

Abstract
With the emergence of highly pathogenic avian influenza (HPAI) H7N9 and H5N1 strains, there is a pressing need to develop direct-acting antivirals (DAAs) to combat such deadly viruses. The M2-S31N proton channel of the influenza A virus (A/M2) is one of the validated and most conserved proteins encoded by the current circulating influenza A viruses; thus, it represents a high-profile drug target for therapeutic intervention. We recently discovered a series of S31N inhibitors with the general structure of adamantyl-1-NH2+CH2–aryl, but they generally had poor physical properties and some showed toxicity in vitro. In this study, we sought to optimize both the adamantyl as well as the aryl/heteroaryl group. Several compounds from this study exhibited submicromolar EC50 values against S31N-containing A/WSN/33 influenza viruses in antiviral plaque reduction assays with a selectivity index greater than 100, indicating that these compounds are promising candidates for in-depth preclinical pharmacology.
Graphical Abstract

INTRODUCTION
Influenza virus infections are a major global health threat. Despite the availability of influenza vaccines and small molecule antivirals, an estimated 10–15% of the population is infected annually in the United States,1 leading to approximately 36 000 deaths and 200 000 hospitalizations.2,3 Currently, vaccination remains the most effective way to prevent influenza virus infection; however, it is only partially effective and offers 65% protection in the best scenario.4 Moreover, due to the antigenic shift and drift of influenza viruses, influenza vaccines have to be regenerated every year.5,6 Even though more effective broad-neutralizing antibodies are still in development,7–9 there is an immediate need for small molecule drugs, particularly for combating emerging highly pathogenic influenza strains, such as H5N1 and H7N9 for which vaccines were not immediately available in the first few months of influenza outbreak. These highly pathogenic avian influenza strains might become human-to-human transmissible with just a few additional mutations.10 Thus, small molecule antivirals that target the most conserved viral proteins, such as the A/M2 proton channel, are highly desired.11,12
A/M2 is a viral membrane protein that forms a homotetrameric proton-selective channel in the viral envelope.13,14 The recognized function of A/M2 includes acidifying the viral interior after endocytosis, thereby initiating viral uncoating. In certain strains of influenza A viruses, A/M2 also functions to equilibrate the pH across the lumen of the late Golgi apparatus, thus preventing premature conformational change of the viral fusogenic protein–hemagglutinin.15
Interest in understanding the proton conductance and drug inhibition mechanism of A/M2 has been strongly motivated by its involvement in influenza virus infections.14 A/M2 was first discovered as the protein target of the anti-influenza drug amantadine.16,17 However, the use of amantadine was discontinued due to the prevalence of drug-resistant mutants.18 Among the large number of drug-resistant mutants identified in cell culture and amantadine-treated patients, only three major mutants, namely V27A, L26F, and S31N, have been found in transmissible viruses.19,20 The stringency of sequence conservation in M2 reflects tight functional constraints on the pore-lining residues, where a single mutation to a monomer in M2 causes four changes within the highly constricted pore.13 This small set of transmissible mutants suggests that M2 is a highly conserved drug target compared with other viral proteins, rendering it an ideal drug target for the development of anti-influenza drugs.21 However, drug discovery targeting M2 has been hampered by the lack of a reliable high-throughput screening assay and high-resolution structures. Nevertheless, guided by information gathered from molecular dynamics simulations,22,23 X-ray crystallography,24,25 and solution- and solid-state NMR spectroscopy,22,26,27 recent years have witnessed significant progress in this area, and several classes of compounds have been shown to inhibit all three major drug resistant mutants: V27A, L26F, and S31N.22,27–31 Subsequently, through iterative cycles of modeling, medicinal chemistry, electrophysiological testing, and antiviral assaying, the potencies of several lead compounds were further improved to the point where their IC50 values against drug-resistant M2 mutants were better than that of amantadine in inhibiting the wild-type M2 channel (Figure 1). The general structure of an S31N inhibitor contains adamantyl-1-NH2+CH2−aryl.27,31 In this study, we systematically explored various hydrophobic scaffolds and substituted-adamantanes as replacements of adamantane and examined several other heterocycles as the aryl headgroup. This study resulted in several compounds with improved potency and selectivity.
Figure 1.

Chemical structures of inhibitors targeting the drug-resistant influenza A virus M2 proton channels, V27A, L26F, and S31N, and the SAR of S31N inhibitors explored in this study. IC50 values were determined in two-electrode voltage clamp assays. EC50 values were determined from plaque reduction assays. A/Udorn/72, A/Udorn/72-V27A, and A/WSN/33 influenza strains were used in plaque reduction assays for determining drug sensitivity to WT, V27A, and S31N inhibitors, respectively. N.T. = not tested.
RESULTS AND DISCUSSION
Chemistry
The synthetic routes to compounds with various hydrophobic scaffolds, 6a–c and 8a–d, are shown in Scheme 1. The synthesis of the adamantyl-benzyl series of compounds was carried out by reductive amination of amines with 2,4-dihydroxybenzaldehyde (Scheme 1A).31 The isoxazole series of compounds was synthesized by direct alkylation (Scheme 1B).27
Scheme 1.

