

Abstract
The objective of this study was to design GE11 peptide (YHWYGYTPQNVI) linked micelles of poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate-graft-gemcitabine-graft-dodecanol (PEG-b-PCC-g-GEM-g-DC) for enhanced stability and target specificity of gemcitabine (GEM) to EGFR-positive pancreatic cancer cells. GE11-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC mixed micelles showed EGFR-dependent enhanced cellular uptake, and cytotoxicity as compared to scrambled peptide HW12-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC mixed micelles and unmodified mPEG-b-PCC-g-GEM-g-DC micelles. Importantly, GE11-linked mixed micelles preferentially accumulated in orthotopic pancreatic tumor and tumor vasculature at 24 h post systemic administration. GE11-linked mixed micelles inhibited orthotopic pancreatic tumor growth compared to HW12-linked mixed micelles, unmodified mPEG-b-PCC-g-GEM-g-DC micelles, and free GEM formulations. Tumor growth inhibition was mediated by apoptosis of tumor cells and endothelial cells as determined by immunohistochemical staining. In summary, GE11-linked mixed micelles is a promising approach to treat EGFR overexpressing cancers.
Graphical abstract
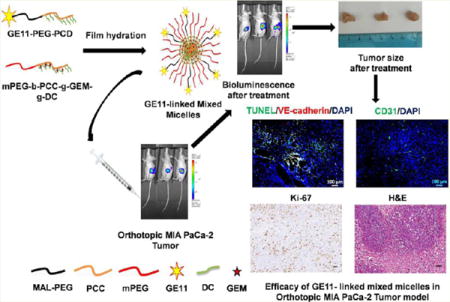
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the United States due to poor prognosis and high metastasis.1–3 Conventional chemotherapy often fails to treat pancreatic cancer and often causes nonspecific toxicity to healthy tissues. Gemcitabine (GEM), a nucleoside analogue, is the first line chemotherapy to treat PDAC with a mere 6.8 month survival rate, and up to 11.1 months as combination therapy.4–6 GEM exhibits its anticancer activity through blocking DNA replication and inducing S-phase cell cycle arrest. However, due to its rapid metabolism into inactive form 2′-deoxy-2′,2′-difluorodeoxyuridine (dFdU) and nonspecific toxicity reduces its therapeutic potential.7,8
Various drug delivery systems like liposomes, nanoparticles and micelles fail to improve GEM plasma stability due to poor drug loading and premature release.9–13 Chemical conjugation of GEM to lipid or polymeric carriers has been shown to improve its loading but has lower aqueous solubility.14,15 To overcome these challenges, squalenoyl derivatives of GEM were developed; however, their efficacy was limited due to the rapid uptake by the reticuloendothelial system (RES).16 Since the incorporation of polyethylene glycol (PEG) onto polymeric systems provides stealth properties and prolonged circulation, we recently synthesized methoxy poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate-graft-gemcitabine-graft-dodecanol (mPEG-b-PCC-g-GEM-g-DC), where PEG corona ensures stealth properties and the presence of carboxyl groups in polymeric backbone enables higher GEM loading. This polymeric system self-assembled into micelles and inhibited subcutaneous xenograft tumor after intravenous administration.17 However, passive targeting is effective only in highly vascularized and rapidly growing tumors, but it is not effective in poorly vascularized and metastatic tumors.18 Since tumor progression, invasion, and metastasis critically depend on endothelial cell-derived angiogenesis, selective delivery of anticancer agents to the tumor endothelial cells is an attractive approach.19,20 To date, very few targeted nanoparticles are reported for drug delivery to the tumor and its vasculature for pancreatic cancer treatment.21
Epidermal growth factor receptor (EGFR) is overexpressed in the pancreatic tumor.22 Full length EGFR natural ligands, such as EGF and monoclonal antibody cetuximab conjugated nanoparticles, have been shown to enhance the receptor-mediated endocytosis of GEM by pancreatic tumor cells.23 However, high mitogenic and pro-angiogenic capability of full length EGFR ligands retard their clinical translation. Alter natively, GE11 peptide (YHWYGYTPQNVI) ligand has been shown its specific binding to EGFR with low mitogenic activity.24 Moreover, GE11 peptide-decorated delivery systems efficiently deliver drugs/genes to EGFR overexpressing cancer cells.25–27
In this study, we conjugated GE11 peptide to the surface of malemido-poly(ethylene glycol)-block-poly(2-methyl-2-carboxyl-propylene carbonate-graft-dodecanol (GE11-PEG-PCD) and synthesized methoxy-poly(ethylene glycol)-block-poly(2-methyl-2-carboxylpropylene carbonate-graft-gemcitabine-graft-dodecanol (mPEG-b-PCC-g-GEM-g-DC) to prepare mixed micelles for enhanced GEM delivery to EGFR expressing pancreatic cancer cells. We showed that GE11 linked mixed micelles exhibited a better therapeutic effect than nontargeted mixed micelles in vitro and in MIA PaCa-2 cells derived orthotopic pancreatic tumor bearing mice.
2. EXPERIMENTAL SECTION
2.1. Materials
N-terminal cysteine containing GE11 peptide was custom synthesized by Shanghai Hanhong Chemical Co. Ltd. (Shanghai, China). Its purity and structure were confirmed by HPLC (purity 98.36%) and 1H NMR. N-terminal cysteine containing CHYPYAHPTHPSW (designated as HW12) peptide was custom synthesized by Biomatik USA, LLC (Delaware, USA). Its purity and structure were confirmed by HPLC (purity 97.58%) and 1H NMR, respectively. Malemide PEG hydroxyl (MW 5000) was purchased from JenKem Technology (Beijing, China). Gemcitabine hydrochloride was purchased from AK Scientific (Union city, CA). Dodecanol (DC), triethylamine (TEA), 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC), hydroxybenzotriazole (HOBT), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), benzyl bromide, 2,2-bis(hydroxymethyl) propionic acid, methoxy poly(ethylene glycol) (mPEG, Mn = 5000, PDI = 1.03), puromycin dihydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). Matrigel matrix basement membrane was procured from Corning (Chicago, IL). Fluorescein cadaverine (FC) dihydrobromide salt was purchased from Life Technology (Chicago, IL). Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) was purchased from Biosynth Chemistry & Biology (Itasca, IL). Polybrene and VE-cadherin (H-72) were purchased from Santa Cruz Biotechnology (Dallas, TX). Rat anti-mouse CD31 was purchased from BD Biosciences (San Diego, CA). Lentiviral particle LP-hLUC-Lv201-0200 was purchased from GeneCopoeia (Rockville, MD). Dead End Fluorometric TUNEL System was purchased from Promega Corporation (Madison, WI). Mouse monoclonal antibodies to EGFR (Phycoerythrin) and mouse IgG1 monoclonal antibodies (Phycoerythrin)-Isotype control were purchased from Abchem (Cambridge, MA). Alexa Fluor 594 F(ab′)2 fragment of goat antirabbit IgG (H+L) was purchased from Invitrogen (Grand Island, NY). All other chemicals were of analytical grade and purchased from Sigma-Aldrich (St. Louis, MO).
2.2. Synthesis of Polymers
2.2.1. Synthesis of Maleimido Poly(ethylene glycol)-block-Poly(2-methyl-2-dodecanoxycarbonylpropylene carbonate) (MAL-PEG-PCD)
2-Methyl-2-benzyloxycarbonylpropylene carbonate (MBC) was synthesized and converted to 2-methyl-2-carboxylpropylene carbonate (MCC) by hydrogenation reaction as reported by Li et al.28 DC was conjugated to the carboxyl group of MCC by carbodiimide chemistry. Briefly, MTC-OH (480 mg) was dissolved in 20 mL of DMF followed by the addition of HOBT (506 mg), EDC (720 mg) and 490 μL of TEA. After 1 h, DC (465 mg) was added to the reaction mixture and allowed to react for 18 h under N2 atmosphere at room temperature (RT). Then, ethyl acetate (20 mL) was added to the reaction mixture, and the organic layer was washed with distilled water, and organic solvent dried over Na2SO4. The organic solvent was evaporated under reduced pressure to yield 2-methyl-2-dodecanoxycarbonylpropylene carbonate (MDC), which was purified by column chromatography.
2.2.2. Synthesis of MAL-PEG-PCD from Copolymerization of MAL-PEG-OH with MDC
MAL-PEG-OH and MDC were dissolved in 15 mL anhydrous CH2Cl2 and stirred under N2 atmosphere at RT. Then, DBU (40 μL) was dissolved in 1 mL anhydrous CH2Cl2 and added to the mixture and left to react under stirring for 3 h. Benzoic acid was added to the reaction mixture at the end of the reaction to neutralize excess DBU, and solvent was removed under reduced pressure. Then, the crude product was dissolved with the minimum amount of CHCl3 and precipitated twice with cold isopropyl alcohol, followed by diethyl ether. The precipitate was dried under high vacuum at RT to obtain purified MAL-PEG-PCD lipopolymer.
2.2.3. Conjugation of GE11 and HW12 Peptide to MAL-PEG-PCD
MAL-PEG-PCD (20 mg) was dissolved in a mixture of DMSO:distilled water (1:1 v/v, 4 mL) and stirred under N2 at RT. GE11 peptide (6.57 mg) and tris(2-carboxyethyl) phosphine hydrochloride (TECP, 100 mM, 100 μL) were dissolved in 1 mL DMSO and added to the reaction. Then, the reaction mixture was left for 24 h at RT under N2 atmosphere with gentle stirring. Finally, the reaction mixture was dialyzed against phosphate buffer saline using 3000 MWCO dialysis tubing to remove unreacted GE11 peptide, and after dialysis, lyophilization was performed to obtain pure GE11-PEG-PCD. The chemical conjugation of GE11 peptide in MAL-PEG-PCD lipopolymer was finally confirmed by 1H NMR analysis. We synthesized HW12-PEG-PCD polymer using the same synthetic route as the synthesis of GE11-PEG-PCD, and finally this polymer was confirmed by 1H NMR analysis.
