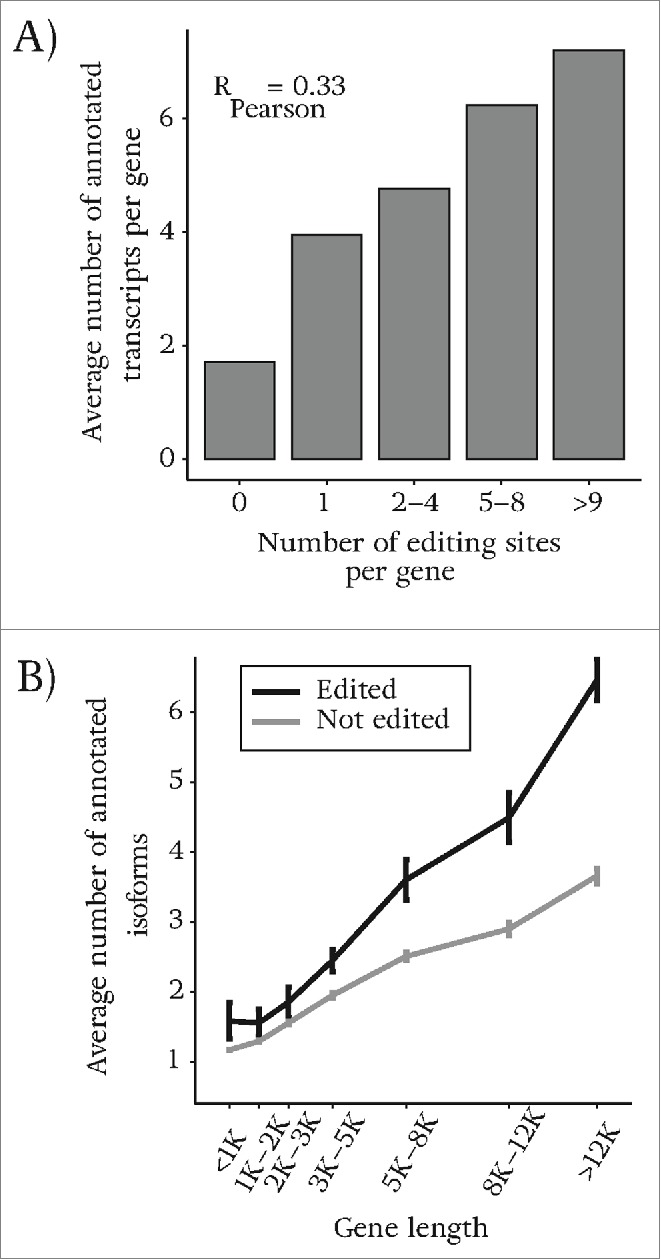
There is a positive correlation between genes that are targets of RNA editing and genes that are alternatively spliced. (A) The number of annotated isoforms vs. the number of predicted sites in our study. The number of detected sites is found to be greater in genes that express more annotated isoforms. (B) We group genes based on their length and compare the average number of annotated isoforms for genes of similar length between those that are edited and those that are un-edited genes. For genes with similar length, edited genes have a higher chance of being alternatively spliced.
Even more interestingly, we find that editing events tend to preferentially occur near exons with multiple splicing donor/acceptor sites (χ2 test, p-value 2.10-15). For this, we classify exons (including UTR exons) into 2 groups, those with multiple known acceptor and/or donor sites and those with unique acceptor and donor sites. Within each group, we count the number of RNA editing sites and normalize by the combined lengths of all exons in that group. Based on the resulting numbers, RNA editing sites are 3.2 times more likely to occur in exons with multiple splicing donor/acceptor sites compared to those with unique acceptor and donor sites (χ2 test, p-value 2.10-15, this p-value is calculated for the null hypothesis of a 1:1 ratio). To further confirm our findings that are based on our set of predicted RNA editing events, we repeated the same analysis for all sites reported by 4 existing high-throughput studies of RNA editing in D. melanogaster17-19,32 and find again that RNA editing is 1.9 times more likely to occur in exons with multiple acceptor/donor sites (χ2 test, p-value 2.10-15).
To drill down further on this finding, we then identified 244 regions where RNA editing and tissue-specific alternative splicing can have reciprocal effect (Table S2). For this, we searched for RNA editing sites in and around exons (between -150 and +150 around each exonic part) that are alternatively spliced when comparing expression for pairs of tissues using DEXSeq.50 Figure 4 shows an example of a region that is predicted to be highly edited and observed to be alternatively spliced. The figure shows that many more editing sites are predicted in the head tissue (blue arrows) compared to digestive system (red arrow). This is also true for the exonic region that is not predicted to be alternatively used (E002).
Figure 4.
