

Abstract
Riboswitches regulate gene expression by rearranging their structure upon metabolite binding. The lysine-sensing lysC riboswitch is a rare example of an RNA aptamer organized around a 5-way helical junction in which ligand binding is performed exclusively through nucleotides located at the junction core. We have probed whether the nucleotides involved in ligand binding play any role in the global folding of the riboswitch. As predicted, our findings indicate that ligand-binding residues are critical for the lysine-dependent gene regulation mechanism. We also find that these residues are not important for the establishment of key magnesium-dependent tertiary interactions, suggesting that folding and ligand recognition are uncoupled in this riboswitch for the formation of specific interactions. However, FRET assays show that lysine binding results in an additional conformational change, indicating that lysine binding may also participate in a specific folding transition. Thus, in contrast to helical junctions being primary determinants in ribozymes and rRNA folding, we speculate that the helical junction of the lysine-sensing lysC riboswitch is not employed as structural a scaffold to direct global folding, but rather has a different role in establishing RNA-ligand interactions required for riboswitch regulation. Our work suggests that helical junctions may adopt different functions such as the coordination of global architecture or the formation of specific ligand binding site.
Keywords: helical junction, lysine, metabolite-sensing, Riboswitch, RNA folding
Introduction
RNA-mediated molecular processes are involved at multiple levels of genetic expression and are found to control complex biochemical pathways. Among recently identified RNA-based regulators, riboswitches are genetic elements controlling gene expression by sensing cellular metabolites. Riboswitches have been shown to control gene expression at various levels and are mostly involved in controlling genes implicated in the biosynthesis or transport of sensed metabolites.1-4 These regulatory RNA consist of 2 domains, the aptamer that is involved in cellular metabolites recognition and the expression platform that performs the regulation of gene expression. In most cases, ligand binding to the aptamer stabilizes P1 formation, which is directly involved in the modulation of the expression platform structure.1 Riboswitch aptamer domains exhibit a large diversity of structural organizations that are presumably important for ligand binding specificity.1-4 The vast majority of riboswitch crystal structures show that aptamer global architectures are organized around helical junctions.5 These highly structured elements are used to coordinate multiple helices to restrict the folding space that RNA molecules must sample to attain biologically relevant active structures. A well-known example is the conserved 4-way helical junction in U1 snRNA that was reported to be important for mRNA splicing.6 Other examples are found in the ribosome, where crystallized subunits revealed an abundance of helical junctions involved in the overall RNA architecture.7 Ribosome crystal structures also show that helical junctions can act as target sites for protein binding, as observed for S15 that was reported to induce junction folding.8-10 Moreover, it was recently shown for several small nucleolytic ribozymes that RNA folding strongly relies on helical junctions to attain their native state.11,12 It was found that mutations perturbing junction folding also disrupt ribozyme activity, consistent with helical junctions being primary determinants for ribozyme folding and catalysis.
Although much less is known about the role of helical junctions in the folding of riboswitches, the ever-expanding repertoire of riboswitch crystal structures enables to deduce structural principles in their architectures.1,13 For instance, a recent structural classification reported that several riboswitches, such as those responding to purine14,15 and lysine,16,17 employ helical junctions to create ligand-binding sites as a way to stabilize the P1 stem upon ligand binding.13 This is in contrast to the thiamine pyrophosphate (TPP),18-20 adenosylcobalamin (AdoCbl),21,22 glycine23 and tetrahydrofolate (THF)24,25 riboswitches, which rely on other structural features such as kissing-loops or internal bulges for ligand sensing. In the case of purine and lysine riboswitches that employ junction core residues to sense ligand, there is little evidence indicating whether these riboswitches use core residues strictly for RNA-ligand interactions or for directing riboswitch folding pathway. In this context, since ligand-binding residues are conserved to specifically interact with cellular metabolites, their additional involvement in global RNA folding would result in additional evolutionary pressure.
The Bacillus subtilis lysC riboswitch is a rare example of an RNA motif organized around a single 5-way junction.26,27 This riboswitch regulates the intracellular level of lysine by controlling the expression of an aspartokinase II.26-28 It was previously shown that ligand binding to the aptamer favors the stabilization of the P1 stem (anti-antiterminator), which in turn promotes terminator stem formation (Fig. 1A).26,27 The 5-way junction of the aptamer domain coordinates the formation of long-range tertiary interactions occurring between loops L2-L3 (L2-L3) and also helix P2 and loop L4 (P2-L4) (Fig. 1A).16,17,29 It was recently shown that interactions L2-L3 and P2-L4 are important for adoption of the riboswitch global structure, where a synergy between these 2 interactions improves their folding efficiency,29 ultimately resulting in a lower Mg2+ requirement for adoption of the native state. Crystal structures have revealed that the lysine aptamer exhibits 2 helical stacking units occurring between stems P1 and P2, and between stems P4 and P5 (Fig. 1B). The lysine-binding site is located in the junction core, where several nucleotides are involved in the formation of the ligand-binding pocket. The binding site comprises nucleotides located in stems P1, P2 and P4, and conserved base pairs G40-C110 and G144•U170, which are specifically involved in recognizing the carboxylate group of the bound lysine (Fig. 1C).26-28 An important binding site feature is the presence of a potassium cation (Fig. 1C) that is mediating the interaction between RNA and a carboxyl oxygen of lysine (Fig. 1C).16 The conserved residue G111 is also involved in lysine recognition by interacting with the lysine ε-amino group (Fig. 1C). Together, these interactions allow recognition of amino acid backbone and side chain, thus ensuring the specificity of ligand binding. However, given that the junction core domain is highly integrated into the lysine binding site, it is not clear how residues involved in ligand binding contribute to the global folding of the lysC aptamer.
Figure 1.
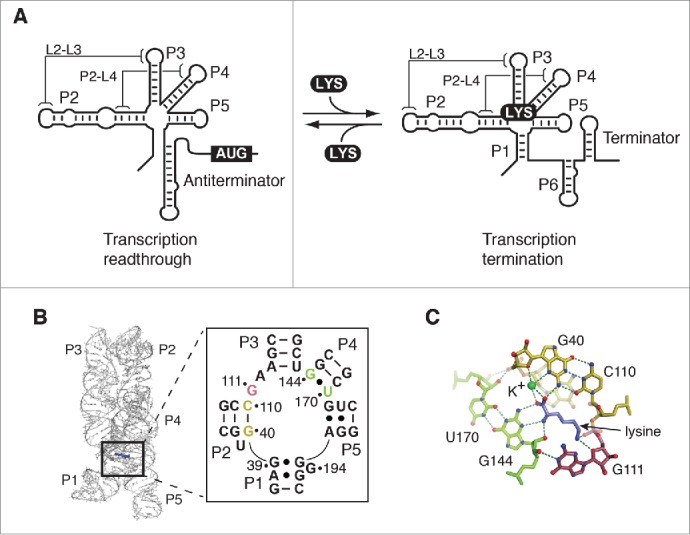
Structure and function of the lysine riboswitch. (A) Schematic representation of the lysine riboswitch regulation mechanism. In absence of lysine, the lysine lysC riboswitch adopts a conformation in which transcription readthrough is allowed. In this configuration, the P1 stem is presumably not formed in the aptamer domain. However, upon lysine (LYS) binding, the P1 stem is stabilized, which in turn allow the formation of a transcription terminator. Tertiary interactions involving L2-L3 and P2-L4 are shown. (B) Structure of the lysine riboswitch aptamer domain. The crystal structure of the lysine aptamer bound to lysine is shown.16,17 Lysine is represented in violet and selected nucleotides involved in the lysine-binding site are shown. The insert shows base pair G40-C110 and the wobble pair G144•U170 in yellow and green, respectively. G111 is represented in magenta. The nucleotide nomenclature is based on previous studies.26,27 (C) Representation of the lysine-binding site. The base pair G40-C110 and the wobble pair G144•U170 are shown in yellow and green, respectively, and G111 is represented in magenta. While pairs G40-C110 and G144•U170 interact with the carboxyl end of lysine, G111 is involved in the recognition of the ε-ammonium group. Lysine is shown in violet. A potassium ion (K+) coordinating lysine is shown in green. Hydrogen bond interactions are shown as dotted lines. Nitrogen and oxygen atoms are shown in blue and red, respectively.