2.2.4. Synthesis of mPEG-b-PCC-graft-Fluorescein Cadaverine-graft-Dodecanol (mPEG-b-PCC-g-FC-g-DC)
mPEG-PCC was synthesized using our previously reported method.17 For FC and DC conjugation, mPEG-PCC (100 mg), HOBT (61 mg) and EDC (77 mg) were dissolved in 6 mL DMF and allowed for stirring for 2 h at RT under N2 atmosphere. FC (13 mg) and DC (56 mg) were added to the reaction mixture and followed by the addition of 120 μL DIPEA. The reaction was allowed to continue for 48 h at RT under N2 atmosphere. The crude product was precipitated twice with an excess of cold isopropyl alcohol and then dissolved in chloroform and precipitated in cold diethyl ether. Finally, the precipitate was dissolved in acetone and dialyzed against water using 2000 MWCO dialysis tubing and after dialysis and lyophilized to yield mPEG-b-PCC-g-FC-g-DC.
2.2.5. Synthesis of GEM and DC Conjugated mPEG-PCC-g-GEM-g-DC copolymer
mPEG-PCC (300 mg), HOBT (223 mg) and EDC (317 mg) were dissolved in 12 mL DMF and stirred for 2 h, and then GEM and DC were added to the reaction mixture, followed by addition of 360 μL N,N-diisopropylethylamine (DIPEA). The reaction was left for 48 h at RT under N2 atmosphere. The crude product was precipitated twice with excess of cold isopropyl alcohol, dissolved in chloroform and precipitated again in the cold diethyl ether. Finally, the precipitate was dissolved in acetone and dialyzed against water using 2000 MWCO dialysis tubing and lyophilized to yield mPEG-b-PCC-g-GEM-g-DC.
2.3. Quantification of GEM Payload in mPEG-b-PCC-g-GEM-g-DC Copolymer
The amount of GEM conjugated to the copolymer was determined by the alkaline hydrolysis method as described previously.17 Briefly, 10 mg of copolymer micelles were subjected to alkaline hydrolysis in NaOH (1 N) at 40 °C for 1 h, and the samples were subjected to HPLC-UV analysis using the HPLC system equipped with photodiode array detector 996 (Waters Corporation); data acquisition and processing were performed with Empower Pro software. A 20 μL volume of the standard and sample was injected into Inertsil ODS 3 column (4.5 × 250 mm). Analytes were eluted isocratically at the flow rate of 1 mL/min with a mobile phase of sodium acetate buffer (20 mM, pH 5.5):methanol (93:07 v/v), and the column temperature was 40 °C.
2.4. Preparation of GE11- or HW12-Linked Mixed Micelles
GE11-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC and HW12-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC mixed micelles were prepared by thin film hydration as reported previously with slight modfication.29,30 Briefly, GE11-PEG-PCD (10–30% weight ratio) and mPEG-b-PCC-g-GEM-g-DC (70–90% weight ratio) were dissolved in 1 mL CHCl3 in a glass vial. The solvent was evaporated under reduced pressure to form thin film and dried film was kept under vacuum overnight to remove residual CHCl3. One milliliter of PBS (pH 7.4) was added to the vacuum-dried film and vortexed for 5 min at RT. Then the resulting solution was centrifuged at 5000g for 5 min and filtrated through a 0.22 μ filter membrane to obtain the micelles. FC-labeled GE11-linked mixed micelles were prepared by keeping FC concentration constant (100 μM) and mixing GE11-PEG-PCD (10–30% weight ratio) with mPEG-PCD/mPEG-b-PCC-g-FC-g-DC (70–90 w/w). Similarly, HW12-linked mixed micelles were prepared by mixing HW12-PEG-PCD and mPEG-PCD/mPEG-b-PCC-g-FC-g-DC (70/30 w/w).
2.5. Particle Size and Zeta (ζ) Potential Measurement
Particle size distribution and surface zeta (ζ) potential of GE11-PEGPCD/mPEG-G-b-PCC-g-GEM-g-DC and HW12-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC mixed micelles (10 mg/mL) were determined by photon correlation spectroscopy at a scattering angle of 173° (Malvern Zetasizer).
2.6. Stable Transfection of MIA PaCa-2 Cells with Lentiviral Particles
MIA PaCa-2 cells were kindly gifted by Dr. Rakesh Singh (UNMC, Omaha, NE) and maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% antibiotic in an incubator at 37 °C/5% CO2. MIA PaCa-2 cells were stably transfected with lentiviral vector encoding luciferase and green fluorescent protein (GFP) (hLUC-Lv201-0200) using Polybrene according to the manufacturer’s instructions. Briefly, 2 × 105 MIA PaCa-2 cells were seeded in each well in a six-well plate for 24 h. For each well, 2 μL of lentiviral particles was mixed with 1 μL of Polybrene (10 mg/mL), and the final volume was made to 2 mL using DMEM. The transfection mixture was then incubated for 15 min at RT with occasional agitating and MIA PaCa-2 cells were washed with PBS and were incubated with the transfection mixture overnight. The medium was replaced with 2 mL fresh DMEM for an additional 48 h. The cells were trypsinized and cultured in T-75 flasks for 15 days in 10 mL DMEM containing 2 μg/mL puromycin. Every alternative day during these 15 days, the medium was removed with a fresh DMEM containing 2 μg/mL puromycin. MIA PaCa-2 cells were then grown for another 4 days with fresh DMEM containing a lower concentration of puromycin (1 μg/mL). Luciferase and GFP expression from stably transfected cells were confirmed by in vivo IVIS and epifluorescence microscopy, respectively. Finally, cells were sorted to obtain luciferase and a GFP reporter gene-expressing cell population using fluorescence-activated cell sorting (FACS). In vitro and in vivo experiments were performed with stable luciferase and GFP-transfected cells culturing in DMEM without puromycin.
2.7. Cellular Uptake Studies
Cellular uptake study was carried out in MIA PaCa-2 cells using FC-labeled mixed micelles. Briefly, 2 × 105 cells were seeded per well in six-well plates for 24 h. GE11-PEG-PCD/mPEG-b-PCC-g-FC-g-DC, HW12-PEG-PCD/mPEG-b-PCC-g-FC-g-DC mixed micelles and mPEG-b-PCC-g-FC-g-DC unmodified micelles were added to each well. After 12 h, the cells were washed with PBS and observed under an epifluorescence microscope. Further, for quantitative estimation of FC dye in the cellular uptake study, cells were washed with PBS, trypsinized, again washed with cold PBS, and centrifuged. Then, the cell pellet was resuspended with PBS containing 4% paraformaldehyde and kept for 10 min at RT. Cells were centrifuged, washed with PBS, resuspended with 1 mL of PBS, and analyzed by a flow cytometer (BD LSRFortessa X-20). For competition studies, excess of free GE11 peptide was diluted with medium and added to cells 2 h prior to the addition of GE11-PEG-PCD/mPEG-b-PCC-g-FC-g-DC mixed micelles. After 12 h, the medium was discarded, and cells were washed with PBS (pH 7.4) and observed under an epifluorescence microscope (Zeiss, Germany).
2.8. Cytotoxicity Study
Cytotoxicity of GE11-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC, HW12-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC mixed micelles, mPEG-b-PCC-g-GEM-g-DC unmodified micelles, and free GEM were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MIA PaCa-2 cells were seeded at a density of ~5000 cells per well in 96-well plates for 18 h and were treated with GE11-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC, HW12-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC mixed micelles, mPEG-b-PCC-g-GEM-g-DC unmodified micelles, and free GEM having 60–500 nM concentration of GEM. The final volume in each well was adjusted to 200 μL using complete media and left for 72 h, and 10 μL of MTT solution (5 mg/mL in PBS) was added to each well and incubated for 4 h. The formazan crystals were solubilized with 200 μL of DMSO, and the absorbance in each well was measured with a microplate reader at 560 nm wavelength; background correction was done by subtracting absorbance at 655 nm. Results were calculated as percent cell viability = [A560 (treated cells) – background/A560 (untreated cells) – background] × 100.
2.9. In Vivo Distribution and Targeting Study
All animal experiments were performed in accordance with the NIH animal use guidelines and protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska Medical Center, Omaha, NE. The orthotopic pancreatic cancer mouse model was generated by implantation of MIA PaCa-2 cells using PBS:matrigel (1:1 v/v) into the pancreas of 6–8 week-old athymic nude mice (n = 3). GE11-PEG-PCD/mPEG-b-PCC-g-FC-g-DC, HW12-PEG-PCD/mPEG-b-PCC-g-FC-g-DC mixed micelles and mPEG-b-PCC-g-FC-g-DC unmodified micelles were injected into mice via the tail vein. Mice were sacrificed 24 h post injection of mixed micellar formulations, and tumors were harvested. Five micromolar tumor cryosections were made using a cryostat instrument (Leica) and fixed with 10% formaldehyde in PBS, and the fixed tumor sections were washed with PBS and visualized under a fluorescent microscope. For tumor vasculature targeting study, the same tissue sections were again fixed in 4% paraformaldehyde in PBS and stained for rabbit polyclonal anti-VE-cadherin antibody (endothelial cells, red) (1:50 in 0.5% bovine serum albumin in PBS). Then, sections were counterstained with Alexa Fluor 594 F(ab′)2 fragment of goat antirabbit IgG (H+L) secondary antibody (10 μg/mL in PBS). Finally, sections were mounted with SlowFade Gold antifade reagent with DAPI, and snaps were taken under a inverted fluorescence microscope.
2.10. Efficacy of Mixed Micelle Formulations in Orthotopic Pancreatic Tumor Bearing Mice
After 12 days post implantation of luciferase and GFP stably expressing 3 × 106 MIA PaCa-2 into athymic nude mice, 100 mg/kg of D-luciferin was administered into each animal via intraperitoneal injection, and bioluminescent was measured using the IVIS Imaging System (Caliper Life Sciences). These tumor-bearing mice were then randomly sorted into five groups (n = 5): (1) vehicle control i.e., normal saline, (2) GE-linked mixed micelles, (3) HW12-linked mixed micelles, (4) mPEG-b-PCC-g-GEM-g-DC unmodified micelles, and (5) free GEM at an equivalent dose of 40 mg/kg GEM. Treatment was started at days 12 via intravenous administration once a week for 3 weeks. Bioluminescent was measured at day 12, 16, 20, 24, 26, and 30. Mouse body weight was measured at day 12, 15, 18, 21, 24, 27, and 30. After final bioluminescent measurement, mice were sacrificed, and tumors were excised. Then excised tumors were frozen in a cryostat at −30 °C for 2 h, cryosections were fixed with 10% formaldehyde in PBS. Fixed tumor sections were washed with PBS and immunostained for cell proliferation marker (Ki-67), apoptosis (cleaved caspase 3) and hematoxylin and eosin (H&E). To detect tumor cells as well as tumor vasculature apoptosis induced by GEM treatment, orthotopic MIA PaCa-2 bearing mice (n = 3) were intravenously administered with GE11-linked mixed micelles, HW12-linked mixed micelles, mPEG-b-PCC-g-GEM-g-DC unmodified micelles, and free GEM once a week for 3 weeks post tumor inoculation. Mice were sacrificed 72 h post last injection, tumors excised. Tumors were cryosectioned, and fixed cryosections were immunostained with a TUNEL assay kit according to the manufacturer’s instructions and counterstained with DAPI and imaged under a fluorescence microscope. To measure the tumor endothelial microvessels density, tumor sections were immunostained with CD31 primary antibody.