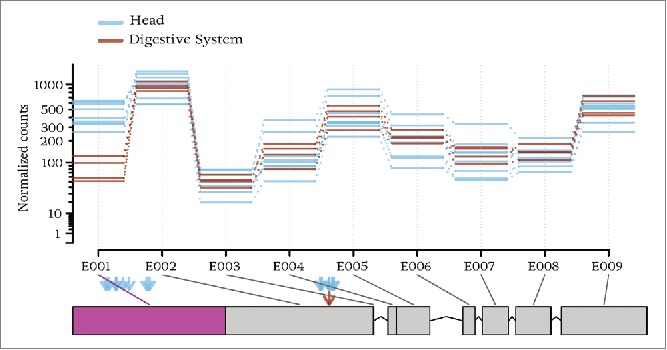
An example of a region where RNA editing and alternative splicing may affect each other. Rectangles at the bottom represent exonic parts of gene CG5850 located on the reverse strand of the left arm of chromosome 2. Exonic parts are numbered by E001, E002, …, E009, where E001 is the 3′ most exonic part and E009 is the 5′ most exonic part. The Y axis shows the number of reads aligned to each exonic bin, normalized by library size. Blue lines correspond to the number of reads from the libraries of the head tissue and red lines correspond to libraries from the digestive system tissue. The purple rectangle shows the rectangle that is predicted to be alternatively expressed between the 2 tissue types. In this region, multiple arrows are shown for identified editing sites for head (blue arrows) and digestive system (red arrows). Figure generated using DEXSeq50
One reason for the alternative splicing of the 3′ exon could be the formation of double stranded structure; or the binding of ADAR could prevent splicing machinery from detecting splicing signals and splice out the last exonic part. We should mention that the predicted editing level is low even in head tissue, and that the low editing level and random sampling may have caused the editing events not to be predicted in the digestive system samples. Dedicated follow-up experiments are required to understand how the 2 mechanisms affect each other.
To discover potential mechanisms regulating the interplay between alternative splicing and RNA editing, we also searched for statistically significant conserved RNA secondary-structure features in the vicinity of exons where we found RNA editing and alternative splicing to co-occur. For this, we employed Transat51 on input alignments of 15 fly species downloaded from UCSC52 (We also added OregonR sequence to the alignment; see supplementary text for more details) around splice sites of alternatively spliced exonic parts where editing sites are also predicted (extended by 150 nucleotides on either side, a total of 167 regions). There already exist quite a few computational methods to predict evolutionarily conserved RNA secondary-structure.53-55 These programs, however, expect the input alignment to contain one more or less global secondary-structure, i.e., a structure spanning the entire alignment. As there a priori no reason to expect secondary-structure features relevant for RNA editing to involve the entire transcript – especially not longish fly pre-mRNAs in vivo – we use Transat as this program has been specifically designed to identify local, conserved RNA secondary-structure features such as the double-stranded regions needed for ADAR binding and RNA editing. Transat takes a set of aligned sequences and an evolutionary tree as input; potential helices in the alignment, and assigns a p-value to each of these helices. For 96 of the 167 regions (57%) where alternative splicing and RNA editing co-occur in our data we find one or more conserved RNA secondary-structure features (when we filter helices with p-value greater than 0.05 and helices shorter than 8 nucleotides). Figure 5 shows an example of these regions and the corresponding, conserved RNA secondary-structure detected by RNAalifold54 in this region. Multiple compensatory mutations for conserved base-pairs provide evolutionary evidence for a likely functional role of this double-stranded region. Finally, we applied RNAalifold to assess the stability of the global structures in these regions. Table S3 presents the list of the identified regions sorted by the energy of the global structure calculated by RNAalifold.
Figure 5.
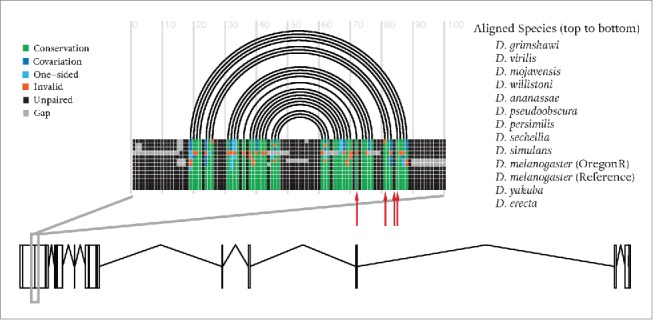
An example of a region where a conserved RNA secondary structure feature detected by investigating editing events can potentially influence alternative splicing. Rectangles at the bottom of the figure show exonic parts of Cip4 gene located on the reverse strand of the left arm of chromosome 3. The figure shows the structure predicted using RNAalifold in a region of 100 nucleotides around the splice site of an exonic region which is predicted to be alternatively used between tissues. Red arrows show predicted editing sites. Black arcs indicate alignment columns that are predicted to be base-paired, and black columns correspond to un-paired nucleotides. Green squares within the alignment show valid base-pairs and orange squares invalid base-pairs. Dark blue squares represent valid base-pairs with 2-sided mutations (compared to the most common base-pair in the pair of columns), probably in order to retain base-pairing potential. Likewise, light blue color represents single mutations to retain base-pairing potential. The existence of multiple compensatory mutations provides evidence for its functional importance throughout evolution. Figure generated using R-CHIE68
Discussion
We identify 2879 A-to-I RNA editing sites in different tissues of D. melanogaster with high precision. More than half of these have not been identified previously. The high ratio of A-to-G conversion type among the detected DNA/RNA discrepancies shows that most of our predictions are true editing events and not the result of potential experimental or computational artifacts. Also, our study suggests that other types of possible RNA editing, apart from A-to-I RNA editing, are very rare or do not happen at all in the investigated tissues of D. melanogaster.