Here, we employ a combination of biophysical and biochemical assays to characterize the role of ligand binding residues in the aptamer folding pathway and riboswitch gene regulation. Since it was previously shown that long-range peripheral interactions are important to pre-fold the aptamer global structure into a ligand-binding competent conformation,29 we sought to establish whether junction core residues are involved in aptamer global folding and/or in lysine recognition. Our results show that mutating base pairs G40-C110 or G144•U170 disrupts riboswitch activity, consistent with the strict requirement of both base pairs for genetic control. Similarly, we find that the junction core residue G111 is important for riboswitch transcriptional control, in agreement with its role in ligand binding recognition.16,17 In contrast, we show using FRET and SHAPE assays that mutating these residues does not perturb the adoption of key Mg2+-dependent riboswitch tertiary interactions, suggesting that such junction core residues are not the primary determinants directing the formation of the riboswitch global architecture. However, FRET assays show that ligand binding results in a specific conformational change involving stems P1 and P5, suggesting that lysine may also participate in aptamer folding. Taken together, in contrast to findings obtained in the context of ribozymes and rRNA junctions, our work suggests that the lysC riboswitch junctions is not used as a structural scaffold in RNA global folding, but rather has a different role in providing key interactions to perform metabolite sensing. This most likely results from the fact that the core domain of the lysC riboswitch has evolved to favor high ligand affinity and discrimination, ensuring a high degree of specific genetic regulation.
Results
The lysine riboswitch relies on the junction core domain to control transcription termination
We have previously shown that ligand binding and riboswitch regulation of B. subtilis lysC riboswitch strongly rely on formation of long-range tertiary interactions such as the L2-L3 and P2-L4 interactions.29 We employed mutagenesis to explore the importance of lysC junction core residues for riboswitch activity. We used single-round in vitro transcriptions with a DNA template consisting of the B. subtilis glyQS promoter fused to the lysC lysine riboswitch containing a 95-nt terminator downstream sequence.29,30 We first performed transcription reactions using the wild-type riboswitch as a function of lysine concentration. In presence of low lysine concentrations, a transcription termination ratio of ∼0.32 was observed (Fig. 2A). However, when transcription reactions were done using higher lysine concentrations, the transcription termination efficiency increased and reached a plateau of ∼0.68 (Fig. 2A), consistent with previous studies.26,27,30,31 Fitting our data to a 2-state model yielded a T50 value of 3.8 ± 0.7 mM, which is similar to our previously determined value.30 However, this T50 value is higher than reported by other groups,31,32 most likely reflecting variations in buffer compositions and NTP concentrations. Nevertheless, our data clearly indicate that lysine binding to lysC riboswitch promotes transcription termination.
Figure 2.
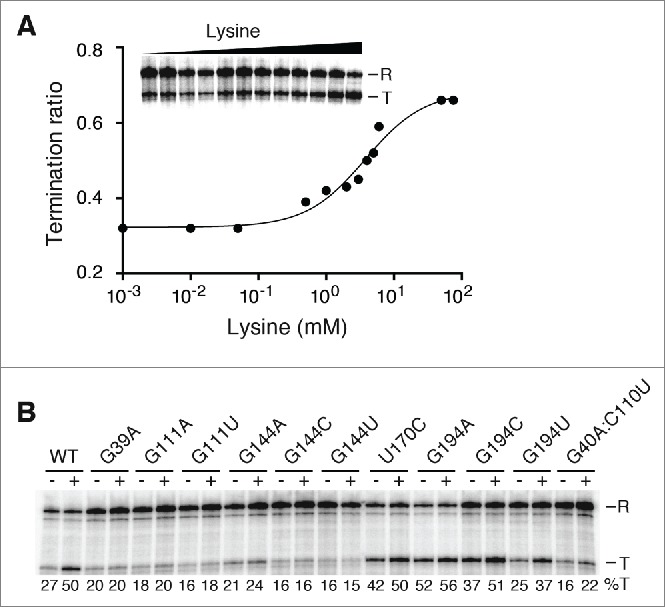
Lysine-induced transcription termination of riboswitch junction core variants. (A) Single-round transcriptions as a function of lysine concentration. Transcriptions were performed in presence of 1 µM, 10 µM, 50 µM, 500 µM, 1 mM, 2 mM, 3 mM, 4 mM, 5 mM, 6 mM, 50 mM and 75 mM of lysine. Readthrough (R) and prematurely terminated (T) products are shown on the right. Reactions were quantified and plotted as a function of lysine concentrations. Fitting the data to a 2-state model gave a T50 value of 3.8 ± 0.7 mM. (B) Junction core residues are important for riboswitch transcription termination. Single-round transcriptions were performed in absence (-) and presence (+) of 10 mM lysine for each riboswitch variant. Readthrough (R) and prematurely terminated (T) products are shown on the right. The percentage of terminated product for each reaction is indicated below.
To characterize the importance of junction core residues for lysC riboswitch regulation, we introduced a series of mutations in the riboswitch junction and assessed their effect on riboswitch activity using single-round transcriptions in absence and in presence of 10 mM lysine (Fig. 2B). As a control, we first mutated position G39, which was previously shown to directly interact with lysine via its 2′-OH group.16,17 The introduction of a G39A mutation completely abolished ligand-dependent transcription termination (Fig. 2B), as previously reported.27,30 We then verified the importance of residue G111, known to interact with both the lysine ε-amino group and the sugar moiety of G144 (Fig. 1C). When G111 was replaced with either an adenine (G111A) or a uracil (G111U), no increase in transcription termination was observed in presence of lysine, suggesting that the identity of G111 is crucial for riboswitch function. Similarly to G111, replacement of G144 with adenine (G144A), cytosine (G144C) or uracil (G144U) strongly altered riboswitch regulation, indicating the importance of G144 for transcription regulation. The low regulation level of G144A is particularly striking, as it should allow formation of a canonical A144-U170 Watson-Crick base pair, suggesting that the wobble configuration of the G144•U170 base pair is critical for lysC riboswitch function. In agreement with the importance of the wobble configuration, the introduction of a U170C mutation allowing formation of a G144-C170 base pair also dramatically perturbed riboswitch function (Fig. 2B). However, in this case, a pronounced effect on transcription termination was observed regardless of the presence of lysine, suggesting that the base pair G144-C170 favors transcription termination. Importantly, for both G144 and U170 mutants, the alteration of the wobble configuration might result in a suboptimal positioning of G144 thereby precluding its interaction with lysine. The identity of base pair G40-C110 is also important for riboswitch activity since the introduction of a G40A:C110U mutation completely abolished the ligand-dependent transcription termination (Fig. 2B).
Previous studies using the B. subtilis lysC riboswitch showed that introduction of a 2-aminopurine fluorophore in the junction core domain, at position 194 (Fig. 1B), results in a fluorescence signal that is increased in presence of Mg2+ or lysine.29-31 The fluorescence increase can be rationalized from available Thermatoga maritima lysine riboswitch crystal structures showing that residue 194 is exposed to solvent in the lysine-bound riboswitch complex.16,17 These crystal structures also show that residue 194 is involved in a long-range interaction with a bulged nucleotide located in P3 stem.16,17 Given that such a bulged residue in the P3 stem is not present in the B. subtilis lysC riboswitch, thus not involving residue 194 in a long-range interaction, we investigated whether the identity of G194 is important for riboswitch transcription termination. Substitution for an adenine (G194A) strongly increased transcription termination regardless of the presence of lysine (52% and 56% with and without lysine, respectively, Fig. 2B), suggesting that G194A mutation is deleterious for riboswitch regulatory activity. However, introduction of either G194C or G194U had a less drastic effect on riboswitch regulation since lysine addition resulted in higher transcription terminations for both mutants (Fig. 2B). Furthermore, while the G194C mutant showed higher transcription termination in absence of lysine, the G194U mutant had lower termination with lysine (Fig. 2B), in agreement with both mutants exhibiting disrupted riboswitch regulation mechanism. Thus, these results indicate that while a guanine at position 194 is important for optimal riboswitch regulation, its identity is not essential given that the presence of lysine promotes transcription termination for both G194C and G194U riboswitch mutants.