2.11. Statistical Analysis
Data are represented as means ± SEM. The statistical comparisons of the experiments were performed by two-tailed Student’s test. P < 0.05 was considered statistically significant.
3. RESULTS AND DISCUSSION
Bioconjugation to a polymeric carrier is an attractive strategy to enhance the in vivo stability and delivery of GEM to the tumor. Several PEGylated conjugates, lipid conjugates, and squalenoyl derivatives of GEM have been demonstrated to enhance its bioavailability.14–16 However, the clinical translation of these delivery systems is limited by poor solubility, uptake by RES, and lower GEM payload. We synthesized mPEG–PCC copolymer having several carboxyl pendant groups for conjugating GEM.17 This copolymer could self-assemble into micelles and significantly inhibited subcutaneous MIA PaCa-2 cells implanted in a xenograft tumor after systemic administration. In the present study, we used GE11 peptide as a targeting ligand that efficiently binds to EGFR and has low mitogenic activity.24 We synthesized GE11-PEG-PCD using GE11 peptide, Mal-PEG-PCD, and TCEP (Figure 1A). To confirm GE11 and not HYPYAHPTHPSW (designated as HW12) is an EGFR ligand, HW12-PEG-PCD was synthesized using the same synthetic route as mentioned for synthesizing GE11-PEG-PCD (Figure 1B). We also synthesized mPEG-b-PCC-g-FC-g-DC and mPEG-b-PCC-g-GEM-g-DC copolymers (Figure 2A,B). It is important to note that the two components of this copolymer are a biocompatible PEG block and a biodegradable polycarbonate (PCC) block. Polycarbonates are biodegradable and have low toxicity, as their degradation products are carbon dioxide and alcohol, which are less acidic, have less effect on microenvironment pH, and thus will not result in local inflammation.31
Figure 1.
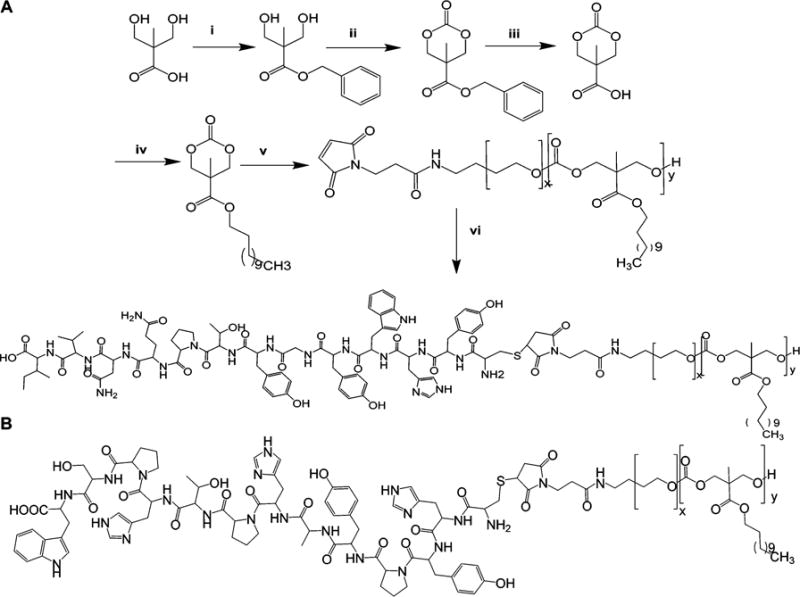
Synthesis scheme of GE11-PEG-PCD (A), and chemical structure of HW12-PEG-PCD (B). Reagents: (i) KOH, DMF, benzyl bromide, 100 °C, 15 h; (ii) triphosgene, pyridine, CH2Cl2, −78 to 0 °C; (iii) 10% Pd/C, H2, ethyl acetate, RT, 40 psi, 3 h; (iv) EDC, HOBT, TEA, DMF, dodecanol, RT, 18 h; (v) DBU, CH2Cl2, MAL-PEG5000-OH, RT, 3 h.; (vi) MAL-PEG-PCD, DMSO: distilled water (1:1 v/v), GE11 peptide (CYHWYGYTPQNVI), TECP (100 mM), RT, N2, 24 h.
Figure 2.
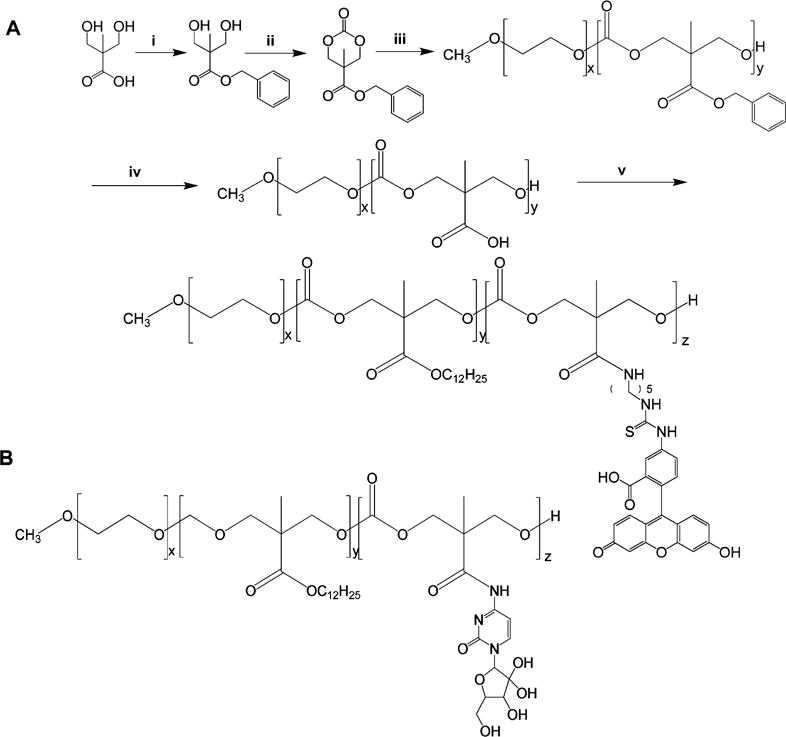
Synthesis scheme of mPEG-PCC-g-FC-g-DC (A); chemical structure of mPEG-PCC-g-GEM-g-DC (B). Reagents: (i) KOH, DMF, benzyl bromide, 100 °C, 15 h; (ii) triphosgene, pyridine, CH2Cl2, −78 to 0 °C; (iii) DBU, CH2Cl2, methoxy-PEG5000-OH, RT, 3 h; (iv) 10% Pd/C, H2, ethyl acetate, RT, 40 psi, 3 h; (v) EDC, HOBT, DIPEA, DMF, dodecanol, fluorescein cadaverine (FC), RT, 48 h.
3.1. Synthesis and Characterization of GE11-PEG-PCD, HW12-PEG-PCD, mPEG-b-PCC-g-GEM-g-DC, and mPEG-b-PCC-g-FC-g-DC copolymers
MBC monomer was characterized by 1H NMR (500 MHz, DMSO-d6), which showed peaks corresponding to phenyl proton at δ7.5, and methylene protons present in the carbonate ring at δ4.2 and δ4.7. Methyl and benzyl protons were present at δ1.3 and δ5.2, respectively (data not shown). MCC was obtained by hydrogenation of MBC and characterized by 1H NMR (500 MHz, chloroform-d) δ13.35 (s, 1H), 4.57 (d, 2H), 4.32 (d, 2H) and 1.18 (s, 3H) (data not shown).