Furthermore, our results show that editing occurs in multiple tissues, with many of the sites being edited exclusively in brain and central nervous system where ADAR expression is also higher than in other tissues. Moreover, patterns of editing differ significantly between tissues, implying a tissue-specific underlying regulatory mechanism.
Our study demonstrates how the appropriate use of ADAR specific features enhances the detection of RNA editing events when DNA reads are not available. A previous study by Ramaswami et al.18 shows that evolutionary information can be used to detect editing sites in the absence of DNA reads. Here, we explicitly capture ADAR specific features - in particular the requirement for the formation of local RNA secondary structures around target sites and clustering of editing sites - in addition to utilizing large number of selected data sets to distinguish editing events from artifacts and SNPs.
We identify more than 200 regions exist where RNA editing and alternative exon usage between tissues co-occur when comparing libraries. Many of the identified regions have been identified in multiple pair-wise comparisons of tissues. Studies showed the co-occurrence of RNA editing and alternative splicing in same genes,32,35 similar to what we find in this analysis. Solomon et al. reported the enrichment of editing events in cassette exons in human, although they reported most of the sites are far from exon boundaries.35 We here show that editing events tend to happen much more abundantly in exons with multiple known acceptor or donor sites, or 3′ and 5′ UTRs that contain alternative splicing potential. Further, we find 96 regions around splice sites with significant statistical evidence for the overlap of evolutionarily conserved, local RNA secondary-structures. The actual formation of these RNA structure features in vivo is supported by both computational RNA secondary-structure prediction programs and predicted RNA editing sites.
RNA editing thus has the potential to regulate alternative splicing via changes of local RNA secondary structures. This suggests a potential, tissue-specific molecular mechanism of regulation for alternative splicing whose potential mediation via changes of local RNA structure we showed earlier.45
Overall, we find strong evidence for our hypothesis that RNA editing and alternative splicing mechanisms directly influence each other in specific regions of the transcriptome. Both, RNA editing and alternative splicing are abundant in the CNS and are both known to be temporally and spatially regulated.37 Also, target genes of the 2 mechanisms correlate well. These mechanisms may influence each other in several ways. First, the splicing machinery may compete with ADAR for common substrates. This is plausible given that RNA editing and splicing can happen at the same time co-transcriptionally in D. melanogaster.19,56 Targeting of a specific location by one machinery limits the access of the other machinery and can thereby affect its functionality. Second, considering the potential importance of RNA secondary structures in regulating alternative splicing, ADAR may edit and thereby alter local secondary structures which can in turn change exon usage. Blow et al.20 showed earlier that RNA editing of double-stranded regions has the overall effect of destabilising these features. Finally, editing of splicing silencers and enhancers or splice site motifs could additionally affect splicing. Our results suggest that the altering secondary structure type of local co-regulation happens predominantly within exons with multiple acceptor or donor sites. In these regions, splicing signals may be weak, and these weak signals may prevent the splicing machinery from always making the same decision.
The formation and RNA editing-mediated modification of local RNA secondary structures therefore has the potential to significantly alter splicing patterns in these genes as local RNA structure features can be “encoded” in a transcript-specific way. In fact, the necessity for encoding RNA structure features that are involved in regulating the alternative splicing of their own transcript may explain why introns tend to be longer in more complex organisms: these RNA structure features are (at least partly) encoded in introns thus imposing no undue additional evolutionary constraints on the protein-coding exons.
Previous studies suggest that the dominant way in which editing regulates splicing is by editing RNA-binding proteins.35 This would, however, imply a more indirect and global way of regulating alternative splicing and could not easily happen in a gene-specific way. Our results support a gene-specific mechanism where alternative splicing can be directly regulated via tissue-specific changes of RNA editing. Detailed follow-up experiments, e.g. ADAR knockdowns and mutational studies of specific genes, are now required to experimentally confirm our results.
Materials and Methods
Data set
To study tissue-specific RNA editing events, we selected tissue-specific RNA-seq libraries of Drosophila melanogaster from the modENCODE project.40,41 These libraries correspond to paired-end, strand-specific RNA-seq reads of 74–120 nucleotides length. The strand-specificity of the reads allows us to assess the correct conversion types in overlapping or incompletely annotated parts of the genome,16 whereas the paired-ends improve the alignment of reads to repeat-rich regions of the genome which would otherwise easily result in incorrectly aligned reads or the false positive prediction of SNPs or RNA editing sites. The 29 selected libraries are classified into 10 tissues (Fig. S1). Some of these libraries are extracted from multiple tissues. For each library there exist 2 to 5 technical replicates. All libraries derive from the OregonR strain of D. melanogaster which is, however, not the strain of the D. melanogaster reference genome.