Synthetic Routes to M2 Inhibitors Bearing Various Hydrophobic Scaffolds
Synthesis of compounds 14 with various substitutions at the 5 position of isoxazole is shown in Scheme 2A.32 The synthesis started with a condensation reaction of methyl ketone 9 and dimethyl oxalate using potassium tert-butoxide as the base (Scheme 2A). Subsequent cyclization with hydroxylamine hydrochloride under mild heating at 50 °C in CH3OH gave the isoxazole ester intermediate 11. Yields for these two steps range from 61% to 82%. The ester was next reduced to the alcohol 12 by addition of sodium borohydride and converted to bromide 13 by triphenylphosphine and carbon tetrabromide in dichloromethane. Overall yields for these two steps are 68–83%. Bromination using PPh3 and CBr4 significantly improved the yield compared to phosphorus tribromide.27 The final N-alkylation of amantadine was performed under refluxing condition in 2-propanol with cesium iodide as the catalyst and triethylamine as the base in yields ranging from 62% to 91%.
Scheme 2.

Synthetic Routes to Adamantane Derivatives Bearing an Isoxazole or Oxadiazole Headgroup
The synthesis of compound 18 bearing 1,2,4-oxadiazole is shown in Scheme 2B.33 Condensation of carboxylic acid 15 with 2-chloro-N-hydroxyethanimidamide gave the hydro-xylamine ester 16, which was cyclized under heating in DMF to give the oxadiazole methylene chloride 17. Subsequent N-alkylation gave oxadiazole analogues 18 with 30–65% overall yields. 1,2,4-Oxadiazoles (20a–c) with the adamantylamino methylene at the 5 position were synthesized by alkylation (Scheme 2C).
The synthesis of 1,3,4-oxadiazole and 1,3,4-thiadiazole compounds 26a,b is shown in Scheme 3.34 Treatment of the 2-thiophenecarboxylic acid hydrazide 21 with methyl oxalyl chloride gave methyl 2-oxo-2-(thiophen-2-ylformohydrazido)-acetate 22 in nearly quantitative yield. This intermediate 22 was cyclized upon treatment with p-toluenesulfonyl chloride (TsCl) or Lawesson’s reagent to give 1,3,4-oxadiazole ester 23a and 1,3,4-thiadiazole ester 23b, respectively.34 The yields for 23a and 23b were 80% and 75%, respectively. The esters were subsequently reduced to hydroxyl 24, followed by bromination and alkylation to give the final compounds 26a,b. Other 1,3,4-oxadiazole analogues 26c,d and thiazoles 26e–j were synthesized by reductive amination or alkylation using commercially available aldehydes and halides as shown in Scheme 1.27
Scheme 3.

Synthetic Route to Adamantane Derivatives Bearing 1,3,4-Oxadiazole or 1,3,4-Thiadiazole Headgroup
Compounds with substituted-adamantanes or adamantane analogues as the hydrophobic scaffolds were synthesized from a common precursor, 4-oxoadamantane-1-carboxylic acid (27) (Scheme 4A). One-pot Curtius rearrangement followed by treatment with tert-butyl alcohol gave tert-butyl N-(4-oxoadamantan-1-yl)carbamate (28) in 78% yield.35 Subsequent deprotection with hexafluoro-2-propanol (HFIP) under microwave irradiation gave 5-aminoadamantan-2-one (29). Alkylation of 29 with 3-(bromomethyl)-5-(thiophen-2-yl)-1,2-oxazole gave the key intermediate 30a in 75% yield. The ketone in 30a was next either reduced by NaBH4 or alkylated by MeMgBr, or oxidized by the in situ-generated Corey–Chaykovsky reagent36 to give 30b, 30c, and 30d, respectively.
Scheme 4.

Synthetic Route to M2-S31N Inhibitors Bearing Adamantane Analogues
The ring-expanded 4-oxatricyclo [4.3.1.13,8]undecan-1-amine (32) was synthesized by Baeyer-Villiger oxidation of 28 with m-chloroperoxybenzonic acid (mCPBA), followed by reduction with indium bromide and triethylsilane and deprotection with HFIP. Finally, installation of the 5-(thiophen-2-yl)-1,2-oxazole was achieved through alkylation using CsI and DIEA under reflux condition in 2-propanol to give 30e in 27% overall yield. Compounds 30f,g were synthesized through the same alkylation procedure starting from 3-aminoadamantan-1-ol (33). Compound 30h with 3,5-dihydroxyladamantane was synthesized from 3-bromo-1-adamantanecarboxylic acid (34). Oxidation of 34 using KMnO4 gave 3,5-dihydroxyl-1-adamantanecarboxylic acid (35). Subsequent Curtius rearrangement followed by treatment with tert-butyl alcohol gave 36. Next deprotection of 36 followed by alkylation afforded the final compound 30h. The overall yield was 17%.
STRUCTURE-ACTIVITY RELATIONSHIP (SAR) STUDY
Exploring the Hydrophobic Scaffold as a Replacement for Adamantane in S31N Inhibitors
Our previous studies of the S31N inhibitors revealed two classes of potent S31N inhibitors (4 and 5), both of which share the same general structure of adamantyl-1-NH2+CH2−aryl (Figure 1).27,31 The adamantyl-benzyl series of compounds, a representative example of which is compound 5, are active against both the WT and the S31N mutant of M2,31 while the adamantyl-isoxazole series, such as 4, show selective inhibitory against S31N.27 SAR studies of the adamantyl-benzyl series revealed that the −NH2+CH2− linker is essential for this activity.31 However, the adamantyl scaffold has not been explored. It has been shown that inhibitors with a wide variety of hydrophobic scaffolds, such as linear alkyl, monocyclic alkyl, bicyclic alkyl, and spiroalkyl, are potent channel blockers of the WT M2.28,37–41 Thus, we sought to explore various hydrophobic scaffolds in both series of S31N inhibitors and examine how this modification would affect their inhibition against both WT and S31N. The hydrophobic scaffolds include adamantane analogues such as ring-contracted adamantane (6a, 8b), ring-expanded adamantane (6b, 8c), and spiroamine (6c) (Table 1). All of the hydrophobic scaffolds examined (without aryl headgroup) are potent inhibitors against WT-M2.37
Table 1.