DC was conjugated to the carboxyl group of MCC by EDC/HOBT coupling, and the final product was purified by column chromatography and characterized by 1H NMR (500 MHz, chloroform-d) with the following characteristic peaks: δ4.50 (d, 2H), 4.2 (d, 4H), 1.65 (m, 2H), 1.4–1.2 (m, 21H) and 0.85 (t, 3H) (data not shown). Ring opening polymerization of MDC and Mal-PEG in the presence of 1,8-diazabicycloundec-7-ene (DBU) catalyst at RT for 3 h under N2 atmosphere yielded MAL-PEG-PDC copolymer. 1H NMR (500 MHz, DMSO-d6) spectra of Mal-PEG-PDC showed peaks corresponding to PEG (–CH2–CH2–O) at δ3.5, PCC (–CO–O–CH2– and CH3– C(CO)–CH2) at δ4.2 (m, 4H), CH3–(CH2)9– at δ0.9 (t, 3H), CH3–(CH2)9– at δ1.2–1.45 (bs, 18H), CH3–(CH2)9– CH2 and –NH–CH2–CH2 at δ1.5–1.8 (m, 4H), CH3– (CH2)9–CH2–CH2– at δ4.02 (m, 2H), –NH–CH2–CH2 and –NH–CO–CH2–CH2 at δ2.5–3.2 (m, 4H), –NH–CO– CH2–CH2 at δ4.6 (m, 2H), –NH–CO–CH2–CH2 at δ8.4 (m, 1H), –CO–CH=CH–CO– at δ7.2–7.5 (t, 2H) (Figure S1). GE11 peptide was conjugated to MAL-PEG-PDC via the addition reaction. GE11-PEG-PCD copolymer was lyophilized and characterized using 1H NMR (500 MHz, DMSO-d6), which showed peaks corresponding to PEG (–CH2–CH2–O) at δ3.5, PCC (–CO–O–CH2– and CH3–C(CO)–CH2) at δ4.2 (m, 4H), CH3–(CH2)9– at δ0.9 (t, 3H), CH3–(CH2)9– at δ1.2–1.45 (bs, 18H), Ile Cβ–H at δ 2.6 (m, 1H), Cα–H of (Cys,Tyr, His, Trp, Thr, Pro, Gln, Asn, Val, and Ile) amino acids at δ4.2–4.9 (m, 12H), Gly –CH2–NH at δ 4.4 (m, 2H), Ile Cγ–H2 at δ 1.5 (m, 2H), Ile Cδ–H3 at δ 0.9 (t, 3H), Ile Cβ– H–CH3 at δ 1.1 (m, 3H), Val Cβ–H at δ 2.5 (m, 1H), Val CβH–CH3 and Val Cγ–H3 at δ 0.9 (t, 6H), Asn Cβ–H2 at δ 2.5–2.9 (m, 2H), Gln CβH2 and Gln Cγ–H2 at δ 2.1–2.3 (m, 4H), Pro Cβ–H2 at δ 2.0–2.2 (m, 2H), Pro Cγ–H2 at δ 1.9–2.1 (m, 2H), Pro Cδ–H2 at δ 3.3–3.5 (m, 2H), Thr –CH– CH(CH3)–OH at δ 4.6 (m, 1H), Thr –CH–CH(CH3)–OH at δ 1.2 (m, 3H), Tyr Cβ–H2 at δ 3.2–3.45 (m, 2H), Tyr– CH2–C6H4–OH at δ 6.5–6.9 (m, 4H), Trp Cβ–H2 at δ 3.0– 3.4 (m, 2H), Trp –CH2–C8H5NH at δ 7.1–8.3 (m, 5H), His Cβ–H2 at δ 2.9–3.15 (m, 2H), His –CH2–C3H2N2H at δ 8.2– 8.7 (m, 2H), Cys Cβ–H2 at δ 2.9–3.2 (m, 2H) (Figure S2). The peaks at 6.5–9.3 ppm confirmed the successful conjugation of GE11 peptide to the copolymer.25 Similarly, HW12-PEG-PCD was synthesized and characterized using 1H NMR (500 MHz, DMSO-d6), which showed peaks corresponding to PEG (–CH2–CH2–O) at δ3.5, PCC (–CO–O–CH2– and CH3–C(CO)–CH2) at δ4.2 (m, 4H), CH3–(CH2)9– at δ0.9 (t, 3H), CH3–(CH2)9– at δ1.2–1.45 (bs, 18H), Ala CαH–CH3 at δ1.3 (s, 3H), Cα–H of (Cys, Tyr, His, Trp, Thr, Pro, Ser, and Ala) amino acids at δ3.4–4.9 (m, 12H), Ser Cβ– H2 at δ 3.2–4.12 (m, 2H), Pro Cβ–H2 at δ 2.0–2.2 (m, 2H), Pro Cγ–H2 at δ 1.9–2.1 (m, 2H), Pro Cδ–H2 at δ 3.3–3.5 (m, 2H), Thr –CH–CH(CH3)–OH at δ 4.6 (m, 1H), Thr – CH–CH(CH3)–OH at δ 1.2 (m, 3H), Tyr Cβ-H2 at δ 3.2– 3.45 (m, 2H), Tyr –CH2–C6H4–OH at δ 6.5–6.9 (m, 4H), Trp Cβ–H2 at δ 3.0–3.4 (m, 2H), Trp –CH2–C8H5NH at δ 7.1–8.3 (m, 5H), His Cβ–H2 at δ 2.9–3.15 (m, 2H), His –CH2–C3H2N2H at δ 8.2–8.7 (m, 2H), Cys Cβ–H2 at δ 2.9–3.2 (m, 2H) (Figure S3).
Fluorescence cadaverine (FC) and dodecanol (DC) were conjugated to the carboxyl groups of mPEG–PCC by EDC/HOBT coupling reaction. At the end of the reaction, FC-conjugated copolymer was purified using isopropanol and diethyl ether and by extensive dialysis and lyophilized. 1H NMR (500 MHz, DMSO-d6) spectra of mPEG-b-PCC-g-FC-g-DC showed peaks corresponding to PEG (–CH2–CH2–O) at δ3.5, PCC (–CO–O–CH2– and CH3–C(CO)–CH2) at δ4.2 (m, 4H), CH3–(CH2)9– at δ0.9 (t, 3H), CH3–(CH2)9– at δ1.2–1.45 (bs, 18H), –CO–NH–CH2–(CH2)3– at δ1.2–1.6 (m, 6H), –CS–NH–CH2– and –CO–NH–CH2– at δ2.6–3.2 (m, 4H), –CS–NH–CH2– and –CO–NH–CH2– at δ (m, 2H), –NH–C6H3(CO2H)–C13H6O2(OH) at δ6.1–7.9 (m, 9H) (Figure S4).
GEM and DC were conjugated to the carboxyl groups of mPEG–PCC to get GEM-conjugated copolymers. 1H NMR spectra of mPEG–PCC showed peaks corresponding to PEG (–CH2–CH2–O) at δ53.5 and PCC (–CH2–) at δ4.2. The absence of benzene protons at δ7.3 confirmed complete hydrogenation along with the presence of –COOH peaks at δ12–14 as reported earlier. Based on the peak integrals of mPEG and PCC protons, an average molecular weight of mPEG–PCC was calculated to be ~9600 Da with 29 PCC units. GEM and DC were conjugated onto the carboxyl groups of mPEG–PCC by EDC/HOBT coupling reaction, and GEM conjugated copolymer was purified by precipitation in isopropanol and diethyl ether followed by extensive dialysis and lyophilization. 1H NMR (500 MHz, DMSO-d6) of mPEG-b-PCC-g-GEM-g-DC showed peaks corresponding to PEG (–CH2–CH2–O) at δ3.5, PCC (–CO–O–CH2– and CH3– C(CO)–CH2) at δ4.2 (m, 4H), CH3–(CH2)9– at δ0.9 (t, 3H), CH3–(CH2)9– at δ1.2–1.45 (bs, 18H), GEM 5′- and 3′-OH groups at δ6.18–6.39 (m, 2H), –CO–NH– at δ8.26 (m, 1H), –CO–N–CH=CH– at δ8.15 (m, 1H) and –CO–N– CH=CH– at δ5.35 (m, 1H) (Figure S5).
3.2. Formulation Development and Characterization
Mixed micellar formulations have been successfully applied for drug delivery application.32,33 Various block copolymers at a certain molar or weight ratios are used for mixed micelle formation, and they formed a single polymeric micellar system rather than two separate micelles.34,35 In our previous report, we synthesized mPEG-b-PCC-g-GEM-g-DC copolymer for GEM delivery by passive targeting only. In the present study, we have conjugated GE11 peptide to our polymeric micelles for EGFR receptor mediated active targeting. However, GE11 peptide, GEM and DC conjugation in the same micellar system created synthetic complexity and reduced drug loading (data not shown). To avoid these obstacles, we synthesized GE11-PEG-PCD and mPEG-b-PCC-g-GEM-g-DC copolymers separately and prepared EGFR targeted mixed micelles by combining GE11-PEG-PCD (10–30% weight ratio) and mPEG-b-PCC-g-GEM-g-DC (70–90% weight ratio) copoly-mers by film hydration. In addition, we used HW12-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC (30/70 w/w) mixed micelles. mPEG-b-PCC-g-GEM-gg-DC had a mean particle size of 26 ± 3 nm with a PDI of 0.27. We measured particle size and surface ζ-potential of GE11-linked mixed micelles (10–30% weight ratios) and HW12-linked (30/70 w/w ratio) mixed micelles to determine the effect of GE11 or HW12 conjugation on size and surface charge of mixed micelles. Unmodified micelles had a mean particle size of 26 ± 3 nm (PDI-0.27), whereas GE11-linked mixed micelles (10–30% weight ratio) and HW12-linked mixed micelles (30% weight ratio) had a mean particle size of 51 ± 5 (PDI-0.229), and 53 ± 0.8 (PDI-0.188), respectively, indicating the conjugation of GE11 or HW12 peptide did not significantly change the size of mixed micelles than mPEG-b-PCC-g-GEM-g-DC micelles. We observed an increase in surface ζ-potential of GE11-linked mixed micelles (10–30% weight ratio) −6.8 ± 2 mV and HW12-linked mixed micelles (30% weight ratio) −5.9 ± 0.2 mV as compared to unmodified micelles −4.6 ± 0.2 mV. GEM loading in mPEG-b-PCC-g-GEM-g-DC was calculated by 1H NMR, which showed GEM 5′-and 3′-OH protons at δ5.90 and δ6.2, respectively. The presence of amide proton at δ8.26 confirmed the formation of an amine bond with the pendant carboxyl group of mPEG–PCC, which is in agreement with the literature.7,36,37 GEM loading in mPEG-b-PCC-g-GEM-g-DC was ~14.8% w/w by integrating and calculating amide protons peak at δ8.26 (Figure S5). However, 1H NMR analysis can only confirm the chemical conjugation of GEM to the polymer backbone but cannot predict accurate drug loading. Therefore, GEM loading in mPEG-b-PCC-g-GEM-g-DC was further determined by alkaline hydrolysis and then analyzed by the HPLC-UV method as reported previously.17 GEM loading in micelles was found ~12% w/w (data not shown). Although, our mPEG-b-PCC-g-GEM-g-DC copolymer (12% w/w drug loading) showed higher drug loading than previously reported PEGylated GEM derivatives (0.98–6.79% w/w drug loading range), but it was lower than reported GEM–squalene nanoassemblies (GEM loading of 41% w/w).7,8,38–40 PEGylated gemcitabine nanoassemblies were achieved by addition of PEG–squalene or PEG–cholesterol (1:0.7 w/w), which showed a decrease in gemcitabine loading but higher efficacy than gemcitabine–squalene nanoassemblies. Therefore, our formulation is a balance between PEG coating (55%) and GEM loading (12% w/w). Although our formulation needs improvement to increase GEM payload but, this polymeric drug conjugate offers numerous advantages over the squalenoylated nanoassemblies. Gemcitabine–squalene nano-assemblies are rapidly cleared from the bloodstream by the reticulo-endothelial system (RES) due to the absence of PEG coating.16 PEG coating in the outer shell is needed for stealth effect and to reduce opsonization and blood clearance. Our micelles had a much smaller particle size (25–50 nm) than gemcitabine–squalene nanoassemblies (140 nm), and the presence of PEG (~55%) coating in our formulation can provide stealth effect. In our previous study, we found a controlled release pattern of GEM (39% GEM release in 10 days) from the micelles even in the presence of proteolytic enzyme Cathepsin B.17 Based on that, we assume that all our formulations had similar drug release patterns.