Since we do not have genomic DNA sequencing reads in our data, it is essential to align the short transcriptome reads to the reference genome of the OregonR strain when searching for DNA/RNA discrepancies; otherwise, genomic differences between the genome of the OregonR strain and the D. melanogaster's reference genome could be misinterpreted as RNA editing events. We therefore generate an annotation for the OregonR genome by aligning the genome of the OregonR strain to the D. melanogaster's reference genome. We first use Mummer57,58 to find a set of consecutive matches of at least 20 nucleotides long. Next, we align the remaining parts between these matches using the Needleman-Wunsch algorithm59 with default parameter values. Finally, we convert the coordinates of the reference annotation of D. melanogaster in Ensembl60 to the corresponding coordinates of the resulting OregonR genome.
Prediction pipeline
Figure 6 gives an overview of the steps of our computational analysis pipeline for identifying RNA editing events using multiple RNA-seq libraries and the reference genome as input. Considering the potential challenges in reliably detecting RNA editing events,42 we designed a probabilistic pipeline to achieve the following in an efficient manner: (1) filter variants against artifacts due to mapping and sequencing errors; (2) explicitly capture ADAR-specific features such as the requirement for double-stranded region to distinguish RNA editing events from other types of observed variants; and (3) leverage the statistical power derived from the size and number of our input data sets. In the following, we briefly explain the steps of our pipeline.
Figure 6.
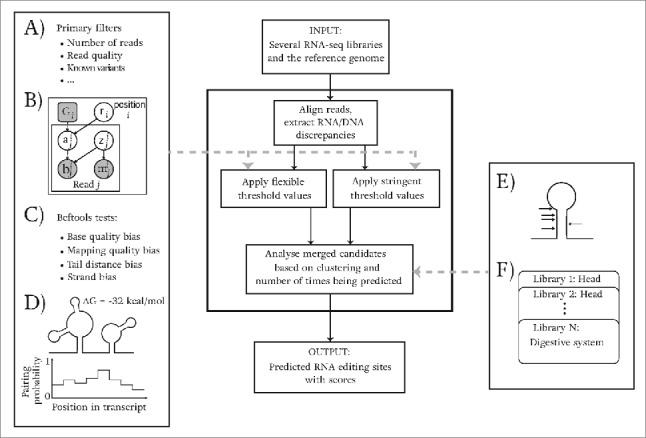
Outline of the computational analysis pipeline for identifying editing events from multiple RNA-seq libraries. The input consists of several RNA-seq libraries and the reference genome. As shown, first, reads are aligned and RNA/DNA mismatches are extracted. Then, 2 sets of values (for flexible and stringent filtering) are used for several filters ((A) - (D)) to remove potential experimental artifacts. Finally, our pipeline considers clustering of identified candidates and the number of times they are detected in multiple libraries to output a final set of predicted editing events. (A) - (D) show the statistical tests and filters used in our pipeline. (A) A set of primary filters used to assess the initial requirements for candidate sites. (B) The statistical graphical model (modified from62) that we use to find the maximum editing ratio, and to compute a log likelihood ratio score. Shaded circles are the random variables that are observed in data and unshaded circles are the ones that are inferred. The rounded square is fixed to represent the reference genotype. is a binary variable which indicates whether or not a read aligned to a position comes from an edited molecule. z is also a binary variable that indicates whether the read is aligned correctly. The editing ratio of position i is presented with node r; and nodes m and b present mapping and base qualities. (C) Statistical tests in samtools/bcftools63 to check the potential biases in reads. (D) The energy of local structures and base pairing probabilities of nucleotides in close vicinity of candidate sites are used to ensure the structural requirements of candidates are met. (E) We use the fact that editing events occur in clusters to improve our predictions. (F) For less confident sites, the site requires to be detected in multiple libraries in order to be reported in our final set.
We use TopHat261 to align short reads to the genome in a splice-aware manner. We allow up to 5 mismatches in the alignment step to permit TopHat2 to successfully align reads that have been RNA-edited multiple times. Next, we employ Picard-tools (http://broadinstitute.github.io/picard) to remove duplicates from each technical replicate. These duplicate reads may be generated during RT-PCR as a result of amplification bias.12,13 Finally, technical replicates are merged and positions showing DNA/RNA discrepancies are extracted for further analysis.
Our analysis pipeline combines a set of statistical and deterministic filters that apply 2 sets of threshold values, one set called the flexible set and one set called the stringent set (Fig. 6). By employing these 2 sets of threshold values and leveraging the large size of the input data, it is possible to simultaneously lower the false positive and the false negative error rates. If only the stringent threshold values were used to distinguish genuine RNA editing sites from mapping and sequencing errors, filtering the potential artifacts could result in discarding many true RNA editing events, i.e. a high false negative error rate. On the other hand, using only the set of flexible threshold values could lead to an increased false positive error rate. To overcome this issue, our pipeline combines the 2 sets of threshold values. The potential editing sites that pass the flexible threshold values are only reported in the final output if they are detected in multiple samples and are close to other predicted sites.