To investigate the increased transcription termination of riboswitches bearing mutations at positions 170 and 194, we performed additional single-round transcription assays by modulating the concentration of nucleotides. Since it was previously reported that nucleotide concentration influences transcription readthrough at terminators,31,33 we increased the NTP concentration to test whether transcription readthrough could be increased in order to improve lysine-induced transcription regulation. As expected, single-round transcription assays in presence of 5 mM NTP (Fig. S1) resulted in a general decrease in termination efficiencies (compare with data obtained at 1 mM NTP in Fig. 2B) for the wild-type as well as for mutants at positions 170 and 194, regardless of the presence of lysine. However, since the proportion of terminated transcripts was also diminished with lysine, the resulting efficiency of transcription regulation was not improved for any tested riboswitch construct. For example, using a U170C mutant, transcription termination efficiencies of 20% and 24% were obtained at 5 mM NTP in absence and presence of lysine, respectively (Fig. S1). Similar increase in riboswitch regulation efficiency was observed at 5 mM NTP using riboswitches carrying mutations at position 194 (Fig. S1), suggesting that increasing the transcription rate does not restore regulation efficiency. Together, our results suggest that the increased transcription termination ratio observed for riboswitches mutated at positions 170 and 194 is most probably caused by stabilization of the aptamer domain, which cannot be altered by modulating the rate of transcription. These results emphasize the importance of junction core residues for ligand binding and riboswitch function, in agreement with their sequence conservation and involvement in lysine-binding site formation.16,17,26,27
Global folding of the lysC aptamer in presence of Mg2+ ions and lysine
It was previously shown that the presence of Mg2+ ions induces stems P1 and P5 to exert a “scissor-like” movement in which both helices come into proximity,29,34 consistent with their respective location in crystal structures.16,17 To gather structural information regarding the role of junction core residues, we employed the FRET technique that is unique in terms of giving quantitative information about nucleic acid conformational changes.35-38 It was previously reported, using small-angle X-ray scattering (SAXS) and crystal structures, that the lysine aptamer does not experience significant structural changes upon ligand binding.16,17,39 However, FRET studies showed that Mg2+ and lysine cofactors both have a strong influence on the energy transfer occurring between fluorophores located in stems P1 and P5, indicating that the aptamer global folding is modulated upon binding of either cofactor.29,34 In addition, we previously showed that P1-P5 folding transition is strongly dependent on the formation of peripheral interactions L2-L3 and P2-L4, suggesting that P1-P5 juxtaposition is intimately linked to the formation of lysC aptamer long-range interactions.29
By introducing fluorescein and Cy3 fluorophores at the end of P1 and P5 helices, respectively (Fig. 3A), we monitored the folding transition of the P1-P5 FRET vector as a function of Mg2+ ions. In absence of lysine, addition of Mg2+ resulted in an increase of FRET efficiency (EFRET), indicating the presence of a folding transition bringing stems P1 and P5 into close juxtaposition. By assuming an all-or-none conformational transition, we fitted the data to a binding isotherm describing the folding transition, and calculated the magnesium ion concentration for which 50% of the molecules have undergone the transition ([Mg2+]1/2). In absence of lysine, values of [Mg2+]1/2 = 0.27 ± 0.08 mM and Hill coefficient (n) = 0.94 ± 0.17 were obtained, suggesting that the P1-P5 transition folds non-cooperatively in a low millimolar range, in agreement with previous studies.29 However, in presence of lysine, a different folding transition was observed in which EFRET values reached a higher plateau. Values of [Mg2+]1/2 = 0.04 ± 0.02 mM and n = 1.38 ± 0.29 were calculated in presence of lysine, indicating that the required magnesium ion concentration for the P1-P5 folding transition is decreased by ~6-fold in presence of lysine (see ratio for wild-type, Table 1). Interestingly, the difference in maximum EFRET values observed in absence and presence of lysine was not observed in our previous study,29 which is most likely due to the different location used for the fluorescein donor position.34 Thus, these results suggest that lysine binding to the lysC aptamer facilitates the Mg2+-dependent P1-P5 folding transition.
Figure 3.
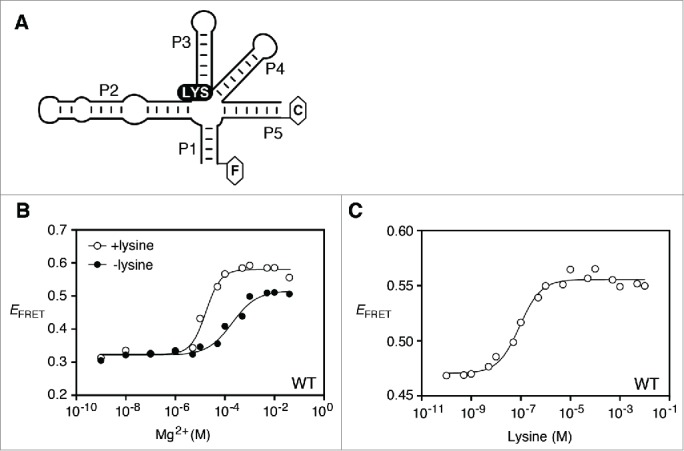
Folding of the P1-P5 transition monitored by FRET assays. (A) Schematic of the lysC aptamer construct used to monitor the P1-P5 FRET vector. Donor (fluorescein; F) and acceptor (Cy3; C) fluorophores are shown as hexagons linked to the 3′ and 5′ ends of P1 and P5 stems, respectively. The construct is based on a previous study that has shown that the removal of the P5 loop does not perturb riboswitch folding.29 The location of both fluorophores was chosen according to crystal structures.16,17 (B) Efficiency of FRET (EFRET) as a function of Mg2+ concentration in absence (black circles) or presence (white circles) of 5 mM lysine. The fluorescence data were fitted (lines) by regression using a 2-state model where Mg2+ binding to the RNA induces P1-P5 folding. (C) Efficiency of FRET (EFRET) as a function of lysine in the context of 10 mM Mg2+ ions. Data were fitted to a 2-state model assuming that lysine binding to the aptamer domain induces the folding of the P1-P5 FRET vector.
Table 1.
Folding transitions of FRET vector P1-P5.
| |
[Mg2+]1/2 (mM) |
|
Hill coefficient (n) |
||
|---|---|---|---|---|---|
| Variants | No lysine | 5 mM lysine | Ratio1 | No lysine | 5 mM lysine |
| Wild-type | 0.27 ± 0.08 | 0.04 ± 0.02 | 6.4 | 0.96 ± 0.17 | 1.38 ± 0.29 |
| G39A | 0.09 ± 0.03 | 0.08 ± 0.01 | 1.1 | 0.90 ± 0.14 | 0.92 ± 0.21 |
| G111A | 0.17 ± 0.01 | 0.17 ± 0.01 | 1.0 | 1.21 ± 0.17 | 0.82 ± 0.13 |
| G111U | 0.19 ± 0.03 | 0.22 ± 0.02 | 0.9 | 0.92 ± 0.15 | 1.06 ± 0.18 |
| G144A | 0.20 ± 0.07 | 0.11 ± 0.08 | 1.9 | 1.00 ± 0.28 | 1.11 ± 0.25 |
| G144C | 0.34 ± 0.16 | 0.26 ± 0.02 | 1.3 | 1.00 ± 0.17 | 0.88 ± 0.18 |
| G144U | 0.07 ± 0.03 | 0.11 ± 0.02 | 0.6 | 1.29 ± 0.25 | 0.98 ± 0.20 |
| U170C | 0.16 ± 0.01 | 0.09 ± 0.01 | 1.7 | 1.19 ± 0.22 | 1.27 ± 0.21 |
| G40U/C110A | 0.10 ± 0.01 | 0.09 ± 0.01 | 1.1 | 0.96 ± 0.19 | 1.33 ± 0.29 |
This value corresponds to the ratio of [Mg2+]1/2 obtained in the absence and presence of 5 mM lysine.