Exploring Various Hydrophobic Scaffolds in S31N a
| R | Compound ID | %S3 IN inhibition* | %WT inhibition* | |
|---|---|---|---|---|
| AMT (1) | 36 | 91 | ||
| 5 | 67 | 64 | ||
| 6a | 40 | 64 | ||
| 6b | 0 | 74 | ||
| 6c | 3 | 65 | ||
 |
8a | 79 | 5 | |
| 8b | 71 | 19 | ||
| 8c | 75 | 11 | ||
| 8d | 34 | 14 |
Values represent the mean of three independent measurements. We typically see no more than 5% variation in the percent inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. Data listed is percent inhibition when tested at 100 µM drug concentration. N.T. = not tested.
The synthesized compounds were first tested in two-electrode voltage clamp (TEVC) electrophysiological assays, in which M2 was expressed in Xenopus laevis oocytes and current conductance across the cell membrane was recorded in a whole cell format.29 All compounds were initially tested at 100 µM drug concentration against both WT and S31N. The antiviral activity of potent compounds with greater than 80% M2 channel inhibition at 100 µM in TEVC assays were further confirmed in plaque reduction assays. For the 2,4-dihydrox-ylbenzyl series of compounds, replacing the adamantane scaffold in 5 with noradamantane (6a), aza-adamantane (6b), or spiropiperidine substituent (6c) decreased the M2-S31N channel blockage (Table 1). However, the resulting compounds (6a–c) all displayed similar or higher inhibitory activity against WT-M2. Similar SAR was also observed for the isoxazole series of compounds: replacing the adamantane in 8a with other hydrophobic scaffolds ameliorated activity against S31N, and all compounds in this series (8a–d) remained inactive against WT. Insertion of a methylene linker between the adamantyl and ammonium gave 8d, which is significantly less active than the parent compound 8a in inhibiting the S31N channel. Collectively, adamantane (5 and 8a) and ring-expanded adamantane (8c) appeared to be the optimal hydrophobic scaffolds for S31N inhibition, while a wide variety of hydrophobic scaffolds are tolerated for WT-M2 inhibition. Noradamantane scaffold was also tolerated for S31N inhibition, although corresponding compounds (6a and 8b) were less active than their adamantane counterparts. A secondary amine group appeared to be essential for S31N inhibition, as compounds with a tertiary amine (6b and 6c) were completely inactive.
Exploring Hydrophobic Substitutions of Isoxazole and 1,2,4-Oxadiazole
Solution NMR structure of 4 bound to M2-S31N (19–49) shows that the distal thienyl ring of the drug forms hydrophobic interactions with V27 side-chain methyl groups (Figure 2),27 suggesting that other hydrophobic. alkane substituents can be similarly accommodated at the same position Table 2.
Figure 2.

Drug binding mode of the A/M2-S31N mutant. (a) Solution NMR structure of A/M2-S31N bound with the isoxazole inhibitor M2WJ332 (4) (PDB: 2LY0). (b) Representation of drug–protein interactions
Table 2.
Exploring Hydrophobic Substitutions of Isoxazole and 1,2,4-Oxadiazole b
| R | z | Compound ID | % S3 IN inhibition* | % WT inhibition* | % plaque formation at 1 uM (A/WSN/33)** |
|
|---|---|---|---|---|---|---|
| C | 14aa | 49/N.T. | 55 | N.T. | ||
| N | 18aa | 60/N.T. | 41 | N.T. | ||
| C | 14b | 81/49 | 16 | 73±7 | ||
| N | 18b | 77/19 | 19 | N.T. | ||
| C | 14ca | 85/65 | 19 | 42±3 | ||
| N | 18ca | 81/60 | 18 | 82±5 | ||
| C | 14d | 84/N.T. | 30 | 42±2 | ||
| C | 14e | 39/N.T. | 18 | N.T. | ||
| C | 14f | 80/55 | 18 | 66±2 | ||
| N | 18da | 70/N.T. | 10 | N.T. | ||
| N | 18e | 62/N.T. | 12 | N.T. | ||
| C | 14g | 70/N.T. | 14 | N.T. | ||
| C | 14h | 90/N.T. | 13 | 57±2 |
Percent inhibition in TEVC assays was previously reported.
Values represent the mean of three independent measurements. We typically see no more than 5% variation in the percent inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. All compounds were initially tested at 100 µM. Compounds that showed greater than 80% inhibition at 100 µM were further tested at 30 µM. The data are presented as percent inhibition at 100 µM/percent inhibition at 30 µM. N.T. = not tested.
Plaque reduction assays were performed against the M2-S31N containing A/WSN/33 virus in duplicates; percent of plaque formation is defined as the ratio of plaque numbers in the presence of compounds divided by the plaque numbers in the absence of compounds.