3.3. EGFR-Mediated Uptake of GEM-Conjugated GE11-Modified Mixed Micelles
To convert a mixed micellar system into a cell surface probe as well as to verify receptor-mediated GEM delivery in vitro and in vivo, we conjugated FC as a fluorescent tag to carboxyl-terminated mPEG–PCC copolymer via an amide bond. Various fluorescein molecules are widely used to label nanoparticles for intracellular and intratumoral distribution of nanoparticles.41,42 We selected FC as a fluorescein substance since prior reports showed that carboxylic acids of proteins, water-soluble biopolymer, and difluorinated cyclooctyne (DIFO) chemically conjugated with the primary aliphatic amine of FC through EDAC coupling for subcellular distribution, interaction between cell surface receptor and ligand, and in vivo imaging.43,44 Therefore, we synthesized FC-conjugated mPEG-b-PCC-g-FC-g-DC copoly-mer. We prepared FC-labeled GE11-linked mixed micelles (10–30% weight ratios) and unmodified micelles. Before cellular uptake of labeled mixed micelles in MIA PaCa-2 cells, we determined EGFR expression levels in MIA PaCa-2 cells by flow cytometry. MIA PaCa-2 cells showed high expression of EGFR (Figure S6). To determine whether GE11 peptide enhanced the cellular uptake by EGFR expressing MIA PaCa-2 cells, cells were incubated with FC-labeled GE11-linked mixed micelles and unmodified micelles for 12 h. The fluorescein images are shown in Figure S7 for qualitative analysis of cellular uptake. MIA PaCa-2 cells treated with GE11-linked mixed micelles at 30% w/w ratios showed significantly higher fluorescence than GE11-linked mixed micelles at 10 and 20% weight ratios and unmodified micelles (Figure S7A–D). To confirm that GE11 and not HW12 is an EGFR ligand, we also prepared FC-labeled HW12-linked mixed micelles at 30% weight ratio, and cells were incubated with FC-labeled GE11-linked mixed micelles and HW12-linked mixed micelles at 30% weight ratio for 12 h, respectively. The fluorescein images of MIA PaCa-2 cells treated with GE11-linked mixed micelles at 30% weight ratio showed stronger fluorescence compared to HW12-linked mixed micelles, suggesting that mixed micelles containing GE11 peptide could facilitate cellular delivery of conjugated drugs (Figure S7E). To further confirm the ligand receptor interaction and specific role of GE11 peptide, we presaturated MIA PaCa-2 cells with free GE11 peptide prior to incubation with GE11-linked mixed micelles at 30% weight ratio. Results depicted in (Figure S7F) showed significant reduction of the fluorescence signal and comparable fluorescence signal of HW12-linked mixed micelle-treated cells (Figure S7E), supporting EGFR receptor-mediated uptake. Cellular uptake results were further analyzed by flow cytometry for quantitative analysis. Results delineated in Figure 3A–D clearly indicated that the mean fluorescence intensity of GE11-linked mixed micelle (30% w/w) treated cells were 1.2-fold and 3.1-fold higher than unmodified micelles and HW12-linked mixed micelles, respectively. However, we observed that the cellular uptake of HW12-linked mixed micelles was lower than unmodified micelles. The exact mechanism for this lower uptake of peptide-linked mixed micelles requires additional investigation, but it may be due to the increase in hydrophilicity of the HW12 peptide. To illustrate this point, we calculated hydrophobicity for GE11 (YHWYGYTPQNVI) peptide, which was 50, whereas for HW12 (HYPYAHPTHPSW) it was 33.33, suggesting that HW12-linked micelles are more hydrophilic compared to GE11-linked mixed micelles. This increase in hydrophilicity is expected to decrease cellular uptake as shown in Figure 3.45 When cells are pretreated with free GE11 peptide prior to incubation of cells with GE11-linked mixed micelles (30% w/w), the mean fluorescence intensity decreased significantly (Figure 3B,D).
Figure 3.
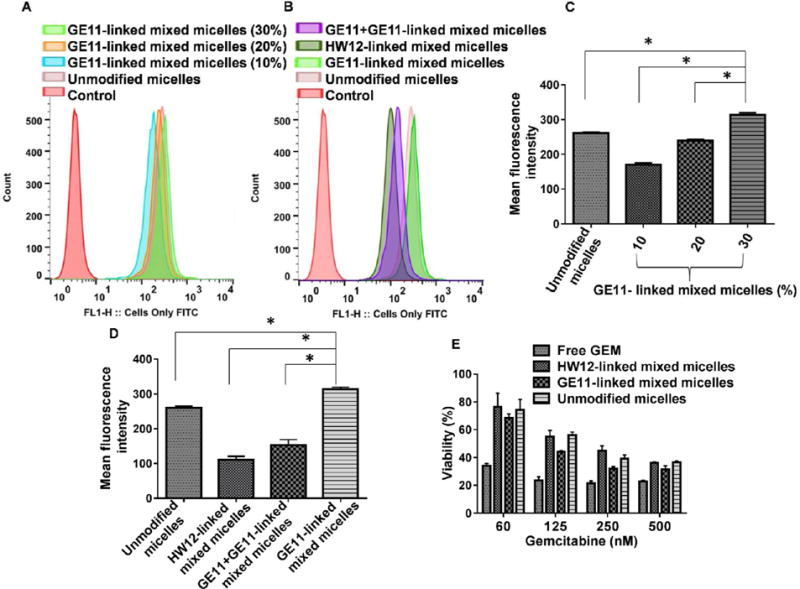
Flow cytometry analysis of cellular uptake of FC-labeled micelles showing the histogram and mean fluorescence intensity of MIA PaCa-2 cells treated with GE11-linked mixed micelles (10–30% w/w), unmodified micelles (mPEG-b-PCC-g-FC-g-DC) (A,C), *p < 0.001; GE11-linked mixed micelles (30% w/w), preincubation of MIA PaCa-2 cells with free GE11 prior incubation of cells with GE11-linked mixed micelles (30/70 w/w), unmodified micelles and HW12-linked mixed micelles (30/70 w/w) (B,D). Effect of GEM on MIA PaCa-2 cell viability (E) *p < 0.001 compared to GE11-linked mixed micelles. Effect of GEM on MIA PaCa-2 cell viability (E).
3.4. In Vitro Cytotoxicity Studies
Density of surface ligand is critical to target cell binding in vitro as well as pharmacokinetic profiles in vivo. Our cellular uptake study clearly indicated that 30% w/w ratios of GE11-linked mixed micelles was superior to HW12-linked mixed micelles and unmodified micelles. Therefore, we prepared GE11-linked mixed micelles (GE11-PEG-PCD/mPEG-b-PCC-g-GEM-g-DC) at 30% w/w ratios to enhance receptor-mediated cellular uptake. Similarly, we prepared HW12-linked mixed micelles using HW12-PEG-PCD and mPEG-b-PCC-g-GEM-g-DC copolymers at 30% weight ratio. For cytotoxicity studies, MIA PaCa-2 cells were treated with GE11-linked mixed micelles, HW12-linked mixed micelles, unmodified micelles, and free GEM. Free GEM exhibited highest cytotoxicity within the studied concentration range compared to GE11-linked mixed micelles, HW12-linked mixed micelles, unmodified micelles, indicated by lowering cell viability (Figure 3E). Although free GEM showed maximum toxicity, its plasma instability and lack of target specificity would restrict its in vivo tumor inhibition capability. GE11-linked mixed micelles showed a higher cytotoxicity than HW12-linked mixed micelles and unmodified micelles at all GEM concentrations tested, further demonstrating that GE11 peptide play important role for antitumor efficacy in GE11 modified mixed micelles (Figure 3E). The IC50 value was calculated for GE11-linked mixed micelles (95.32 nM), which was 1.2-fold and 1.3-fold lower than unmodified micelles (116.91 nM) and HW12-linked mixed micelles (126.58 nM), respectively. Our results are in good agreement with the literature. For example, Chen et al. demonstrated that a GE11 density of 10% is enough to enable EGFR-ligand binding.25 In their study, the cell viability for liposomes with a 15% GE11 density was similar to that for liposomes with a 10% GE11 density.
3.5. GE11 Peptide Conjugated Mixed Micelles for Targeting EGFR Expressing Pancreatic Tumor As Well As Tumor Neovasculature
After in vitro efficacy of GE11-linked mixed micelles, we determined targeting efficiency of GE11-linked mixed micelles in an orthotopic pancreatic tumor bearing athymic nude mice by systemic administration of FC-labeled GE11-linked mixed micelles, HW12-linked mixed micelles and unmodified micelles. FC-labeled GE11-linked mixed micelles injected mouse tumor sections showed higher fluorescence intensity at 24 h post injection than FC-labeled HW12-linked mixed micelles and unmodified micelles injected tumor sections (Figure 4), supporting that GE11 peptide facilitated distribution and accumulation of drug molecule into the tumor. Our results are in good agreement with the work of Xu et al., who demonstrated enhanced accumulation of GE11 peptide conjugated gelatin nanoparticles in a subcutaneous Panc-1 pancreatic tumor model especially at early time points.26 Previous reports showed that tumor cells as well as tumor-derived endothelial cells express EGFR.46 Toward confirming that GE11-linked mixed micelles can target tumor as well as tumor neovaculature under in vivo condition, we stained the same tumor sections with rabbit polyclonal anti-VE-cadherin antibody (markers of tumor endothelial cells) and counter-stained with Alexa Fluor 594 F(ab′)2 fragment of goat antirabbit IgG (H+L) secondary antibody. When tumor sections were observed under a fluorescent microscope, the merged image of FC-labeled GE11-linked mixed micelles mouse tumor sections showed that targeted mixed micelles deliver FC dye to tumor as well as tumor vasculature. In contrast, FC-labeled HW12-linked mixed micelles and unmodified micelles did not deliver FC dye to tumor vasculature. Therefore, our result in Figure 4A clearly indicated that GE11-linked mixed micelles are not only capable of delivering drug to tumor cells but also effective in distributing drug to tumor vasculature.