After the alignment step, a set of primary filters are applied to reduce the identified DNA/RNA discrepancies and remove those that are likely to be due to mapping and sequencing errors (Fig. 6A). These primary filters examine, for example, the number of reads covering a candidate RNA editing site, the read and mapping qualities of the input data, and also the distance to both ends of the read. In addition, any known variants listed in the Ensembl fly variant files are removed. Some of these variants may correspond to genuine RNA editing events – similar to what has been observed in human SNP data bases26 – yet we decided to be conservative and to remove all known variants in the absence of any corresponding DNA sequencing reads.
Figure 6B shows the graphical model we use to compute the maximum likelihood editing ratio, and to apply a log-likelihood ratio test. The model is a modification of the model introduced in SNVMix2.62 The original model considers both mapping and base qualities of the reads and takes uncertainties of bases and alignments into account. We took a part of the model and added a new node (shown with “ri” in the figure) that presents the editing ratio (r). This can take values ranging from 0 (not edited) to 1 (always edited) with uniform prior. We model aij (which indicates wether read j aligned to position i comes from an edited RNA molecule) to have a Bernoulli distribution with its parameter set to the editing ratio r. The conditional probability distribution for the other 3 nodes (“z”, “b” and “m”) are the original ones used in SNVMix2.62 Using this statistical model, the null hypothesis of a position having an RNA editing level of zero is compared to the hypothesis of the position being edited with the inferred maximum likelihood level of editing. More precisely, for each candidate position i, we compute the following log-likelihood ratio score:
where ri is the editing ratio; Di presents the observed reads overlapping position i; Bi and are the base and the mapping qualities of reads, respectively, overlapping position i.
In the following step, the pipeline applies samtools/bcftools63 tests to identify and remove positions that are discovered as a result of potential biases (Fig. 6C). These tests have been used in the literature to improve the quality of variant calls.16 Base quality and mapping quality tests gauge the bias of the corresponding scores between the reads showing the reference allele and the reads showing the variant allele. Two additional other tests evaluate the strand bias and the tail distance bias. The strand bias gauges the bias between the distribution of the strand of reference reads and the distribution of the strand of non-reference reads. The tail distance bias investigates whether nucleotide reads from one allele tend to occur closer to read ends compared to nucleotide reads from the other allele.
Unlike most other existing prediction methods for RNA editing sites, our analysis pipeline explicitly utilises the requirement for the existence of double-stranded regions in potential ADAR target regions64 to further improve our predictions (Fig. 6D). Long double-stranded regions constitute perfect potential target sites for ADARs,7 and structured regions also recruit ADARs to nearby sites that are not in the same double-stranded region.65 Consequently, the stability of potential structures has been used to rank output candidates,22 although edited double-stranded region have been observed to have a wide range of stabilities.27 Also, the vicinity of complementary nucleotide regions which allows the formation of RNA secondary-structures was used to improve prediction results.23 Most of the double-stranded regions bound by ADAR have been shown to correspond to intramolecular interactions, i.e., RNA structure features in the same transcript.20 We therefore use local RNA secondary-structure prediction algorithms in our pipeline. We employ RNAfold66 on a sequence interval of 200 nucleotides length around each candidate editing site to calculate the minimum-free-energy (MFE) RNA structure predicted for this region. We use the corresponding minimum free energy as an indicator of the stability of all potential local RNA structures that can be formed in that region. Additionally, as ADAR binding and editing predominantly happens in double-stranded regions,7 we use RNAplfold46 to estimate the probability of a potential RNA editing site being in a double-stranded region. For this we examine sequence intervals of 5 nucleotides length around the candidate editing site. Finally, the 2 sets of thresholds (stringent and flexible) introduced above are applied to these potential RNA editing sites in order to incorporate structural information in our pipeline.
It is well known that ADAR tends to edit several sites in the same double-stranded region upon binding27 which we explicitly judge by our analysis pipeline (Fig. 6E). In addition, we expect true RNA editing events to show up in several libraries due to the large amount of input reads. To use these features, we first include all candidate editing sites that pass the stringent threshold values. Any remaining candidate sites are then added if: a) The same position passes the stringent threshold values in another sample, or b) the position has been predicted (passes the flexible threshold values) at least twice and there is another identified site showing the same conversion type within a distance of 25 nucleotides.
To summarise, by using a large number of samples as input, by explicitly capturing ADAR specific features and requirements and by combining 2 distinct sets of threshold values, we create an analysis pipeline that has a low false positive as well as a high true positive rate (see results section). Assuming similar mutation rates for transitions and similar mutation rates for transversions, we can use the ratio of A-to-G conversions in our predictions to estimate the false positive ration.16,18 Based on this estimate, we chose a set of pipeline parameters that result in a decent overall number of predictions and also a high ratio of A-to-G conversion type. Details of the pipeline including parameter values are explained in supplementary text.