Replacing a small methyl substituent at the 5 position of isoxazole or 1,2,4-oxadiazole (14a and 18a) with a bulkier propyl (14b and 18b), cyclopropyl (14c and 18c), isopropyl (14f and 18d), cyclobutyl (14d), or 2-methyl-2-(methylsulfanyl)propyl (14g) group dramatically increased the activities against S31N. Compound 14e with a terminal methoxy group was much less active than its methylene analogue 14b, highlighting the importance of hydrophobic interactions with the V27 side chains in high-affinity binding of drugs. Increasing the size of the substituent from isopropyl (18d) to isobutyl (18e), however, led to a slight decrease in S31N inhibition. The most active compound against S31N in this series is 14h, showing 90% inhibition at 100 µM. In general, isoxazole-containing compounds are more active than 1,2,4-oxadiazole-containing analogues. It was also noticed that increasing the size of the substituents at the 5 position of isoxazole and oxadiazole decreased their inhibition against WT M2. The active compounds were further tested in plaque reduction assays against S31N-containing A/WNS/33 viruses, and two compounds, 14c and 14d, were found to decrease the plaque number more than 50% at 1 µM, indicating their EC50 values are less than 1 µM.
Exploring Substitutions in the Distal Aromatic Ring of Isoxazole and 1,2,4-Oxadiazole
It has been found that S31N inhibition is relatively insensitive to different regioisomers of the distal ring at the 5 position of isoxazole and 1,2,4-oxadiaole: compounds 14i and 18g showed activities similar to those of 4 and 18f, respectively. This finding suggests that the sulfur atom in the distal thienyl ring is not involved in specific interactions with the M2 channel, which is consistent with the solution NMR structure showing that the thienyl group of 4 forms hydrophobic interactions with the Val27 side-chain methyls.27 Compounds with a furanyl substitution, 14j and 18h, were slightly less active, which might due to decreased hydrophobicity (the clogP for 4 and 14j is 3.74 and 2.37, respectively, as calculated from ChemBioDraw 14). Compound 14k with a methoxyl at the 3 position of the distal thienyl ring was highly active against S31N, showing 87% inhibition at 30 µM. When a phenyl ring was placed at the 5 position of isoxazole and 1,2,4-oxadiazole, the S31N inhibition of the resulting compounds, 14l and 18i, was retained. Halogen substitution in the distal phenyl ring was also examined, and the S31N inhibition was found to be insensitive to these structural modifications. All chloro-substituted (14m and 18j), bromo-substituted (14n), and difluoro-substituted (14o and 14p) compounds showed potency similar to that of 14l and 18i. Monomethoxyl substitution at all three positions of the phenyl ring, ortho (18l), meta (14r), and para (14q and 18k), did not affect the S31N inhibition. Similarly, compounds with monomethylsulfanyl substitutions at the ortho (14s) and para (14a) of the distal phenyl ring retained potent inhibition against S31N. Double-methoxyl substitutions decreased the S31N inhibition of the isoxazole-containing compound 14t but not the 1,2,4-oxadiazole-containing compound 18m. Consistent with previous SAR conclusions, compounds with 1-(1-adamantylamino)methylene substitution were more active when placed at the 3 position of isoxazole and 1,2,4-oxadiazole than at the 5 position (20b versus 18l, 20c versus 14h).27 In plaque reduction assays, several compounds, 14k, 14l, and 14r, showed antiviral activity comparable or even better than that of the parent compound 4 in inhibiting A/WSN/33 virus replication Table 3.
Table 3.
Exploring Aromatic Substitutions of Isoxazole and 1,2,4-Oxadiazoleb
| R | Z | Compound ID | %S31N inhibition* |
%WT inhibition* |
% plaque formation at 1 uM(A/WSN/33)** |
|
|---|---|---|---|---|---|---|
| C | 4a | 90/63 | 11 | 32±2 | ||
| N | 18fa | 83/N.T. | 11 | 66±3 | ||
| C | 14i | 90/N.T. | 28 | 47±5 | ||
| N | 18g | 81/N.T. | 3 | 74±10 | ||
| C | 14j | 84/N.T. | 12 | 47±1 | ||
| N | 18h | 73/N.T. | 24 | N.T. | ||
| C | 14k | 90/87 | 27 | 39±3 | ||
| C | 141a | 91/N.T. | 20 | 26±3 | ||
| N | 18ia | 75/N.T. | 8 | N.T. | ||
| C | 14m | 87/65 | 14 | 84±6 | ||
| N | 181 | 86/72 | 16 | 88±1 | ||
| C | 14n | 77/N.T. | 12 | N.T. | ||
| C | 14o | 78/N.T. | 23 | N.T. | ||
| C | 14p | 85/68 | 8 | 56±7 | ||
| C | 14qa | 87/N.T. | 13 | 59±9 | ||
| N | 18ka | 86/N.T. | 18 | 92±7 | ||
| C | 14r | 87/73 | 12 | 39±1 | ||
| C | 14s | 79/N.T. | 8 | N.T. | ||
| C | 8a | 79/N.T. | 5 | N.T. | ||
| N | 181 | 90/77 | 7 | 66±3 | ||
| C | 14t | 62/N.T. | 10 | N.T. | ||
| N | 18m | 82/N.T. | 18 | 76±5 | ||
| N | 20a | 39/N.T. | 18 | N.T. | ||
| N | 20b | 58/N.T. | 13 | N.T. | ||
| C | 20c | 67/N.T. | 27 | N.T. |
Percent inhibition in TEVC assays was previously reported.
Values represent the mean of three independent measurements. We typically see no more than 5% variation in the percent inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. The data are presented as percent inhibition at 100 µM/percent inhibition at 30 µM. N.T. = not tested.