To characterize in more detail the influence of lysine binding on P1-P5 aptamer folding transition, we performed FRET assays as a function of lysine concentration in presence of 10 mM Mg2+ ions. An increase in EFRET values was observed when titrating lysine concentration, indicating that a folding transition between stems P1 and P5 occurs upon lysine binding (Fig. 3C). Fitting the data to a binding isotherm revealed a [Lysine]1/2 = 89 ± 18 nM, consistent with previous binding assays.16,27,40 The fact that the [Lysine]1/2 value is close to previously determined dissociation constants16,27,40 suggests that the presence of fluorophores does not perturb the ligand binding activity of the lysC aptamer. Furthermore, the low extent of lysine-dependent FRET change is consistent with the absence of significant structural changes obtained in presence of lysine.16,17,39 Together, our results indicate that both Mg2+ and lysine binding cofactors are important for the P1-P5 FRET transition of the lysine aptamer, consistent with previous studies.29,34
Importance of junction core nucleotides for the aptamer P1-P5 folding transition
To determine the influence of junction core residues on the P1-P5 folding transition of the lysC aptamer, we introduced mutations in the junction and analyzed their effect using the P1-P5 FRET vector (all calculated values are reported in Table 1). For all mutants, FRET efficiency was monitored as a function of Mg2+ ions in absence or presence of lysine to determine if introduced mutations alter the adoption of the Mg2+-dependent global structure and if the ligand binding activity is inhibited. In all cases, Hill coefficients were found to be close to unity suggesting that no change in cooperativity was observed. We first introduced a G39A mutation and observed a FRET transition similar to the wild-type in absence of ligand (Fig. 4A). An isotherm fitting gave [Mg2+]1/2 values of 0.09 ± 0.03 mM and 0.08 ± 0.01 mM in absence and in presence of lysine (Fig. 4A), respectively. Since the ratio of both [Mg2+]1/2 values is close to unity (ratio = 1.1, Table 1), lysine does not appear to participate in the P1-P5 folding transition for this mutant. A very similar folding transition to G39A was observed for G111U (Fig. 4B) and G111A (Fig. S2A), where [Mg2+]1/2 ratios close to 1 were also obtained, suggesting the inability of both mutants to perform ligand binding.
Figure 4.
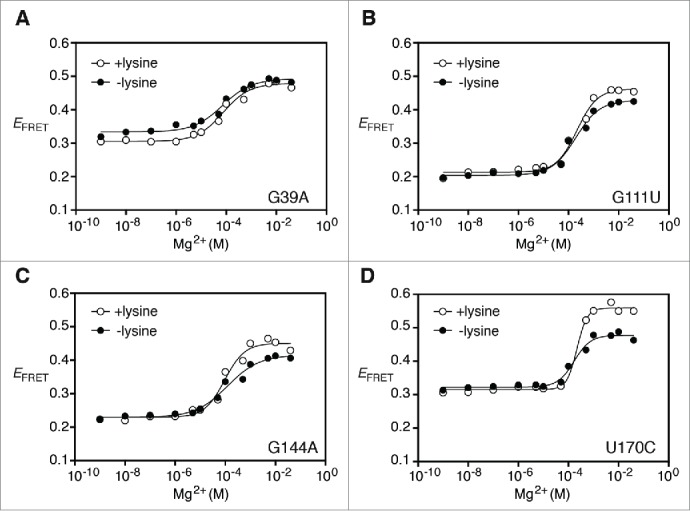
Importance of junction core residues for P1-P5 folding transition. (A) Efficiency of FRET (EFRET) of the G39A riboswitch mutant as a function of Mg2+ concentration in absence (black circles) or presence (white circles) of 5 mM lysine. The fluorescence data were fitted (lines) by regression using a 2-state model where Mg2+ binding to RNA induces P1-P5 folding. (B, C and D) Efficiency of FRET (EFRET) as a function of Mg2+ concentration in absence (black circles) or presence (white circles) of 5 mM lysine. Experiments were carried out for the G111U (B), G144A (C) and U170C (D) riboswitch variants. Data analysis as performed as indicated in (A).
We also established the importance of base pairs G40-C110 and G144•U170 for the P1-P5 folding transition. When introducing a G144A mutation, FRET transitions similar to wild-type were observed (Fig. 4C), with [Mg2+]1/2 values of 0.20 ± 0.07 mM and 0.11 ± 0.08 mM obtained in absence and presence of lysine, respectively (Table 1). Since the ratio obtained between [Mg2+]1/2 values corresponds to 1.9, it suggests that the P1-P5 transition for the G144A mutant is still modulated by the presence of lysine. However, the addition of lysine did not alter the [Mg2+]1/2 value when mutating G144 for a cytosine (G144C) or a uracil (G144U) (Fig. S2B and S2C), indicating that these mutations are detrimental for the lysine-induced folding transition. The introduction of a U170C mutation, allowing the formation of a G144-C170 base pair, resulted in a reproducible lysine effect on the P1-P5 folding transition (Fig. 4D). In this case, fitting of binding isotherms revealed a [Mg2+]1/2 ratio of 1.7 (Table 1), consistent with lysine binding promoting the folding of the lysC aptamer, albeit to a lower degree than the wild-type (Table 1). Furthermore, the importance of base pair G40-C110 identity was also assessed using the P1-P5 FRET vector. In this case, the introduction of a G40U-C110A mutation yielded [Mg2+]1/2 values of 0.10 ± 0.01 mM and 0.09 ± 0.01 mM in absence and presence of lysine, respectively (Table 1). Given that the [Mg2+]1/2 ratio is close to 1 (ratio = 1.1, see Table 1), it suggests that the G40U-C110A mutation completely abolishes the ligand binding activity of the aptamer.
Taken together, our FRET results show that all tested mutants undergo Mg2+-induced folding transition similar to the wild-type in absence of lysine (Table 1), suggesting that mutated residues are not crucial for adoption of the Mg2+-induced folding transition. However, our data also indicate that junction core residues are highly important for the lysine-induced P1-P5 conformational change as most of the tested mutants exhibited [Mg2+]1/2 ratios close to 1 (Table 1), consistent with their inability to bind lysine. For base pair mutants A144-U170 and G144-C170 ratios of 1.9 and 1.7 were obtained (Table 1), respectively, suggesting their ability to bind ligand, but with reduced efficiency when compared to wild-type (ratio of 6.4, Table 1). Such low ligand binding efficiency is in agreement with a strongly reduced lysine ability to promote transcription termination in the context of corresponding riboswitch mutants (Fig. 2B).
Junction core mutations do not perturb lysC aptamer peripheral interactions
To determine the influence of junction core mutations on the global folding of the lysC aptamer (Fig. 5A), we employed the selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) technique.41 This assay allows the determination of flexible regions in RNA molecules because 2′-OH located in such regions are prone to react with N-methylisatoic anhydride (NMIA). SHAPE is thus a powerful assay to monitor riboswitch conformational changes as a function of experimental conditions, as previously done for the lysC aptamer.17,29,31 In particular, we have previously used SHAPE analysis to demonstrate the existence of a folding synergy occurring between long-range interactions L2-L3 and P2-L4 leading to a decrease in magnesium ions requirement for lysC aptamer folding.29
Figure 5.