Figure 4.
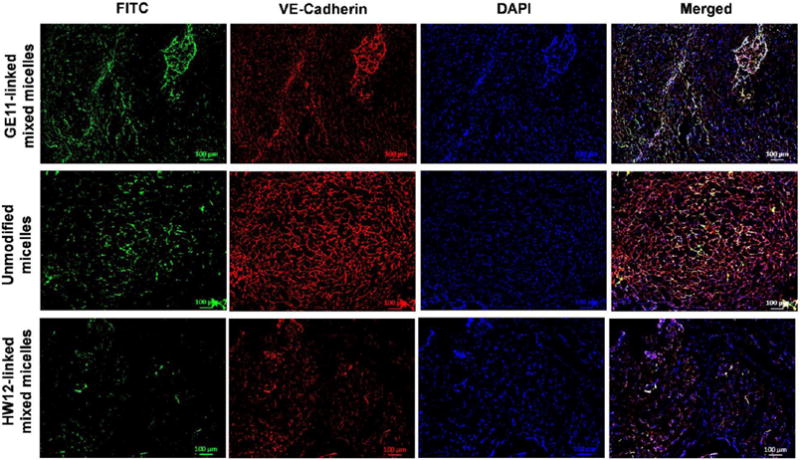
In vivo tumor as well as tumor vasculature targeting of labeled micellar system. Male athymic nude mice were orthotopically implanted with MIA PaCa-2 tumor. FC-labeled GE11-linked mixed micelles, unmodified micelles, and HW12-linked mixed micelles were intravenously administered at day 21. After 24 h, the mice were sacrificed; tumors were removed, for sectioning and immunostaining with anti-VE-cadherin (marker of endothelial cells). GE11-linked mixed micelles significantly accumulated in tumor neovasculature, whereas unmodified micelles and HW12-linked mixed micelles poorly accumulated in the tumor neovasculature. Scale bar, 100 μm.
3.6. In Vivo Efficacy in Orthotopic Pancreatic Tumor Model
After demonstrating the ability of GE11-linked mixed micelles to target EGFR-expressing tumor as well as tumor endothelial cells, we investigated whether GE11-linked mixed micelles could inhibit orthotopic pancreatic tumor growth. After orthotopically implanting MIA PaCa-2/luciferase tumor cells in the pancreas, mice were intravenously administered GE11-linked mixed micelles, HW12-linked mixed micelles, unmodified micelles, and free GEM at an equivalent dose of 40 mg/kg GEM three times in a week after 12 days of cell implantation. We selected this dose according to our previous report, and this dose is lower than the maximum tolerated dose (MTD) of GEM.17 Our in vivo efficacy of GEM in mice, and numerous other reports showed a wide range of its dose ranging from 7 mg/kg to 500 mg/kg for intraperitoneal route and intravenous route of administration.40,47–49 At day 12, all mice in different groups were noninvasively imaged for luciferase bioluminescence to determine the tumor growth and randomized for treatment. Tumor growth inhibitions of different formulations were monitored by measuring luciferase bioluminescence at different time points during the treatment (day 12, 16, 20, 24, 26, and 30), and photon counts were calculated by Living Image 3.2 software (Caliper Life Science). Final bioluminescence images were taken at day 30, and images depicted in Figure 5A clearly showed significant tumor growth inhibition in mice treated with GE11-linked mixed micelles compared to HW12-linked mixed micelles, unmodified micelles, and free GEM. We compared photon counts between different groups after plotting photon value versus days after treatment (Figure 5B). This result further strengthened our in vitro observation and signified the important role of GE11 for EGFR targeting. HW12-decorated HW12-linked mixed micelles showed minimal tumor growth effect, supporting that GE11 peptide is the EGFR-targeting ligand, not HW12 peptide. Unmodified micelles showed better tumor growth inhibition than free GEM. We removed all tumors from different groups, measured tumor volume and weight and took images of the tumor after sacrificing the mice at the end of the experiment. Tumor images as shown in Figure 5C confirmed remarkable tumor growth inhibition capability of GE11-linked mixed micelles compared to HW12-linked mixed micelles, unmodified micelles, and free GEM. This was further confirmed by measuring the tumor volume and weight (Figure 6A,B). Mouse body weight was taken during the treatment course to monitor the adverse effect of GEM. Figure 6C clearly indicated that our in vivo GEM dose (40 mg/kg) did not show any abnormal body weight change in all the experimental groups. Immunohistochemical analysis was done for Ki-67, cleaved caspase-3 and H&E staining of tumor cryosections obtained from all treated groups. Unmodified micelle-treated mice showed reduction in Ki-67 staining and increase in cleaved caspase-3 staining compared to the free GEM.
Figure 5.
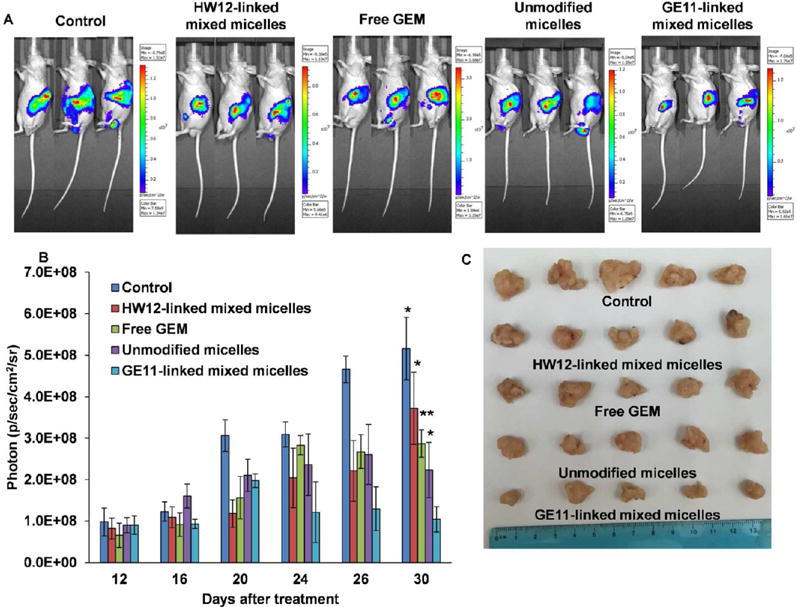
In vivo efficacy of GE11-linked mixed micelles after systemic administration in orthotopic pancreatic tumor bearing mice. A representative bioluminescence image from the mice treated with a saline, free GEM, HW12-linked mixed micelles, unmodified micelles, and GE11-linked mixed micelles (n = 5) (A). Relative photon intensity plot of all groups were measured from day 12 to day 30 (B). Data represented as the mean ± SEM *p < 0.05; **p < 0.001 compared to GE11-linked mixed micelles. Representative tumor sizes of each group were taken after sacrificing the mice at the end of the experiment (C).
Figure 6.
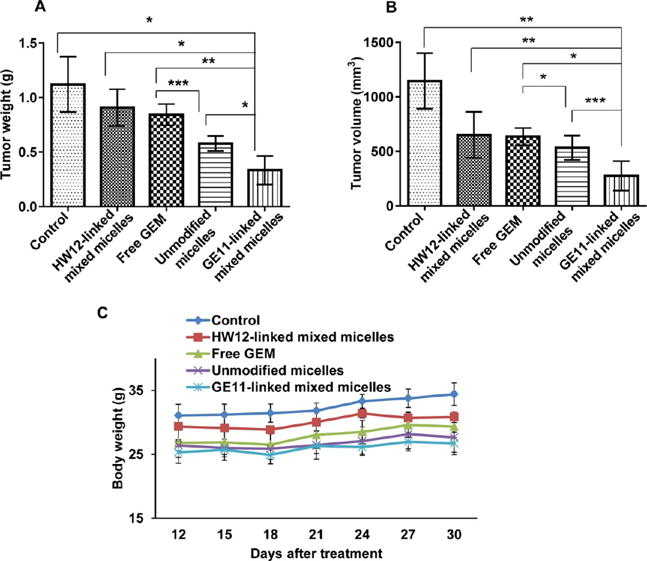
Tumor volume (A) and tumor weight (B) were measured after sacrificing the mice at the end of the experiment. For all graphs, each data point represents the mean ± SEM (n = 5) *p < 0.001, **p <.0001, ***p < 0.05 (A) and *p < 0.001, **p < 0.05, ***p < 0.005 (B). Body weight of all groups were measured from day 12 to day 30 (C).
Mice treated with GE11-linked mixed micelles showed the least Ki-67 staining and highest cleaved caspase-3 staining (Figure 7A,B). GE11-linked mixed micelles and unmodified micelle-treated groups showed more necrotic areas compared to HW12-linked mixed micelles, free GEM, and control (Figure 7C). GE11-linked mixed micelle treatment showed more necrotic areas compared to unmodified micelles, supporting the beneficial effect of GE11 peptide-mediated active targeting over passive targeting. After three doses of treatment, major organs, such as heart, liver, and kidney were collected from all treated groups. Histological staining of these organs did not show any pathological changes after treatment in all the groups (Figure S8). Tumor growth inhibition potential of GE11-linked mixed micelles and unmodified micelles were confirmed by TUNEL assay. Mice treated with GE11-linked mixed micelles showed enhanced apoptosis of cancer cells compared to control, free GEM, HW12-linked mixed micelles, and unmodified micelles treated mice (Figure 8). Solid tumor growth and metastatic potential to distant organs critically depend on angiogenesis, sprouting new blood vessels from preexisting blood vessels.19 The newly formed blood vessels are structurally and functionally abnormal compared to normal blood vessels. Delivery of antiangiogenic agent specifically to tumor derived endothelial cells is an attractive strategy for antiangiogenic cancer therapy. GEM shows its anticancer activity by mainly cytotoxic action on cancer cells.50 In addition to its cytotoxic action, recent studies showed that it also has antiangiogenic activity.51,52 Previous in vitro studies showed that GEM is more sensitive to endothelial cells than pancreatic cancer cells.51 GEM is a hydrophilic drug and its plasma instability hinders its delivery into tumor endothelial cells in vivo. We previously described that conjugation of GEM to polymer increased its in vivo stability. Further, we showed in our model drug distribution study using an FC-conjugated polymeric system that GE11-linked mixed micelles deliver FC dye in tumor-derived endothelial cells. We coimmunostained the same TUNEL-stained tumor sections with rabbit polyclonal anti-VE-cadherin (marker of endothelial cells) to determine the apoptosis of tumor endothelial cells. The result delineated in Figure 8 showed mice treated with GE11-linked mixed micelles induced significant apoptosis of tumor endothelial cells, supporting the significant tumor growth inhibition potential of this mixed micellar system.