Plaque reduction assays were performed against the M2-S31N containing A/WSN/33 virus in duplicates. The percent of plaque formation is defined as the ratio of plaque numbers in the presence of compounds divided by the plaque numbers in the absence of compounds.
Exploring 1,3,4-Oxadiazole-, 1,3,4-Thiadiazole-, and Thiazole-Containing M2 Inhibitors
To investigate whether isoxazole and 1,2,4-oxadiazole can be replaced by other heterocycles, several 1,3,4-oxadiazole-containing compounds (26a, 26c, and 26d) were synthesized. All three 1,3,4-oxadiazole-containing compounds were less active than their isoxazole and 1,2,4-oxadiazole counterparts (26a versus 4 and 18f; 26c versus 14l and 18i; 26d versus 14q and 18k). Encouragingly, a 1,3,4-thiadiazole-containing compound, 26b, was similarly active as the isoxazole analogue (4) in both TEVC and plaque reduction assays. 26b also showed no apparent toxicity up to 200 µM. Compound 26e with a 1-(1-adamantylamino)methylene substitution at the 5 position of thiazole was also potent, showing 75% inhibition at 100 µM concentration. Replacement of the distal thienyl ring at the 2 position of thiazole in 26e with methyl (26g) and tert-butyl (26f) dramatically decreased S31N inhibition. Compounds containing another thiazole regioisomer with 1-(1-adamantylamino)methylene at the 4 position of thiazole were much less active. All compounds were found to have minimal inhibition against WT M2, except 26j (57% inhibition at 100 µM) Table 4.
Table 4.
Exploring 1,3,4-Oxadiazole, 1,3,4-Thiadiazole, and Thiazoleb
| R | Compound ID | %S31N inhibition* |
%WT inhibition* |
% plaque formation at 1 µM (A/WSN/33)** |
CC50 (µM) |
|
|---|---|---|---|---|---|---|
| 26a | 52/N.T. | 16 | N.T. | N.T. | ||
| 26b | 83/N.T. | 21 | 39±2 | >200 | ||
| 26c | 61/N.T. | 18 | N.T. | N.T. | ||
| 26d | 58/N.T. | 3 | N.T. | N.T. | ||
| 26e | 75/N.T. | 13 | N.T. | 113 | ||
| 26fa | 38/N.T. | 5 | N.T. | N.T. | ||
| 26ga | 28/N.T. | 22 | N.T. | N.T. | ||
| 26h | 37/N.T. | 7 | N.T. | N.T. | ||
| 26i | 24/N.T. | 0 | N.T. | N.T. | ||
| 26ja | 36/N.T. | 57 | N.T. | N.T. |
Percent inhibition in TEVC assays was previously reported.
Values represent the mean of three independent measurements. We typically see no more than 5% variation in the percent inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. The data are presented as percent inhibition at 100 µM/percent inhibition at 30 µM. N.T. = not tested.
Plaque reduction assays were performed against the M2-S31N containing A/WSN/33 virus in duplicates. The percent of plaque formation is defined as the ratio of plaque numbers in the presence of compounds divided by the plaque numbers in the absence of compounds
Exploring Adamantane Analogues as Potent M2 Inhibitors
Having identified the favorable aryl auxiliary substitutions on M2-S31N inhibitors, we decided to revisit the hydrophobic scaffolds. Our preliminary data suggested that adamantane is the optimal scaffold for S31N inhibition; we therefore focused on substituted adamantanes, such as 4-hydroxyadamantane (30b), 3-hydroxyadamantane (30f,g), 3,-5-dihydroxyadamantane (30h), 4-hydroxy-4-methyladamantane (30c), spiro[adamantane-2,2′-oxirane] (30d), and 4-oxatricyclo[4.3.1.13,8]undecane (30e). 5-(Thiophen-2-yl)-1,2-oxazole and 5-(3-methoxythiophen-2-yl)-1,2-oxazole were chosen as the aryl auxiliary substitutions, as they are among the most favorable aryl substitutions for S31N inhibition. Compared with the parent compound 4, all modifications except 30d retained potent channel blockage against M2-S31N, showing more than 70% inhibition at 100 µM. In plaque reduction assays, compounds with hydroxylation on the adamantane ring, 30b, 30f, and 30g, were similarly potent as the parent compound 4, all showing EC50s less than 1 µM (Figure 3). However, further increasing the hydrophilicity by adding an additional hydroxyl group led to a significant loss of activity (compound 30h versus compound 30f). Notably, hydroxylation of the adamantane ring greatly decreased the cellular cytotoxicity of the parent compound (compound 30f versus compound 4) Table 5.
Figure 3.

Antiviral activity of potent S31N inhibitors. Compounds are tested in duplicate with serial dilutions against A/WSN/33 (M2-S31N).
Table 5.