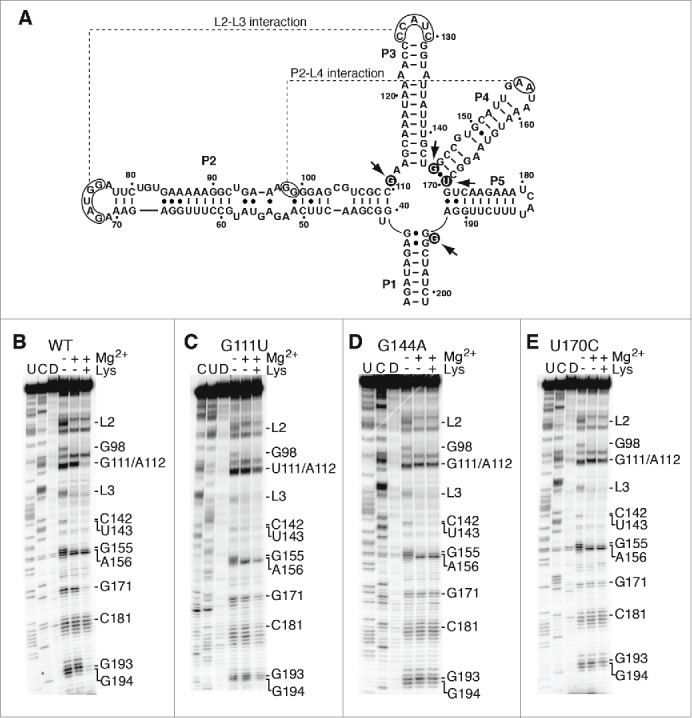
Junction core mutations are not important for the global folding of the aptamer. (A) Secondary structure representing the lysC aptamer. The regions participating in the formation of L2-L3 and P2-L4 interactions are indicated (dotted lines). Arrows indicate nucleotides involved in the corresponding tertiary interactions. Residues mutated are shown in black circles. The nucleotide nomenclature is based on previous studies.26,27 (B, C, D and E) SHAPE modification of selected lysC aptamers performed in the context of 10 mM NaCl. Experiments were done as a function of 10 mM Mg2+ and 5 mM lysine and analysis was performed using the wild-type aptamer (B) and variants G111U (C), G144A (D) and U170C (E). In each case, cytosine (C) and uracil (U) sequencing lanes were added to resolve the sequence. NMIA was replaced by DMSO (D) in control reactions. Nucleotides or regions exhibiting changes either in Mg2+ or lysine are indicated on the right.
Compared to results obtained in presence of only monovalent ions, SHAPE analysis revealed that the addition of Mg2+ ions resulted in the protection of L2, L3 and L4 regions, as well as protection of residue G98 located in stem P2 (Fig. 5B). These results are in agreement with the formation of long-range interactions L2-L3 and P2-L4 in presence of Mg2+ ions, consistent with previous findings.29 The addition of 5 mM lysine yielded additional protection for junction core residues G111/A112, G171, G193 and G194 (Fig. 5B), in agreement with the aptamer junction being involved in lysine recognition.17,29 These results indicate that, while the binding of Mg2+ ions promote the formation of peripheral interactions, lysine binding to the pre-folded aptamer structure induces a reorganization of the junction core domain, thereby leading to the native structure.17,29
Next, we performed SHAPE analysis on the lysC aptamer carrying a G111U mutation. For this variant, the Mg2+-dependent SHAPE reaction profile revealed that both L2-L3 and P2-L4 long-range interactions are formed (Fig. 5C), suggesting that the global structure of the lysC aptamer is preserved. However, an incomplete NMIA protection was observed for residues G111/A112 in presence of lysine, indicating a reduced ability of G111U to perform ligand binding (Fig. 5C). These results are in agreement with transcription and FRET data showing the inability of variant G111U to perform both transcription regulation and ligand-induced folding, respectively (Figs. 2B and 4B). A similar analysis was performed on aptamer variants disrupting base pair G144•U170, in which G144A (Fig. 5D) or U170C (Fig. 5E) mutations were introduced. When analyzing the SHAPE profile of both mutants in presence of Mg2+, NMIA protections were observed for regions L2-L3 and P2-L4, consistent with both long-range interactions not being perturbed in the context of these aptamer variants. However, in each case, no noticeable ligand-binding effect could be observed (Figs. 5D and 5E), suggesting the inability of aptamers to perform ligand binding. Disruption of wobble pair G144•U170 was also achieved by introducing a G144C mutation (Fig. S3A). As expected, the SHAPE profile revealed that the mutation G144C does not prevent long-range tertiary interactions L2-L3 and P2-L4, as obtained for G144A and U170C mutants. Thus, as observed for G111, residues G144 and U170 are not involved in the Mg2+-dependent global folding of the lysC aptamer, but are rather critically important for lysine sensing.
Given that G194 responds to the lysine-dependent folding of the lysC aptamer (Fig. 5B), we also examined the influence of G194 mutations (G194C and G194A) on the aptamer structure using SHAPE analysis (Figs. 3B and 3C). As observed for the wild-type aptamer, long-range interactions L2-L3 and P2-L4 were found to occur in an Mg2+-dependent manner. However, in contrast to junction core mutations introduced at positions G111, G144 and U170, a very efficient lysine-dependent protection was observed for residues G111, A112, G171, G193 and position 194, indicating that mutations at position 194 do not disrupt the aptamer ligand binding activity. These results suggest that the reduced transcriptional control for G194 riboswitch mutants (see G194A in Fig. 2B) is not caused by a decreased ability of the aptamer domain to perform ligand binding.
Discussion
Current literature describing folding mechanisms of small nucleolytic ribozymes and rRNA domains show that the folding of helical junctions is determinant for the overall RNA architecture.8,11,12 A similar role for helical junction is also likely to be found in riboswitches, given the high preponderance of helical junctions in their secondary structures.1 For example, centrally located junctions such as in TPP riboswitches are expected to be of primary importance for aptamer folding.18-20 Indeed, since residues located in the helical junction are not involved in ligand binding but are nevertheless highly conserved,5 it suggests that they are implicated in aptamer global folding. However, in the case of riboswitches using junction core domains to build ligand binding sites, such as the lysine riboswitch,1 it is expected that ligand binding residues undergo an evolutionary pressure to maintain a high affinity and specific binding site, thus relying on additional tertiary interactions to allow efficient architecture folding. In agreement with this idea, our work indicates that lysC junction core residues are not involved in RNA global folding but rather in lysine sensing.
Our study suggests that the lysC aptamer adopts a Mg2+-dependent folding state (FMg) characterized by the formation of long-range tertiary interactions L2-L3 and P2-L4, as well as a structural reorganization involving stems P1 and P5 (Fig. 6). Since we previously demonstrated that formation of L2-L3 and P2-L4 interactions influences the P1-P5 folding transition,29 it indicates that adoption of the lysC aptamer global structure is important to promote the transition involving stems P1 and P5. Based on SHAPE and FRET data, the FMg state is not perturbed by mutations introduced at the level of junction core nucleotides, showing that these residues are not participating in interactions important for the adoption of the FMg structure. Thus, our study suggests that the FMg state provides a rigid scaffold allowing the formation of the lysine-binding site. We also observed that lysine binding to the FMg state results in a distinct FRET P1-P5 transition (Fig. 3C) as well as SHAPE protection of junction core residues (Fig. 5A), leading to the adoption of the ligand-bound native state (FNS), similarly to a recent FRET analysis.34 Based on our results, it is not possible to directly determine whether the FNS state is conformationally-captured or induced-fit. Recent sm-FRET analysis has reported the presence of a lysine-dependent FRET state34 but more work is required to fully differentiate between both folding models. However, the observation of an exclusive FRET state in presence of ligand suggests an induced-fit model operating at physiological magnesium concentrations. It is likely that the aptamer global architecture is conserved in absence of lysine (FMg) and that lysine binding only results in the folding of the junction core domain (FNS). Indeed, since L2-L3 and P2-L4 interactions involve multiple base pairs and H-bond interactions (Fig. 6, insert), it is expected that they remain folded in a saturating Mg2+ concentration. However, since it was previously proposed that base pair G39•G193 is stabilized upon lysine binding,16,17 the lysine-induced FRET transition (Fig. 3C) could result from G39•G193 base pair formation, which would either cause the P1 stem to have a rotational movement along its axis, as recently observed for the SAM-I aptamer.42 The reorganization of base pair G39•G193 is also supported by our SHAPE assays showing that residues G193 and G194 become protected in presence of lysine (Fig. 5B). This structural change is also in agreement with previous studies showing that a 2-aminopurine introduced at position 194 exhibits high fluorescence upon lysine binding,29-31 in agreement with the P1 stem being reorganized in the ligand-bound aptamer.
Figure 6.