Finding alternatively spliced exons
To find alternatively expressed exons between pairs of tissues, we use DEXSeq.50 DEXSeq applies a generalized linear model to detect exonic regions that are differentially expressed between 2 conditions. We consider libraries from the same tissue as replicates as required by DEXSeq. Furthermore, we only consider genes that show an expression higher than a pre-defined threshold in both conditions in our analysis (by applying a threshold on expression predicted by Cufflinks67). Additionally, we discard genes for which many of the exonic parts are predicted to be alternatively used, keeping only those genes for which the number of alternatively used exons is smaller than max (2,(1/4) number of exons. The main reason doing so is to focus our analysis of the potential interplay between alternative splicing and RNA editing on genes that are more likely to be regulated locally.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplemental data for this article can be accessed on the publisher's website.
Acknowledgments
We thank Brenton Graveley for sharing the sequence of the OregonR fly genome with us.
Funding
This work was supported by Natural Sciences and Engineering Research Council (NSERC) of Canada and Canada Foundation for Innovation (CFI) [to I.M.M.], and UBC Four Year Doctoral Fellowship [to A.M.]. Funding for open access charge: NSERC.
References
- 1.Baltimore D. Our genome unveiled. Nature 2001; 409:814-6; PMID:11236992; http://dx.doi.org/ 10.1038/35057267 [DOI] [PubMed] [Google Scholar]
- 2.Crick F. Central dogma of molecular biology. Nature 1970; 227:561-3; PMID:4913914; http://dx.doi.org/ 10.1038/227561a0 [DOI] [PubMed] [Google Scholar]
- 3.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. TRENDS Genet 2001; 17:100-7; PMID:11173120; http://dx.doi.org/ 10.1016/S0168-9525(00)02176-4 [DOI] [PubMed] [Google Scholar]
- 4.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008; 40:1413-5; PMID:18978789; http://dx.doi.org/ 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- 5.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2004; 2:e391; PMID:15534692; http://dx.doi.org/ 10.1371/journal.pbio.0020391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng Z, Cheng Y, Tan BC-M, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, et al.. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 2012; 30:253-60; PMID:22327324; http://dx.doi.org/ 10.1038/nbt.2122 [DOI] [PubMed] [Google Scholar]
- 7.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 2010; 79:321-49; PMID:20192758; http://dx.doi.org/ 10.1146/annurev-biochem-060208-105251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benne R, Van Den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, Tromp MC. Major transcript of the frameshifted coxll gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 1986; 46:819-26; PMID:3019552; http://dx.doi.org/ 10.1016/0092-8674(86)90063-2 [DOI] [PubMed] [Google Scholar]
- 9.Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al.. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 2014; 24:365-76; PMID:24347612; http://dx.doi.org/ 10.1101/gr.164749.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li JB, Levanon EY, Yoon J-K, Aach J, Xie B, LeProust E, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 2009; 324:1210-3; PMID:19478186; http://dx.doi.org/ 10.1126/science.1170995 [DOI] [PubMed] [Google Scholar]
- 11.Park E, Williams B, Wold BJ, Mortazavi A. RNA editing in the human ENCODE RNA-seq data. Genome Res 2012; 22:1626-33; PMID:22955975; http://dx.doi.org/ 10.1101/gr.134957.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, Li JB. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods 2012; 9:579-81; PMID:22484847; http://dx.doi.org/ 10.1038/nmeth.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahn JH, Lee J-H, Li G, Greer C, Peng G, Xiao X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 2012; 22:142-50; PMID:21960545; http://dx.doi.org/ 10.1101/gr.124107.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekdahl Y, Farahani HS, Behm M, Lagergren J, Öhman M. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res 2012; 22:1477-87; PMID:22645261; http://dx.doi.org/ 10.1101/gr.131912.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu T, Buaas FW, Simons AK, Ackert-Bicknell CL, Braun RE, Hibbs MA. Canonical A-to-I and C-to-U RNA editing is enriched at 3′ UTRs and microRNA target sites in multiple mouse tissues. PloS ONE 2012; 7:e33720; PMID:22448268; http://dx.doi.org/ 10.1371/journal.pone.0033720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danecek P, Nellaker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol 2012; 13:26; PMID:22524474; http://dx.doi.org/ 10.1186/gb-2012-13-4-r26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al.. The developmental transcriptome of Drosophila melanogaster. Nature 2011; 471:473-9; PMID:21179090; http://dx.doi.org/ 10.1038/nature09715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. Identifying RNA editing sites using RNA sequencing data alone. Nature Methods 2013; 10:128-32; PMID:23291724; http://dx.doi.org/ 10.1038/nmeth.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez J, Menet JS, Rosbash M. Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell 2012; 47:27-37; PMID:22658416; http://dx.doi.org/ 10.1016/j.molcel.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blow M, Futreal PA, Wooster R, Stratton MR. A survey of RNA editing in human brain. Genome Res 2004; 14:2379-87; PMID:15545495; http://dx.doi.org/ 10.1101/gr.2951204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 2002; 71:817-46; PMID:12045112; http://dx.doi.org/ 10.1146/annurev.biochem.71.110601.135501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maas S, Sie CG, Stoev I, Dupuis D, Latona J, Porman A, Evans B, Rekawek P, Kluempers V, Mutter M, et al.. Genome-wide evaluation and discovery of vertebrate A-to-I RNA editing sites. Biochem Biophys Res Commun 2011; 412:407-12; PMID:21835166; http://dx.doi.org/ 10.1016/j.bbrc.2011.07.075 [DOI] [PubMed] [Google Scholar]
- 23.Neeman Y, Levanon EY, Jantsch MF, Eisenberg E. RNA editing level in the mouse is determined by the genomic repeat repertoire. RNA 2006; 12:1802-9; PMID:16940548; http://dx.doi.org/ 10.1261/rna.165106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoopengardner B, Bhalla T, Staber C, Reenan R. Nervous system targets of RNA editing identified by comparative genomics. Science 2003; 301:832-6; PMID:12907802; http://dx.doi.org/ 10.1126/science.1086763 [DOI] [PubMed] [Google Scholar]
- 25.Rieder LE, Reenan RA. The intricate relationship between RNA structure, editing, and splicing. Semin Cell Dev Biol 2012:281-8; http://dx.doi.org/ 10.1016/j.semcdb.2011.11.004 [DOI] [PubMed] [Google Scholar]
- 26.Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, Rechavi G, Jantsch MF, Eisenberg E. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res 2005; 33:1162-8; PMID:15731336; http://dx.doi.org/ 10.1093/nar/gki239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morse DP, Aruscavage PJ, Bass BL. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc Natl Acad Sci 2002; 99:7906-11; http://dx.doi.org/ 10.1073/pnas.112704299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scadden A. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol 2005; 12:489-96; PMID:15895094; http://dx.doi.org/ 10.1038/nsmb936 [DOI] [PubMed] [Google Scholar]
- 29.Hood JL, Emeson RB. Editing of Neurotransmitter Receptor and Ion Channel RNAs in the Nervous System. Curr Top Microbiol Immunol 2012; 353:61-90; PMID:21796513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He T, Wang Q, Feng G, Hu Y, Wang L, Wang Y. Computational detection and functional analysis of human tissue-specific A-to-I RNA editing. PloS ONE 2011; 6:e18129; PMID:21448465; http://dx.doi.org/ 10.1371/journal.pone.0018129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2011; 2:319; PMID:21587236; http://dx.doi.org/ 10.1038/ncomms1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. St Laurent G, Tackett MR, Nechkin S, Shtokalo D, Antonets D, Savva YA, Maloney R, Kapranov P, Lawrence CE, Reenan RA. Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nat Struct Mol Biol 2013; 20:1333-9; PMID:24077224; http://dx.doi.org/ 10.1038/nsmb.2675 [DOI] [PubMed] [Google Scholar]
- 33.Tariq A, Garncarz W, Handl C, Balik A, Pusch O, Jantsch MF. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res 2013; 41:2581-93; PMID:23275536; http://dx.doi.org/ 10.1093/nar/gks1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wahlstedt H, Daniel C, Ensterö M, Öhman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 2009; 19:978-86; PMID:19420382; http://dx.doi.org/ 10.1101/gr.089409.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solomon O, Oren S, Safran M, Deshet-Unger N, Akiva P, Jacob-Hirsch J, Cesarkas K, Kabesa R, Amariglio N, Unger R, et al.. Global regulation of alternative splicing by adenosine deaminase acting on RNA (ADAR). RNA 2013; 19:591-604; PMID:23474544; http://dx.doi.org/ 10.1261/rna.038042.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson TR, Sansam CL, Emeson RB. Structure and sequence determinants required for the RNA editing of ADAR2 substrates. J Biol Chem 2004; 279:4941-51; PMID:14660658; http://dx.doi.org/ 10.1074/jbc.M310068200 [DOI] [PubMed] [Google Scholar]
- 37.Paro S, Li X, O'Connell M, Keegan L. Regulation and functions of ADAR in drosophila. Curr Top Microbiol Immunol 2011; 353:221-36. [DOI] [PubMed] [Google Scholar]
- 38.de Hoon MJ, Taft RJ, Hashimoto T, Kanamori-Katayama M, Kawaji H, Kawano M, Kishima M, Lassmann T, Faulkner GJ, Mattick JS, et al.. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res 2010; 20:257-64; PMID:20051556; http://dx.doi.org/ 10.1101/gr.095273.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barraud P, Allain F. ADAR Proteins: Double-stranded RNA and Z-DNA Binding Domains. Curr Top Microbiol Immunol 2012; 353:35-60; PMID:21728134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Celniker SE, Dillon LA, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al.. Unlocking the secrets of the genome. Nature 2009; 459:927-30; PMID:19536255; http://dx.doi.org/ 10.1038/459927a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, et al.. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 2010; 330:1787-97; PMID:21177974; http://dx.doi.org/ 10.1126/science.1198374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pachter L. A closer look at RNA editing. Nat Biotechnol 2012; 30:246-7; PMID:22398619; http://dx.doi.org/ 10.1038/nbt.2156 [DOI] [PubMed] [Google Scholar]