Exploring Adamantane Analogues as the Hydrophobic Scaffoldsa
| X,R | Compound ID | %S31N inhibition* |
% plaque formation at 1 µM** |
Plaque Reduction EC50 (µM)** |
CC50 (µM)*** |
clogPΔ | |
|---|---|---|---|---|---|---|---|
 |
 |
4 | 90 | 32±2 | 0.353 | 100 | 4.2 |
 |
30a | 78/N.T. | 31±1 | N.T. | N.T. | 2.6 | |
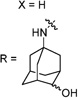 |
30b | 91/56 | 10±3 | 0.727 | 116 | 2.1 | |
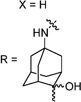 |
30c | 87/48 | 27±2 | N.T. | N.T. | 2.6 | |
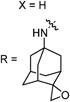 |
30d | 61/N.T. | 72±3 | N.T. | N.T. | 2.5 | |
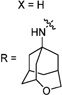 |
30e | 72/N.T. | 58±1 | N.T. | N.T. | 2.8 | |
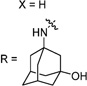 |
30f | 82/N.T. | 40±1 | 0.510 | >200 | 2.7 | |
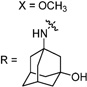 |
30g | 86/N.T. | 33±2 | 0.404 | N.T. | 2.2 | |
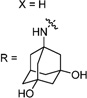 |
30h | 88/45 | 69±4 | 2.5 | >200 | 2.0 |
Values represent the mean of three independent measurements. We typically see no more than 5% variation in the percent inhibition on a given day, or 10% error for measurements made on different days with different batches of oocytes. The data are presented as percent inhibition at 100 µM/percent inhibition at 30 µM. N.T. = not tested.
Plaque reduction assays were performed against the M2-S31N containing A/WSN/33 virus in duplicates. The percent plaque formation is defined as the ratio of plaque numbers in the presence of compounds divided by the plaque numbers in the absence of compounds.
CC50 values were determined using MDCK cells in the MTT assay.
clogP was calculated in ChemBioDraw Ultra 14.0.
CONCLUSIONS
M2 belongs to a family of proteins called viroporins, which are virus-encoded ion channels.42 Viroporins are involved in diverse steps during viral replication, including viral entry, assembly, and release. They are made of small (normally fewer than 100 amino acids), hydrophobic peptides by self-oligomerization and reside in the viral or cellular compartment membranes. Representative examples of viroporins include the hepatitis C virus p7 ion channel, the HIV Vpu ion channel, M2 from the influenza virus, and the 6K proteins from alphavirus. Compounds that block cation flux through viroporins inhibit virus replication, corroborating viroporins as validated antiviral drug targets. The advantage of targeting viroporins compared to other viral proteins is that viroporins are relatively conserved, as one mutation causes multiple changes simultaneously in the constrained homo-oligomeric channel. For A/M2, only three predominant drug-resistant mutations have been identified in transmissible viruses, namely V27A, L26F, and S31N,19,20 all of which have similar levels of specific activity compared to the WT M2.43
Starting from the lead compounds that we identified earlier for S31N inhibition,27,31 we hereby disclose the SAR studies of the S31N inhibitor with the generic structure of adamantyl-1-NH2+CH2−aryl. The adamantane framework was found to be the optimal hydrophobic core, suggesting that it fits snuggly in the S31N channel. We next explored adding polar substitutions to the adamantane framework to increase the drug-like properties of the compounds. Monohydroxylation at the adamantane scaffold was well tolerated (compounds 30b, 30f, and 30g). Systematic substitutions of the heterocycle immediately attached to adamantane revealed that 1-(1-adamantylamino)methylene prefers to be placed at the 3 position of isoxazole and 1,3,4-oxadiazole,27 and the remaining variable 5 position favors hydrophobic substitutions, such as alkyl, thienyl, and substituted phenyl. A wide variety of substitutions are tolerated at the ortho-, meta-, and para-position of the distal ring when it is a phenyl group. Significantly, we have identified several compounds (26b and 30f) with a selectivity index (relative to toxicity toward mammalian cells) much higher than that of the parent compound 4. Collectively, the SAR agrees with the mode of inhibitor binding inside the S31N channel,27,44 in which the adamantane ring is surrounded by hydrophobic G34, the positively charged amine along with the heteroatom from the first aryl ring forms bidentate hydrogen bonds with the N31 side chain amide, and the distal aryl ring fits in the hydrophobic pocket formed by the V27 gate. The lead compounds identified here have improved physical properties and should provide excellent tools for in-depth pharmacological examinations.
EXPERIMENTAL SECTION
Chemistry
All chemicals are commercially available and were used without further purification. Detailed synthesis procedures can be found in the Supporting Information for reactions described in Schemes 1–4. All final compounds were purified by flash column chromatography, and the purity of all compounds tested was ≥95% as determined by LC-MS.
Two-Electrode Voltage Clamp (TEVC) Assay
The inhibitors were tested in a two-electrode voltage clamp assay using Xenopus laevis frog oocytes microinjected with RNA expressing either the WT or the S31N mutant of the A/M2 protein as previously reported.29 The potency of the inhibitors was expressed as percentage inhibition of A/M2 current observed after 2 min of incubation with either 100 or 30 µM of compounds.
Plaque Reduction Assay
Plaque reduction assay was performed as previously reported.27 The virus used for this assay was A/WSN/33, which contains the S31N mutant of M2.
Cytotoxicity Assay
The cytotoxicity of compounds was determined using the CellTiter-Glo Luminescent Cell Viability Assay kit from Promega. Briefly, Madin–Darby canine kidney (MDCK) cells in DMEM + 10% FBS + P/S medium at 50 000 cells/mL were dispensed into opaque 96-well plates (cat. no.: CLS3362) at 100 µL/well. Twenty-four hours later, the growth medium was replaced with fresh DMEM and compounds were added at 2-fold series dilution starting from 300 µM. After incubation for 72 h at 37 °C with 5% CO2 in a CO2 incubator, the plates were equilibrated to room temperature before 100 µL of the CellTiter-Glo solution was added. The plates were shaken for 5 s on a plate reader to allow even mixing. Luminescence signals were recorded on a multimode SpectraMax M5 plate reader, and the signals were normalized against the wells containing cells only. The CC50 values were calculated from best-fit dose-response curves with variable slope.