Figure 7.
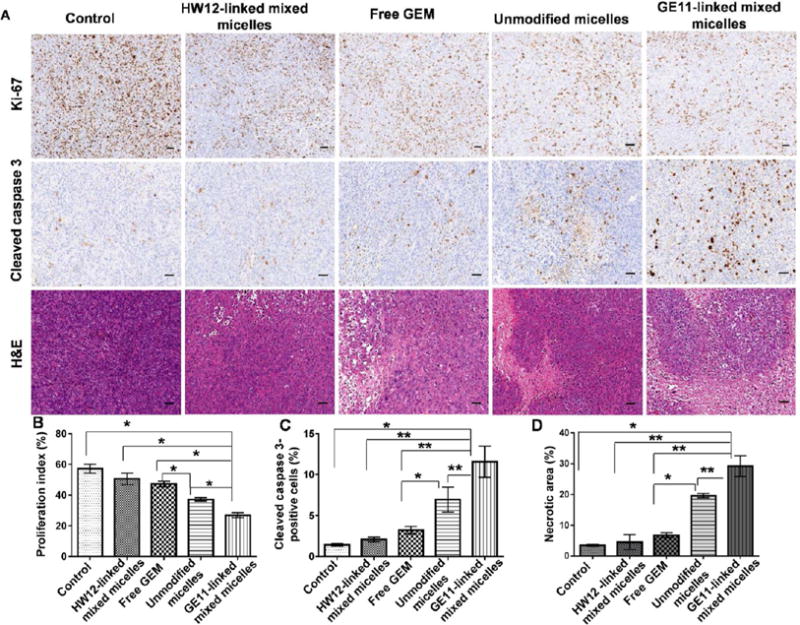
Immunohistochemical analysis of tumor samples for Ki-67 (cell proliferation marker), cleaved caspase 3 (apoptosis marker), and H&E staining (A). Saline, free GEM, HW12-linked mixed micelles, unmodified micelles, and GE11-linked mixed micelle-treated tumor samples were cryosectioned, fixed, and immunostained for Ki-67, cleaved caspase 3, and H&E (A). Statistical analysis was performed for Ki-67 (B), *p < 0.001; cleaved caspase 3 (C), *p < 0.05; **p < 0.01 and H&E (D), *p < 0.01; **p < 0.001. Scale bar, 2 mm.
Figure 8.
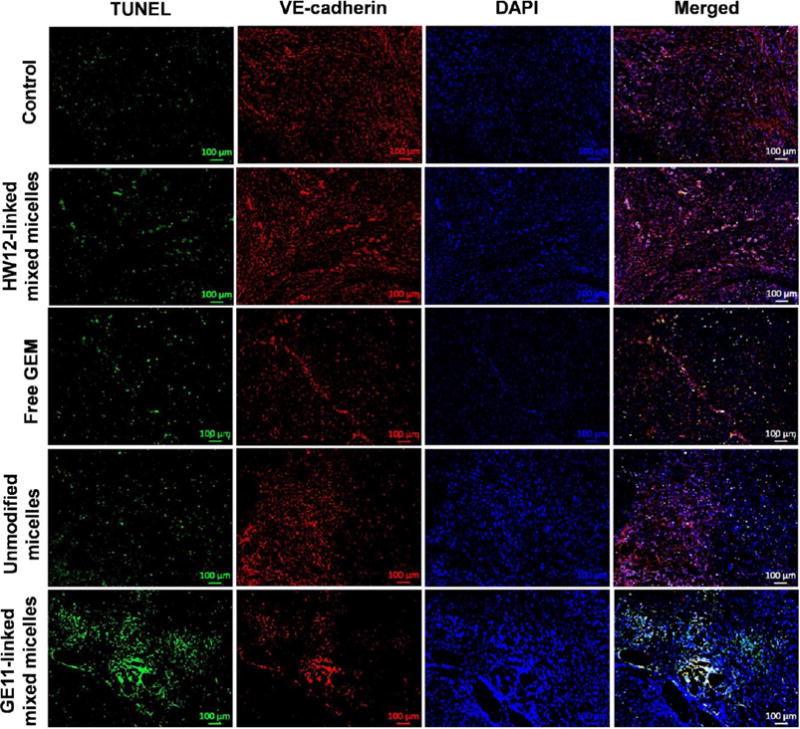
Detection of apoptosis induced by GE11-linked mixed micelles in tumor and tumor endothelial cells of an orthotopic pancreatic tumor. Saline, HW12-linked mixed micelles, free GEM, unmodified micelles, and GE11-linked mixed micelle-treated tumor samples were cryosectioned, fixed, and immunostained for TUNEL-positive nuclei (green), VE-cadherin (endothelial cells, red), and DAPI (blue) stained nuclei. Scale bar, 100 μm.
In an animal model, inhibition of the angiogenesis can efficiently reduce pancreatic tumor growth.47,53–55 Further, vascular abnormality leads to a complex tumor environment with impaired blood flow and interstitial hypertension, which hinders delivery of anticancer drugs and oxygen.56 The reduction of this abnormal vasculature improves delivery of chemotherapy to tumor tissues.57 CD31 is a 130 kDa transmembrane glycoprotein, one of the best markers of benign and malignant tumor vessels.58 Tumor cryosections were immunostained with CD31 antibody to determine the antivascular efficacy of GE11-linked mixed micelles. Our immunohistological staining data showed significantly reduced tumor microvessel density in GE11-linked mixed micelles group compared to control, free GEM, HW12-linked mixed micelles, and unmodified micelle-treated groups (Figure 9). TUNEL assay and microvessel density results clearly showed that GE11-linked mixed micelles target cancer cells as well as tumor vasculature. All these results demonstrate the significant inhibition potential of GEM-conjugated GE11-modified mixed micelles in a orthotopic pancreatic tumor mouse model.
Figure 9.
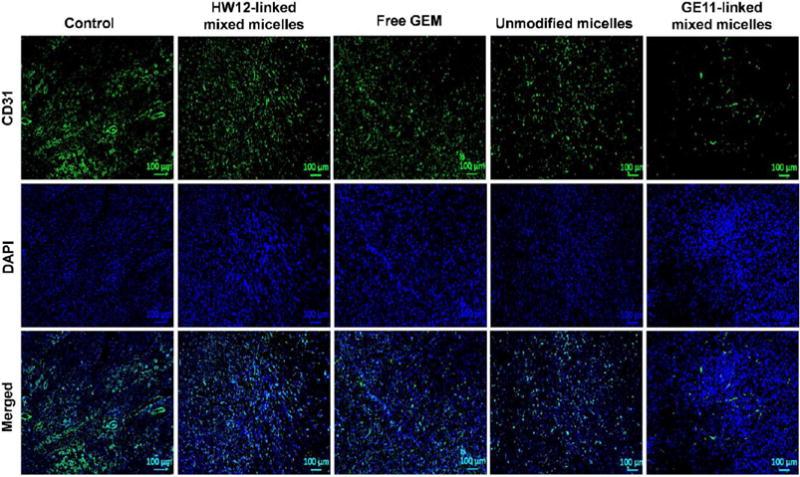
Immunostaining of cryosections for CD31 as a marker for microvessel density. GE11-linked mixed micelle-treated tumor section showed significant reduction of microvessel densities. Saline, HW12-linked mixed micelles, free GEM, unmodified micelles, and GE11-linked mixed micelle-treated tumor samples were cryosectioned, fixed, and immunostained with rat antimouse CD31 antibody (green) and DAPI-stained nuclei (blue). Scale bar, 100 μM.
4. CONCLUSIONS
In summary, we have developed an EGFR-targeted GEM-conjugated polymeric mixed micelles for treating pancreatic cancer. GE11-linked mixed micelles specifically and efficiently delivered GEM to EGFR-expressing pancreatic cancer, interacted with tumor blood vessels, and showed significant inhibition of pancreatic tumor growth in an orthotopic mouse model. Thus, GE11-linked mixed micelles is a promising carrier for drug delivery to treat various EGFR-expressing cancers.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (1R01EB017853), the Fred and Pamela Buffet Cancer Center, and a Faculty Start-up fund to R.I.M.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-mac.5b01419.