- 43.Chawla G, Sokol NS. ADAR mediates differential expression of polycistronic microRNAs. Nucleic Acids Res 2014; 42:5245-55; PMID:24561617; http://dx.doi.org/ 10.1093/nar/gku145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vesely C, Tauber S, Sedlazeck FJ, von Haeseler A, Jantsch MF. Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res 2012; 22:1468-76; PMID:22310477; http://dx.doi.org/ 10.1101/gr.133025.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer IM, Miklós I. Statistical evidence for conserved, local secondary structure in the coding regions of eukaryotic mRNAs and pre-mRNAs. Nucleic Acids Res 2005; 33:6338-48; PMID:16275783; http://dx.doi.org/ 10.1093/nar/gki923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernhart SH, Hofacker IL, Stadler PF. Local RNA base pairing probabilities in large sequences. Bioinformatics 2006; 22:614-5; PMID:16368769; http://dx.doi.org/ 10.1093/bioinformatics/btk014 [DOI] [PubMed] [Google Scholar]
- 47.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2008; 4:44-57; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 48.Palladino MJ, Keegan LP, O'connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 2000; 102:437-49; PMID:10966106; http://dx.doi.org/ 10.1016/S0092-8674(00)00049-0 [DOI] [PubMed] [Google Scholar]
- 49.Jepson JE, Savva YA, Yokose C, Sugden AU, Sahin A, Reenan RA. Engineered Alterations in RNA Editing Modulate Complex Behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J Biol Chem 2011; 286:8325-37; PMID:21078670; http://dx.doi.org/ 10.1074/jbc.M110.186817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res 2012; 22:2008-17; PMID:22722343; http://dx.doi.org/ 10.1101/gr.133744.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiebe NJ, Meyer IM. Transat—a method for detecting the conserved helices of functional RNA structures, including transient, pseudo-knotted and alternative structures. PLoS Comput Biol 2010; 6:e1000823; PMID:20589081; http://dx.doi.org/ 10.1371/journal.pcbi.1000823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al.. The UCSC genome browser database: update 2011. Nucleic Acids Res 2011; 39:D876-D82; PMID:20959295; http://dx.doi.org/ 10.1093/nar/gkq963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knudsen B, Hein J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res 2003; 31:3423-8; PMID:12824339; http://dx.doi.org/ 10.1093/nar/gkg614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 2008; 9:474; PMID:19014431; http://dx.doi.org/ 10.1186/1471-2105-9-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer IM, Miklós I. SimulFold: simultaneously inferring RNA structures including pseudoknots, alignments, and trees using a Bayesian MCMC framework. PLoS Computat Biol 2007; 3:1441-54; http://dx.doi.org/ 10.1371/journal.pcbi.0030149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khodor YL, Rodriguez J, Abruzzi KC, Tang C-HA, Marr MT, Rosbash M. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev 2011; 25:2502-12; PMID:22156210; http://dx.doi.org/ 10.1101/gad.178962.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 2002; 30:2478-83; PMID:12034836; http://dx.doi.org/ 10.1093/nar/30.11.2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL. Alignment of whole genomes. Nucleic Acids Res 1999; 27:2369-76; PMID:10325427; http://dx.doi.org/ 10.1093/nar/27.11.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 1970; 48:443-53; PMID:5420325; http://dx.doi.org/ 10.1016/0022-2836(70)90057-4 [DOI] [PubMed] [Google Scholar]
- 60.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al.. Ensembl 2014. Nucleic Acids Res 2014; 42:D749-D55; PMID:24316576; http://dx.doi.org/ 10.1093/nar/gkt1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14:R36; PMID:23618408; http://dx.doi.org/ 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goya R, Sun MG, Morin RD, Leung G, Ha G, Wiegand KC, Senz J, Crisan A, Marra MA, Hirst M, et al.. SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics 2010; 26:730-6; PMID:20130035; http://dx.doi.org/ 10.1093/bioinformatics/btq040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011; 27:2987-93; PMID:21903627; http://dx.doi.org/ 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 1993; 75:1361-70; PMID:8269514; http://dx.doi.org/ 10.1016/0092-8674(93)90622-W [DOI] [PubMed] [Google Scholar]
- 65.Daniel C, Venø MT, Ekdahl Y, Kjems J, Öhman M. A distant cis acting intronic element induces site-selective RNA editing. Nucleic Acids Res 2012; 40:9876-86; PMID:22848101; http://dx.doi.org/ 10.1093/nar/gks691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981; 9:133-48; PMID:6163133; http://dx.doi.org/ 10.1093/nar/9.1.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7:562-78; PMID:22383036; http://dx.doi.org/ 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lai D, Proctor JR, Zhu JYA, Meyer IM. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res 2012; 40:e95; PMID:22434875; http://dx.doi.org/ 10.1093/nar/gks241 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.