Supplementary Material
Acknowledgments
This research is supported by NIH Grant AI119187 and the PhRMA foundation 2015 research starter grant in pharmacology and toxicology to J.W., and NIH Grants GM056423 and AI74571 to W.F.D.
ABBREVIATIONS USED
- WT
wild type
- SAR
structure-activity relationship
- DMEM
Dulbecco’s modified eagle medium
- MDCK
Madin-Darby canine kidney
- TEV
two-electrode voltage clamps
- HPAI
highly pathogenic avian influenza
- DAA
direct-acting antiviral
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01910.
1H NMR, 13C NMR, and mass spectra of intermediates and final products (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Cox N, Subbarao K. Global epidemiology of influenza: Past and present. Annu. Rev. Med. 2000;51:407–421. doi: 10.1146/annurev.med.51.1.407. [DOI] [PubMed] [Google Scholar]
- 2.Thompson W, Shay D, Weintraub E, Brammer L, Cox N, Anderson L, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 3.Thompson W, Shay D, Weintraub E, Brammer I, Bridges C, Cox N, Fukuda K. Influenza-associated hospitalizations in the United States. JAMA. 2004;292:1333–1340. doi: 10.1001/jama.292.11.1333. [DOI] [PubMed] [Google Scholar]
- 4.Osterholm M, Kelley N, Sommer A, Belongia E. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012;12:36–44. doi: 10.1016/S1473-3099(11)70295-X. [DOI] [PubMed] [Google Scholar]
- 5.Lambert L, Fauci A. Influenza Vaccines for the Future. N. Engl. J. Med. 2010;363:2036–2044. doi: 10.1056/NEJMra1002842. [DOI] [PubMed] [Google Scholar]
- 6.Wong S, Webby R. Traditional and New Influenza Vaccines. Clin. Microbiol. Rev. 2013;26:476–492. doi: 10.1128/CMR.00097-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laursen NS, Wilson IA. Broadly neutralizing antibodies against influenza viruses. Antiviral Res. 2013;98:476–483. doi: 10.1016/j.antiviral.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pica N, Palese P, Caskey C. Toward a Universal Influenza Virus Vaccine: Prospects and Challenges. Annu. Rev. Med. 2013;64:189–202. doi: 10.1146/annurev-med-120611-145115. [DOI] [PubMed] [Google Scholar]
- 9.Jin H, Chen Z. Production of live attenuated influenza vaccines against seasonal and potential pandemic influenza viruses. Curr. Opin. Virol. 2014;6:34–39. doi: 10.1016/j.coviro.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Qi X, Qian YH, Bao CJ, Guo XL, Cui LB, Tang FY, Ji H, Huang Y, Cai PQ, Lu B, Xu K, Shi C, Zhu FC, Zhou MH, Wang H. Probable person to person transmission of novel avian influenza A (H7N9) virus in Eastern China, 2013: epidemio-logical investigation. BMJ [Br. Med. J.] 2013;347:f4752. doi: 10.1136/bmj.f4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Preziosi P. Influenza pharmacotherapy: present situation, strategies and hopes. Expert Opin. Pharmacother. 2011;12:1523–1549. doi: 10.1517/14656566.2011.566557. [DOI] [PubMed] [Google Scholar]
- 12.Webster RG, Govorkova EA. Continuing challenges in influenza. Ann. N. Y. Acad. Sci. 2014;1323:115–139. doi: 10.1111/nyas.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Qiu JX, Soto C, DeGrado WF. Structural and dynamic mechanisms for the function and inhibition of the M2 proton channel from influenza A virus. Curr. Opin. Struct. Biol. 2011;21:68–80. doi: 10.1016/j.sbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Clercq E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discovery. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong M, DeGrado WF. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 2012;21:1620–1633. doi: 10.1002/pro.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies WL, Grunert RR, Haff RF, McGahen JW, Neumayer EM, Paulshock M, Watts JC, Wood TR, Hermann EC, Hoffmann CE. Antiviral Activity of 1-Adamantanamine (Amantadine) Science. 1964;144:862–863. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- 17.Pinto LH, Holsinger LJ, Lamb RA. Influenza-Virus M2 Protein Has Ion Channel Activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 18.Dong G, Peng C, Luo J, Wang C, Han L, Wu B, Ji G, He H. Adamantane-Resistant Influenza A Viruses in the World (1902–2013): Frequency and Distribution of M2 Gene Mutations. PLoS One. 2015;10:e0119115. doi: 10.1371/journal.pone.0119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuse Y, Suzuki A, Kamigaki T, Oshitani H. Evolution of the M gene of the influenza A virus in different host species: Large-scale sequence analysis. Virol. J. 2009;6:67. doi: 10.1186/1743-422X-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furuse Y, Suzuki A, Oshitani H. Large-scale sequence analysis of M gene of influenza A viruses from different species: Mechanisms for emergence and spread of amantadine resistance. Antimicrob. Agents Chemother. 2009;53:4457–4463. doi: 10.1128/AAC.00650-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Li F, Ma C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Biopolymers. 2015;104:291–309. doi: 10.1002/bip.22623. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Ma C, Fiorin G, Carnevale V, Wang T, Hu F, Lamb RA, Pinto LH, Hong M, Klein ML, DeGrado WF. Molecular Dynamics Simulation Directed Rational Design of Inhibitors Targeting Drug-Resistant Mutants of Influenza A Virus M2. J. Am. Chem. Soc. 2011;133:12834–12841. doi: 10.1021/ja204969m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khurana E, Dal Peraro M, DeVane R, Vemparala S, DeGrado W, Klein M. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acharya R, Carnevale V, Fiorin G, Levine BG, Polishchuk AL, Balannik V, Samish I, Lamb RA, Pinto LH, DeGrado WF, Klein ML. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2010;107:15075–15080. doi: 10.1073/pnas.1007071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Cady SD, Balannik V, Pinto LH, DeGrado WF, Hong M. Discovery of Spiro-Piperidine Inhibitors and Their Modulation of the Dynamics of the M2 Proton Channel from Influenza A Virus. J. Am. Chem. Soc. 2009;131:8066–8076. doi: 10.1021/ja900063s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 2013;110:1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Ma C, Wu Y, Lamb RA, Pinto LH, DeGrado WF. Exploring organosilane amines as potent inhibitors and structural probes of influenza a virus M2 proton channel. J. Am. Chem. Soc. 2011;133:13844–13847. doi: 10.1021/ja2050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balannik V, Wang J, Ohigashi Y, Jing X, Magavern E, Lamb RA, Degrado WF, Pinto LH. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry. 2009;48:11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Canturk B, Jo H, Ma C, Gianti E, Klein ML, Pinto LH, Lamb RA, Fiorin G, Wang J, DeGrado WF. Flipping in the Pore: Discovery of Dual Inhibitors That Bind in Different Orientations to the Wild-Type versus the Amantadine-Resistant S31N Mutant of the Influenza A Virus M2 Proton Channel. J. Am. Chem. Soc. 2014;136:17987–17995. doi: 10.1021/ja508461m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Ma C, Wang J, Jo H, Canturk B, Fiorin G, Pinto LH, Lamb RA, Klein ML, DeGrado WF. Discovery of Novel Dual Inhibitors of the Wild-Type and the Most Prevalent Drug-Resistant Mutant, S31N, of the M2 Proton Channel from Influenza A Virus. J. Med. Chem. 2013;56:2804–2812. doi: 10.1021/jm301538e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider JW, Gao Z, Li S, Farooqi M, Tang TS, Bezprozvanny I, Frantz DE, Hsieh J. Small-molecule activation of neuronal cell fate. Nat. Chem. Biol. 2008;4:408–410. doi: 10.1038/nchembio.95. [DOI] [PubMed] [Google Scholar]
- 33.McBriar MD, Clader JW, Chu I, Del Vecchio RA, Favreau L, Greenlee WJ, Hyde LA, Nomeir AA, Parker EM, Pissarnitski DA, Song LX, Zhang LL, Zhao ZQ. Discovery of amide and heteroaryl isosteres as carbamate replacements in a series of orally active gamma-secretase inhibitors. Bioorg. Med. Chem. Lett. 2008;18:215–219. doi: 10.1016/j.bmcl.2007.10.092. [DOI] [PubMed] [Google Scholar]
- 34.Garfunkle J, Ezzili C, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. Optimization of the central heterocycle of alpha-ketoheterocycle inhibitors of fatty acid amide hydrolase. J. Med. Chem. 2008;51:4392–4403. doi: 10.1021/jm800136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corey EJ, Chaykovsky M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2), Dimethylsulfonium Methylide ((CH3)2SCH2)Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965;87:1353–1364. [Google Scholar]
- 37.Wang J, Ma C, Balannik V, Pinto LH, Lamb RA, Degrado WF. Exploring the Requirements for the Hydrophobic Scaffold and Polar Amine in inhibitors of M2 from Influenza A Virus. ACS Med. Chem. Lett. 2011;2:307–312. doi: 10.1021/ml100297w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duque MD, Ma C, Torres E, Wang J, Naesens L, Juarez-Jimenez J, Camps P, Luque FJ, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J. Med. Chem. 2011;54:2646–2657. doi: 10.1021/jm101334y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X, Jie Y, Rosenberg MR, Wan J, Zeng S, Cui W, Xiao Y, Li Z, Tu Z, Casarotto MG, Hu W. Design and synthesis of pinanamine derivatives as anti-influenza A M2 ion channel inhibitors. Antiviral Res. 2012;96:91–99. doi: 10.1016/j.antiviral.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Zhao X, Li C, Zeng S, Hu W. Discovery of highly potent agents against influenza A virus. Eur. J. Med. Chem. 2011;46:52–57. doi: 10.1016/j.ejmech.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 41.Hu WH, Zeng SG, Li CF, Jie YL, Li ZY, Chen L. Identification of Hits as Matrix-2 Protein Inhibitors through the Focused Screening of a Small Primary Amine Library. J. Med. Chem. 2010;53:3831–3834. doi: 10.1021/jm901664a. [DOI] [PubMed] [Google Scholar]
- 42.Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat. Rev. Microbiol. 2012;10:563–574. doi: 10.1038/nrmicro2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balannik V, Carnevale V, Fiorin G, Levine B, Lamb R, Klein M, DeGrado W, Pinto L. Functional Studies and Modeling of Pore-Lining Residue Mutants of the Influenza A Virus M2 Ion Channel. Biochemistry. 2010;49:696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams JK, Tietze D, Wang J, Wu Y, DeGrado WF, Hong M. Drug-Induced Conformational and Dynamical Changes of the S31N Mutant of the Influenza M2 Proton Channel Investigated by Solid-State NMR. J. Am. Chem. Soc. 2013;135:9885–9897. doi: 10.1021/ja4041412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.