1H NMR of spectra of copolymers, EGFR expression in MIA PaCa-2 cells, epifluorescence microscopic images of cells treated with copolymers, hematoxylin and eosin (H&E) staining of major organs (liver, heart and kidney).(PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Ko AH, Truong TG, Kantoff E, Jones KA, Dito E, Ong A, Tempero MA. Cancer Chemother Pharmacol. 2012;70:875–881. doi: 10.1007/s00280-012-1979-7. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Sengupta R, Banerjee S, Li Y, Zhang Y, Rahman KM, Aboukameel A, Mohammad R, Majumdar AP, Abbruzzese JL, Sarkar FH. Cancer Res. 2006;66:7653–7660. doi: 10.1158/0008-5472.CAN-06-1019. [DOI] [PubMed] [Google Scholar]
- 3.Kumar V, Mondal G, Slavik P, Rachagani S, Batra SK, Mahato RI. Mol Pharmaceutics. 2015;12:1289–1298. doi: 10.1021/mp500847s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberstein PE, Saif MW. JOP J Pancreas. 2011;12:96–100. [PubMed] [Google Scholar]
- 5.Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Semin Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- 6.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 7.Pasut G, Canal F, Dalla Via L, Arpicco S, Veronese FM, Schiavon O. J Controlled Release. 2008;127:239–248. doi: 10.1016/j.jconrel.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Vandana M, Sahoo SK. Biomaterials. 2010;31:9340–9356. doi: 10.1016/j.biomaterials.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Bornmann C, Graeser R, Esser N, Ziroli V, Jantscheff P, Keck T, Unger C, Hopt UT, Adam U, Schaechtele C, Massing U, von Dobschuetz E. Cancer Chemother Pharmacol. 2008;61:395–405. doi: 10.1007/s00280-007-0482-z. [DOI] [PubMed] [Google Scholar]
- 10.Cosco D, Bulotta A, Ventura M, Celia C, Calimeri T, Perri G, Paolino D, Costa N, Neri P, Tagliaferri P, Tassone P, Fresta M. Cancer Chemother Pharmacol. 2009;64:1009–1020. doi: 10.1007/s00280-009-0957-1. [DOI] [PubMed] [Google Scholar]
- 11.Maksimenko A, Mougin J, Mura S, Sliwinski E, Lepeltier E, Bourgaux C, Lepetre S, Zouhiri F, Desmaele D, Couvreur P. Cancer Lett. 2013;334:346–353. doi: 10.1016/j.canlet.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 12.Celia C, Malara N, Terracciano R, Cosco D, Paolino D, Fresta M, Savino R. Nanomedicine. 2008;4:155–166. doi: 10.1016/j.nano.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Moog R, Burger AM, Brandl M, Schuler J, Schubert R, Unger C, Fiebig HH, Massing U. Cancer Chemother Pharmacol. 2002;49:356–366. doi: 10.1007/s00280-002-0428-4. [DOI] [PubMed] [Google Scholar]
- 14.Fan M, Liang X, Li Z, Wang H, Yang D, Shi B. Eur J Pharm Sci. 2015;79:20–26. doi: 10.1016/j.ejps.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Tao XM, Wang JC, Wang JB, Feng Q, Gao SY, Zhang LR, Zhang Q. Eur J Pharm Biopharm. 2012;82:401–409. doi: 10.1016/j.ejpb.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Reddy LH, Khoury H, Paci A, Deroussent A, Ferreira H, Dubernet C, Decleves X, Besnard M, Chacun H, Lepetre-Mouelhi S, Desmaele D, Rousseau B, Laugier C, Cintrat JC, Vassal G, Couvreur P. Drug Metab Dispos. 2008;36:1570–1577. doi: 10.1124/dmd.108.020735. [DOI] [PubMed] [Google Scholar]
- 17.Chitkara D, Mittal A, Behrman SW, Kumar N, Mahato RI. Bioconjugate Chem. 2013;24:1161–1173. doi: 10.1021/bc400032x. [DOI] [PubMed] [Google Scholar]
- 18.Kunjachan S, Pola R, Gremse F, Theek B, Ehling J, Moeckel D, Hermanns-Sachweh B, Pechar M, Ulbrich K, Hennink WE, Storm G, Lederle W, Kiessling F, Lammers T. Nano Lett. 2014;14:972–981. doi: 10.1021/nl404391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carmeliet P, Jain RK. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 20.Yu DH, Lu Q, Xie J, Fang C, Chen HZ. Biomaterials. 2010;31:2278–2292. doi: 10.1016/j.biomaterials.2009.11.047. [DOI] [PubMed] [Google Scholar]
- 21.Valetti S, Maione F, Mura S, Stella B, Desmaele D, Noiray M, Vergnaud J, Vauthier C, Cattel L, Giraudo E, Couvreur P. J Controlled Release. 2014;192:29–39. doi: 10.1016/j.jconrel.2014.06.039. [DOI] [PubMed] [Google Scholar]
- 22.Chitkara D, Singh S, Kumar V, Danquah M, Behrman SW, Kumar N, Mahato RI. Mol Pharmaceutics. 2012;9:2350–2357. doi: 10.1021/mp3002792. [DOI] [PubMed] [Google Scholar]
- 23.Patra CR, Bhattacharya R, Wang E, Katarya A, Lau JS, Dutta S, Muders M, Wang S, Buhrow SA, Safgren SL, Yaszemski MJ, Reid JM, Ames MM, Mukherjee P, Mukhopadhyay D. Cancer Res. 2008;68:1970–1978. doi: 10.1158/0008-5472.CAN-07-6102. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, Zhao R, Wu X, Sun Y, Yao M, Li J, Xu Y, Gu J. FASEB J. 2005;19:1978–1985. doi: 10.1096/fj.05-4058com. [DOI] [PubMed] [Google Scholar]
- 25.Cheng L, Huang FZ, Cheng LF, Zhu YQ, Hu Q, Li L, Wei L, Chen DW. Int J Nanomed. 2014;9:921–935. doi: 10.2147/IJN.S53310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Gattacceca F, Amiji M. Mol Pharmaceutics. 2013;10:2031–2044. doi: 10.1021/mp400054e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang H, Chen X, Rui M, Sun W, Chen J, Peng J, Xu Y. Mol Pharmaceutics. 2014;11:3242–3250. doi: 10.1021/mp5001718. [DOI] [PubMed] [Google Scholar]
- 28.Li F, Danquah M, Mahato RI. Biomacromolecules. 2010;11:2610–2620. doi: 10.1021/bm100561v. [DOI] [PubMed] [Google Scholar]
- 29.Wang HX, Xiong MH, Wang YC, Zhu J, Wang J. J Controlled Release. 2013;166:106–114. doi: 10.1016/j.jconrel.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Shi Y, Chen Y, Ye J, Sha X, Fang X. Biomaterials. 2011;32:2894–2906. doi: 10.1016/j.biomaterials.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 31.Danquah M, Fujiwara T, Mahato RI. Biomaterials. 2010;31:2358–2370. doi: 10.1016/j.biomaterials.2009.11.081. [DOI] [PubMed] [Google Scholar]
- 32.Mu CF, Balakrishnan P, Cui FD, Yin YM, Lee YB, Choi HG, Yong CS, Chung SJ, Shim CK, Kim DD. Biomaterials. 2010;31:2371–2379. doi: 10.1016/j.biomaterials.2009.11.102. [DOI] [PubMed] [Google Scholar]
- 33.O’Neil CP, van der Vlies AJ, Velluto D, Wandrey C, Demurtas D, Dubochet J, Hubbell JA. J Controlled Release. 2009;137:146–151. doi: 10.1016/j.jconrel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Yin H, Lee ES, Kim D, Lee KH, Oh KT, Bae YH. J Controlled Release. 2008;126:130–138. doi: 10.1016/j.jconrel.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bae Y, Diezi TA, Zhao A, Kwon GS. J Controlled Release. 2007;122:324–330. doi: 10.1016/j.jconrel.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 36.Cavallaro G, Mariano L, Salmaso S, Caliceti P, Gaetano G. Int J Pharm. 2006;307:258–269. doi: 10.1016/j.ijpharm.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 37.Khare V, Kour S, Alam N, Dubey RD, Saneja A, Koul M, Gupta AP, Singh D, Singh SK, Saxena AK, Gupta PN. Int J Pharm. 2014;470:51–62. doi: 10.1016/j.ijpharm.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. J Controlled Release. 2007;124:20–27. doi: 10.1016/j.jconrel.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 39.Rejiba S, Reddy LH, Bigand C, Parmentier C, Couvreur P, Hajri A. Nanomedicine. 2011;7:841–849. doi: 10.1016/j.nano.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Reddy LH, Renoir JM, Marsaud V, Lepetre-Mouelhi S, Desmaele D, Couvreur P. Mol Pharmaceutics. 2009;6:1526–1535. doi: 10.1021/mp900099e. [DOI] [PubMed] [Google Scholar]
- 41.Weissleder R, Nahrendorf M, Pittet M. J Nat Mater. 2014;13:125–138. doi: 10.1038/nmat3780. [DOI] [PubMed] [Google Scholar]
- 42.Trehin R, Figueiredo JL, Pittet MJ, Weissleder R, Josephson L, Mahmood U. Neoplasia. 2006;8:302–311. doi: 10.1593/neo.05751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon EJ, Gestwicki JE, Strong LE, Kiessling LL. Chem Biol. 2000;7:9–16. doi: 10.1016/s1074-5521(00)00060-0. [DOI] [PubMed] [Google Scholar]
- 44.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peetla C, Rao KS, Labhasetwar V. Mol Pharmaceutics. 2009;6:1311–1320. doi: 10.1021/mp900011h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Cancer Res. 2006;66:2173–2180. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 47.Bruns CJ, Shrader M, Harbison MT, Portera C, Solorzano CC, Jauch KW, Hicklin DJ, Radinsky R, Ellis LM. Int J Cancer. 2002;102:101–108. doi: 10.1002/ijc.10681. [DOI] [PubMed] [Google Scholar]
- 48.Sun FX, Tohgo A, Bouvet M, Yagi S, Nassirpour R, Moossa AR, Hoffman RM. Cancer Res. 2003;63:80–85. [PubMed] [Google Scholar]
- 49.Bruns CJ, Harbison MT, Davis DW, Portera CA, Tsan R, McConkey DJ, Evans DB, Abbruzzese JL, Hicklin DJ, Radinsky R. Clin Cancer Res. 2000;6:1936–1948. [PubMed] [Google Scholar]
- 50.Storniolo AM, Allerheiligen SR, Pearce HL. Semin Oncol. 1997;24:S7-2–S7-7. [PubMed] [Google Scholar]
- 51.Laquente B, Lacasa C, Ginesta MM, Casanovas O, Figueras A, Galan M, Ribas IG, Germa JR, Capella G, Vinals F. Mol Cancer Ther. 2008;7:638–647. doi: 10.1158/1535-7163.MCT-07-2122. [DOI] [PubMed] [Google Scholar]
- 52.Amoh Y, Li L, Tsuji K, Moossa AR, Katsuoka K, Hoffman RM, Bouvet M. J Surg Res. 2006;132:164–169. doi: 10.1016/j.jss.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 53.Khan AW, Dhillon AP, Hutchins R, Abraham A, Shah SR, Snooks S, Davidson BR. Eur J Surg Oncol. 2002;28:637–644. doi: 10.1053/ejso.2002.1307. [DOI] [PubMed] [Google Scholar]
- 54.Jia L, Zhang MH, Yuan SZ, Huang WG. World J Gastroenterol. 2005;11:447–450. doi: 10.3748/wjg.v11.i3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang X, Galardi E, Duquette M, Lawler J, Parangi S. Clin Cancer Res. 2005;11:5622–5630. doi: 10.1158/1078-0432.CCR-05-0459. [DOI] [PubMed] [Google Scholar]
- 56.Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 57.Jain RK. Nat Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 58.Pusztaszeri MP, Seelentag W, Bosman FT. J Histochem Cytochem. 2006;54:385–395. doi: 10.1369/jhc.4A6514.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.