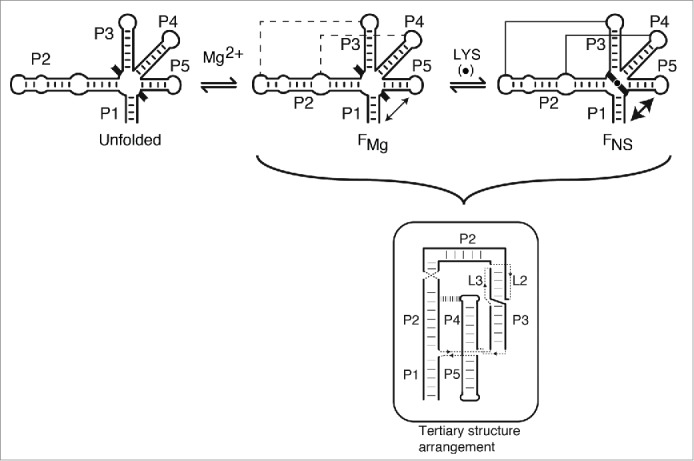
Proposed folding pathway and ligand sensing mechanism of the lysC riboswitch aptamer. In absence of Mg2+ ions, the lysC aptamer is assumed to adopt an unfolded structure (unfolded) in which only secondary structure elements (P1 to P5 stems) are formed. The presence of black braces pointing outside the aptamer domain indicates that junction core residues are not folded at this stage. Upon Mg2+ ions binding, the aptamer is reorganized into a structure (FMg) exhibiting long-range tertiary interactions L2-L3 and P2-L4 (dotted lines), as observed in crystal structures. Mg2+ ions binding also promote the formation of the P1-P5 folding transition (double arrow). Addition of lysine (LYS, black circle) results in the adoption of the native state (FNS) that consists of an additional P1-P5 transition as well as the reorganization of the junction core residues (inward orientation of black braces).
Single-round in vitro transcriptions showed that riboswitches carrying mutations G194A and G194C exhibit higher transcription termination in absence of lysine (Fig. 2B). Since SHAPE data indicate that both mutants perform lysine binding (Fig. S3B and S3C), it suggests that their inability to modulate transcription termination is not likely caused by perturbations of the ligand binding site. Rather, a more plausible explanation is that these mutations stabilize P1 stem formation, therefore promoting transcription termination independently of the presence of ligand, as reported for adenine33 and lysine29 riboswitches. Such an increase in transcription termination was also observed for a U170C riboswitch mutant (Fig. 2B). However, increased transcription termination is unlikely to be caused by a P1 stem direct stabilization. Instead, it is more probable that the U170C substitution stabilizes the junction core domain (Fig. 1C) by forming a G144-C170 base pair. Precedence of such a junction core stabilizing mutation has been described for purine-sensing riboswitches,43 in which NMR and X-ray studies revealed that the mutated sequence adopts a conformation highly similar to the ligand-bound aptamer.44 Furthermore, FRET data reveal that a U170C aptamer shows a lysine-dependent increase in energy transfer similar to the wild-type (Figs. 3B and 4D), indicating that the U170C junction core domain undergoes a similar structural rearrangement. Although we do not currently know whether the U170C mutant exhibits a ligand binding site mimicking the lysine-bound conformation, it is clear that this mutation provides sufficient stability to efficiently terminate transcription in absence of lysine (Fig. 2B).
Consistent with previous findings,32 our study shows that the identity of base pairs G40-C110 and G144•U170 is important for this riboswitch control of transcription termination (Fig. 2B). However, although mutations preventing wobble pairing interaction of G144•U170 completely prevented lysine binding as determined from FRET data (Table 1), the introduction of Watson-Crick base pairs A144-U170 and G144-C170 partially restored lysine binding as [Mg2+]1/2 ratio values of 1.9 and 1.7 were respectively obtained. These results suggest a high importance of the wobble base pair to allow lysine to efficiently decrease the magnesium ions requirement for the P1-P5 folding transition. It was recently reported that a G40A-C110U riboswitch mutant modulates transcription termination in presence of lysinamide or lysine hydroxamate, indicating that ligand binding specificity is altered.32 Furthermore, in addition to G111 mutations, a similar specificity change was also obtained for an A144-U170 base pair mutant, which showed riboswitch regulation in presence of an ethyl ester-carrying lysine derivative (LysEE).32 In agreement with our study, these observations indicate that altering junction core residues does not perturb the aptamer global folding, but can in fact result in ligand specificity variation. These results suggest that aptamer global folding and ligand recognition are uncoupled in the lysC riboswitch, as demonstrated from SHAPE and FRET data showing that junction core mutations do not alter long-range tertiary interactions or the Mg2+-dependent FRET transition (Fig. 4 and 5). In all cases, the folding process occurs in a low millimolar range and exhibits a Hill coefficient close to unity (Table 1). The [Mg2+]1/2 ratio values for G144A and U170C aptamers are close to ~2, suggesting that both variants still exhibit a low level of ligand binding (Figs. 4C and 4D). However, the lysine binding activity of both mutants is not detected by SHAPE assays, perhaps reflecting different sensitivities between these techniques (Figs. 5D and 5E).
The lysC riboswitch is similar to the purine-sensing class as it also uses the junction core domain to pre-fold the ligand binding site. Purine riboswitches contain a single 3-way helical junction exhibiting unpaired nucleotides in the core region, as observed for the vast majority of naturally occurring 3-way junctions.45 The purine riboswitch junction is intimately involved in the formation of a highly specific purine-binding site and a long-range loop-loop interaction has been shown to pre-fold the junction core domain in a configuration productive for ligand binding.46,47 As found for lysC,32 ligand specificity can be altered in purine riboswitches by mutating core residues,48 indicating that both lysC and purine riboswitches share common ligand binding mechanisms using junction core residues. However, riboswitches sensing TPP,18-20 AdoCbl,21,22 glycine23 and THF24,25 mostly rely on kissing-loops or internal bulges for ligand sensing. For the thiM TPP-sensing riboswitch, it was shown that the junction domain is pre-organized prior to TPP binding,49 in agreement with junction residues not interacting with TPP. In contrast to lysC, TPP binding results in stabilization of a long-range tertiary interaction,49 showing that the aptamer global folding is coupled to TPP binding. However, such a strong speciation in ligand sensing versus junction folding is most likely not representative of all junction core residues since ligand-induced local interactions may propagate and influence the formation of long-range tertiary interactions, as observed for the lysine-induced P1-P5 transition (Fig. 3C) and the pbuE adenine aptamer at low magnesium concentrations.46 Whether specific riboswitch representatives employ helical junctions for both global folding and ligand recognition remains to be determined.
Materials and Methods
Single-round in vitro transcription assays
The single-round in vitro transcription assay was adapted from a previous study.29 Briefly, DNA templates for transcription assays were made using PCR from DNA oligonucleotides (IDT). The glyQS promoter was used to initiate transcription using E. coli polymerase (Epicenter Biotechnologies).29 The concentrations of lysine are indicated in the respective figure legends. Experiments were repeated at least 3 times and exhibited similar uncertainties (<10%). The RNA sequence is shown below for the wild-type construct and mutations are indicated in the text.
Transcribed wild-type sequence
GCGUGUUAUGGUGAAGAUAGAGGUGCGAACUUCAAGAGUAUGCCUUUGGAGAAAGAUGGAUUCUGUGAAAAAGGCUGAAAGGGGAGCGUCGCCGAAGCAAAUAAAACCCCAUCGGUAUUAUUUGCUGGCCGUGCAUUGAAUAAAUGUAAGGCUGUCAAGAAAUCAUUUUCUUGGAGGGCUAUCUCGUUGUUCAUAAUCAUUUAUGAUGAUUAAUUGAUAAGCAAUGAGAGUAUUCCUCUCAUUGCUUUUUUUAUUGUGGACAAAGCGCUCUUUCUCCUCACCCGCACGAACCAAAAUGUAAAGGGUGGUAAUACAUGGGUCUUAUUGUACAAAAAUUCGGAGGCACU
FRET analysis
Fluorescence spectroscopy was done as previously reported.29,42 Polarization artifacts were avoided by setting excitation and emission polarizers crossed at 54.75°. EFRET values were calculated using the acceptor normalization method.50,51 Data were measured at 10°C in 90 mM Tris-borate, pH 8.3 and 100 mM KCl. Excitation wavelengths of 490 and 547 nm were used for fluorescein and Cy3, respectively. Titrations using fluorescent dual-labeled lysC aptamers were performed by varying magnesium ions or lysine concentration. The concentrations of lysine are indicated in the respective figure legends. Experiments were performed at least 3 times and error values are reported for calculated folding descriptors ([Mg2Mg2+]1/2 and n) (Table 1).
FRET constructs were made using a T7-transcribed strand
GCGAAGAUAGAGGUGCGAACUUCAAGAGUAUGCCUUUGGAGAAAGAUGGAUUCUGUGAAAAAGGCUGAAAGGGGAGCGUCGCCGAAGCAAAUAAAACCCCAUCGGUAUUAUUUGCUGGCCGUGCAUUGAAUAAAUGUAAGGCUGUCAAGAAGCAUCG
and a 3′ synthetic strand (IDT)
Cy3-CGAUGCUUCUUGGAGGGCUAUCUUCG(5-N-U)
The synthetic strand was labeled using fluorescein (Invitrogen) at the 5′-amino-allyl uridine nucleotide according to the manufacturer protocol. Transcribed and synthetic strands were prepared, annealed together as a complex, and purified as previously described.29
SHAPE probing
Wild-type and aptamer variants were produced as previously described.29,42 Reactions were prepared by mixing 1 pmol of purified aptamer resuspended in a 2:1 mixture of 0.5× TE buffer: 3.3× folding buffer (333 mM K-HEPES, pH 8.0, 333 mM NaCl) and the required concentration of MgCl2 and lysine. Samples were treated with N-methylisatoic anhydride (NMIA, Invitrogen) as previously described.29 Lysine was added to a final concentration of 5 mM. Reverse transcription reactions were performed on reacted RNA and products were separated on 5% denaturing polyacrylamide gel.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of the Lafontaine laboratory for discussion and Drs Alain Lavigueur and Carlos Penedo for critical reading of the manuscript. This work was supported by the Canadian Institutes of Health Research (CIHR). D.A.L. is a Fonds de Recherche Santé Québec Senior Scholar.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Serganov A, Nudler E. A decade of riboswitches. Cell 2013; 152:17-24; PMID:23332744; http://dx.doi.org/ 10.1016/j.cell.2012.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards TE, Klein DJ, Ferre-D'Amare AR. Riboswitches: small-molecule recognition by gene regulatory RNAs. Curr Opin Struct Biol 2007; 17:273-9; PMID:17574837; http://dx.doi.org/ 10.1016/j.sbi.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 3.Schwalbe H, Buck J, Furtig B, Noeske J, Wohnert J. Structures of RNA switches: insight into molecular recognition and tertiary structure. Angewandte Chemie 2007; 46:1212-9; PMID:17226886; http://dx.doi.org/ 10.1002/anie.200604163 [DOI] [PubMed] [Google Scholar]
- 4.Haller A, Souliere MF, Micura R. The Dynamic Nature of RNA as Key to Understanding Riboswitch Mechanisms. Acc Chem Res 2011:1339-48; PMID:21678902; http://dx.doi.org/ 10.1021/ar200035g [DOI] [PubMed] [Google Scholar]
- 5.Barrick JE, Breaker RR. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol 2007; 8:R239; PMID:17997835; http://dx.doi.org/ 10.1186/gb-2007-8-11-r239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duckett DR, Murchie AI, Lilley DM. The global folding of four-way helical junctions in RNA, including that in U1 snRNA. Cell 1995; 83:1027-36; PMID:8521503; http://dx.doi.org/ 10.1016/0092-8674(95)90218-X [DOI] [PubMed] [Google Scholar]
- 7.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000; 289:905-20; PMID:10937989; http://dx.doi.org/ 10.1126/science.289.5481.905 [DOI] [PubMed] [Google Scholar]
- 8.Orr JW, Hagerman PJ, Williamson JR. Protein and Mg(2+)-induced conformational changes in the S15 binding site of 16 S ribosomal RNA. J Mol Biol 1998; 275:453-64; PMID:9466923; http://dx.doi.org/ 10.1006/jmbi.1997.1489 [DOI] [PubMed] [Google Scholar]
- 9.Serganov AA, Masquida B, Westhof E, Cachia C, Portier C, Garber M, Ehresmann B, Ehresmann C. The 16S rRNA binding site of Thermus thermophilus ribosomal protein S15: comparison with Escherichia coli S15, minimum site and structure. RNA 1996; 2:1124-38; PMID:8903343 [PMC free article] [PubMed] [Google Scholar]
- 10.Batey RT, Williamson JR. Interaction of the Bacillus stearothermophilus ribosomal protein S15 with 16 S rRNA: II. Specificity determinants of RNA-protein recognition. J Mol Biol 1996; 261:550-67; PMID:8794876; http://dx.doi.org/ 10.1006/jmbi.1996.0482 [DOI] [PubMed] [Google Scholar]
- 11.Lilley DM. Structures of helical junctions in nucleic acids. Quarterly reviews of biophysics 2000; 33:109-59; PMID:11131562; http://dx.doi.org/ 10.1017/S0033583500003590 [DOI] [PubMed] [Google Scholar]
- 12.Lilley DM. Comparative gel electrophoresis analysis of helical junctions in RNA. Methods Enzymol 2009; 469:143-57; PMID:20946788; http://dx.doi.org/ 10.1016/S0076-6879(09)69007-8 [DOI] [PubMed] [Google Scholar]
- 13.Serganov A, Patel DJ. Metabolite recognition principles and molecular mechanisms underlying riboswitch function. Annu Rev Biophys 2012; 41:343-70; PMID:22577823; http://dx.doi.org/ 10.1146/annurev-biophys-101211-113224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature 2004; 432:411-5; PMID:15549109; http://dx.doi.org/ 10.1038/nature03037 [DOI] [PubMed] [Google Scholar]
- 15.Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ. Structural Basis for Discriminative Regulation of Gene Expression by Adenine- and Guanine-Sensing mRNAs. Chem Biol 2004; 11:1729-41; PMID:15610857; http://dx.doi.org/ 10.1016/j.chembiol.2004.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serganov A, Huang L, Patel DJ. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature 2008; 455:1263-7; PMID:18784651; http://dx.doi.org/ 10.1038/nature07326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem 2008; 283:22347-51; PMID:18593706; http://dx.doi.org/ 10.1074/jbc.C800120200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thore S, Leibundgut M, Ban N. Structure of the Eukaryotic Thiamine Pyrophosphate Riboswitch with Its Regulatory Ligand. Science 2006:1208-11; PMID:16675665; http://dx.doi.org/ 10.1126/science.1128451 [DOI] [PubMed] [Google Scholar]
- 19.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature 2006; 441:1167-71; PMID:16728979; http://dx.doi.org/ 10.1038/nature04740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards TE, Ferre-D'Amare AR. Crystal Structures of the Thi-Box Riboswitch Bound to Thiamine Pyrophosphate Analogs Reveal Adaptive RNA-Small Molecule Recognition. Structure 2006; 14:1459-68; PMID:16962976; http://dx.doi.org/ 10.1016/j.str.2006.07.008 [DOI] [PubMed] [Google Scholar]
- 21.Johnson JE Jr., Reyes FE, Polaski JT, Batey RT. B12 cofactors directly stabilize an mRNA regulatory switch. Nature 2012; 492:133-7; PMID:23064232; http://dx.doi.org/ 10.1038/nature11607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peselis A, Serganov A. Structural insights into ligand binding and gene expression control by an adenosylcobalamin riboswitch. Nat Struct Mol Biol 2012; 19:1182-4; PMID:23064646; http://dx.doi.org/ 10.1038/nsmb.2405 [DOI] [PubMed] [Google Scholar]
- 23.Huang L, Serganov A, Patel DJ. Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch. Mol Cell 2010; 40:774-86; PMID:21145485; http://dx.doi.org/ 10.1016/j.molcel.2010.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trausch JJ, Ceres P, Reyes FE, Batey RT. The structure of a tetrahydrofolate-sensing riboswitch reveals two ligand binding sites in a single aptamer. Structure 2011; 19:1413-23; PMID:21906956; http://dx.doi.org/ 10.1016/j.str.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang L, Ishibe-Murakami S, Patel DJ, Serganov A. Long-range pseudoknot interactions dictate the regulatory response in the tetrahydrofolate riboswitch. Proc Natl Acad Sci U S A 2011; 108:14801-6; PMID:21873197; http://dx.doi.org/ 10.1073/pnas.1111701108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grundy FJ, Lehman SC, Henkin TM. The L box regulon: lysine sensing by leader RNAs of bacterial lysine biosynthesis genes. Proc Natl Acad Sci U S A 2003; 100:12057-62; PMID:14523230; http://dx.doi.org/ 10.1073/pnas.2133705100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev 2003; 17:2688-97; PMID:14597663; http://dx.doi.org/ 10.1101/gad.1140003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Regulation of lysine biosynthesis and transport genes in bacteria: yet another RNA riboswitch? Nucleic Acids Res 2003; 31:6748-57; PMID:14627808; http://dx.doi.org/ 10.1093/nar/gkg900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blouin S, Chinnappan R, Lafontaine DA. Folding of the lysine riboswitch: importance of peripheral elements for transcriptional regulation. Nucleic Acids Res 2010; 39:3373-87; PMID:21169337; http://dx.doi.org/ 10.1093/nar/gkq1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blouin S, Lafontaine DA. A loop-loop interaction and a K-turn motif located in the lysine aptamer domain are important for the riboswitch gene regulation control. RNA 2007; 13:1256-67; PMID:17585050; http://dx.doi.org/ 10.1261/rna.560307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garst AD, Porter EB, Batey RT. Insights into the regulatory landscape of the lysine riboswitch. J Mol Biol 2012; 423:17-33; PMID:22771573; http://dx.doi.org/ 10.1016/j.jmb.2012.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson-Mitchell SN, Grundy FJ, Henkin TM. Analysis of lysine recognition and specificity of the Bacillus subtilis L box riboswitch. Nucleic Acids Res 2012; 40:5706-17; PMID:22416067; http://dx.doi.org/ 10.1093/nar/gks212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemay JF, Desnoyers G, Blouin S, Heppell B, Bastet L, St-Pierre P, Masse E, Lafontaine DA. Comparative Study between Transcriptionally- and Translationally-Acting Adenine Riboswitches Reveals Key Differences in Riboswitch Regulatory Mechanisms. PLoS Genet 2011; 7:e1001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiegland LR, Garst AD, Batey RT, Nesbitt DJ. Single-molecule studies of the lysine riboswitch reveal effector-dependent conformational dynamics of the aptamer domain. Biochemistry 2012; 51:9223-33; PMID:23067368; http://dx.doi.org/ 10.1021/bi3007753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lilley DM. Structure, folding and mechanisms of ribozymes. Curr Opin Struct Biol 2005; 15:313-23; PMID:15919196; http://dx.doi.org/ 10.1016/j.sbi.2005.05.002 [DOI] [PubMed] [Google Scholar]
- 36.Walter NG, Harris DA, Pereira MJ, Rueda D. In the fluorescent spotlight: global and local conformational changes of small catalytic RNAs. Biopolymers 2001; 61:224-42; PMID:11987183; http://dx.doi.org/ 10.1002/bip.10144 [DOI] [PubMed] [Google Scholar]
- 37.St-Pierre P, McCluskey K, Shaw E, Penedo JC, Lafontaine DA. Fluorescence tools to investigate riboswitch structural dynamics. Biochim Biophys Acta 2014; 1839:1005-19; PMID:24863161; http://dx.doi.org/ 10.1016/j.bbagrm.2014.05.015 [DOI] [PubMed] [Google Scholar]
- 38.Shaw E, St-Pierre P, McCluskey K, Lafontaine DA, Penedo JC. Using sm-FRET and denaturants to reveal folding landscapes. Methods Enzymol 2014; 549:313-41; PMID:25432755; http://dx.doi.org/ 10.1016/B978-0-12-801122-5.00014-3 [DOI] [PubMed] [Google Scholar]
- 39.Baird NJ, Ferre-D'Amare AR. Idiosyncratically tuned switching behavior of riboswitch aptamer domains revealed by comparative small-angle X-ray scattering analysis. RNA; 16:598-609; PMID:20106958; http://dx.doi.org/ 10.1261/rna.1852310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blount KF, Wang JX, Lim J, Sudarsan N, Breaker RR. Antibacterial lysine analogs that target lysine riboswitches. Nat Chem Biol 2007; 3:44-9; PMID:17143270; http://dx.doi.org/ 10.1038/nchembio842 [DOI] [PubMed] [Google Scholar]
- 41.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 2005; 127:4223-31; PMID:15783204; http://dx.doi.org/ 10.1021/ja043822v [DOI] [PubMed] [Google Scholar]
- 42.Heppell B, Blouin S, Dussault AM, Mulhbacher J, Ennifar E, Penedo JC, Lafontaine DA. Molecular insights into the ligand-controlled organization of the SAM-I riboswitch. Nature Chem Biol 2011; 7:384-92; http://dx.doi.org/ 10.1038/nchembio.563 [DOI] [PubMed] [Google Scholar]
- 43.Tremblay R, Lemay JF, Blouin S, Mulhbacher J, Bonneau E, Legault P, Dupont P, Penedo JC, Lafontaine DA. Constitutive regulatory activity of an evolutionary-excluded riboswitch variant. J Biol Chem 2011; 286:27406-15; PMID:21676871; http://dx.doi.org/ 10.1074/jbc.M111.229047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delfosse V, Bouchard P, Bonneau E, Dagenais P, Lemay JF, Lafontaine DA, Legault P. Riboswitch structure: an internal residue mimicking the purine ligand. Nucleic Acids Res 2010; 38:2057-68; PMID:20022916; http://dx.doi.org/ 10.1093/nar/gkp1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lilley DM. Folding of branched RNA species. Biopolymers 1998; 48:101-12; PMID:11180044; http://dx.doi.org/ 10.1002/(SICI)1097-0282 (1998)48:2%3c101::AID-BIP2%3e3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 46.Lemay JF, Penedo JC, Tremblay R, Lilley DM, Lafontaine DA. Folding of the adenine riboswitch. Chem Biol 2006; 13:857-68; PMID:16931335; http://dx.doi.org/ 10.1016/j.chembiol.2006.06.010 [DOI] [PubMed] [Google Scholar]
- 47.Dalgarno PA, Bordello J, Morris R, St-Pierre P, Dube A, Samuel ID, Lafontaine DA, Penedo JC. Single-molecule chemical denaturation of riboswitches. Nucleic Acids Res 2013; 41:4253-65; PMID:23446276; http://dx.doi.org/ 10.1093/nar/gkt128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilbert SD, Love CE, Edwards AL, Batey RT. Mutational analysis of the purine riboswitch aptamer domain. Biochemistry 2007; 46:13297-309; PMID:17960911; http://dx.doi.org/ 10.1021/bi700410g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haller A, Altman RB, Souliere MF, Blanchard SC, Micura R. Folding and ligand recognition of the TPP riboswitch aptamer at single-molecule resolution. Proc Natl Acad Sci U S A 2013; 110:4188-93; PMID:23440214; http://dx.doi.org/ 10.1073/pnas.1218062110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murchie AI, Clegg RM, von Kitzing E, Duckett DR, Diekmann S, Lilley DM. Fluorescence energy transfer shows that the four-way DNA junction is a right-handed cross of antiparallel molecules. Nature 1989; 341:763-6; PMID:2797209; http://dx.doi.org/ 10.1038/341763a0 [DOI] [PubMed] [Google Scholar]
- 51.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol 1992; 211:353-88; PMID:1406315; http://dx.doi.org/ 10.1016/0076-6879(92)11020-J [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.