

Abstract
The broad-range phospholipase C (PLC) from Listeria monocytogenes has been expressed using an intein expression system and characterized. This zinc metalloenzyme, similar to the homologous enzyme from Bacillus cereus, targets a wide range of lipid substrates. With monomeric substrates, the length of the hydrophobic acyl chain has significant impact on enzyme efficiency by affecting substrate affinity (Km). Based on a homology model of the enzyme to the B. cereus protein, several active site residue mutations were generated. While this PLC shares many of the mechanistic characteristics of the B. cereus PLC, a major difference is that the L. monocytogenes enzyme displays an acidic pH optimum regardless of substrate status (monomer, micelle, or vesicle). This unusual behavior might be advantageous for its role in the pathogenicity of Listeria monocytogenes.
Keywords: phospholipase C, Listeria monocytogenes, acidic pH optimum, mutagenesis, vesicle binding
Graphical abstract
1. Introduction
Listeria monocytogenes is a Gram-positive, facultative intracellular bacterial pathogen that causes serious infections in humans and other animals. L. monocytogenes can invade a variety of cells through phagocytosis or pathogen-induced endocytosis whose critical events have been reviewed recently [1,2]. After entry into the host cell, the bacteria replicate within the host cell cytosol, and ultimately spread from cell-to-cell. An important feature of L. monocytogenes pathogenicity is its prompt escape from (i) the single-membrane vacuoles that surround the bacteria after initial host cell invasion and (ii) the double-membrane vacuoles resulting from cell-to-cell spread. Failure in vacuolar escape results in abortive and avirulent infection. Multiple L. monocytogenes virulence factors have been identified as responsible for mediating vacuolar escape, including listeriolysin O (LLO1), a pore-forming cytolysin that also has a role in triggering host responses [3], a phosphatidylinositol-specific phospholipase C [4,5], and a broad-range phospholipase C (LmPLC) [6–8]. LLO is essential in mediating phagosomal membrane disruption by forming pores in the target membrane; the PI-specific PLC is associated with evasion of autophagy (9). While less is known about the exact role of LmPLC in vacuolar escape, L. monocytogenes lacking LmPLC activity was found to be 20-fold less virulent in mice and defective in cell-to-cell spread (4).
Despite the number of reports on the contribution of the broad range PLC to L. monocytogenes virulence [4,6–8], with the exception of one report using LmPLC isolated from L. monocytogenes culture supernatant [10], there is limited knowledge about its enzymatic features and mechanism. In this study, the L. monocytogenes broad-range PLC was cloned, expressed as an intein fused protein in Escherichia coli, purified, and characterized in an active and homogeneous form. LmPLC orthologues from other Gram-positive bacteria, in particular the broad-range phospholipase C from Bacillus cereus (BcPLC) [11] and alpha-toxin from Clostridium perfringens [12], have been extensively characterized. LmPLC shares considerable sequence (38.7%) and functional identity with BcPLC, which binds three Zn2+ ions at the enzyme active sites. The zinc ions are critical for enzyme structure and function [12]. The structure of BcPLC has also been determined allowing us to model the LmPLC structure [13].
Systematic kinetic studies were undertaken to probe substrate specificity, Zn2+ dependence, pH dependence, and rate-determining step of LmPLC. Binding of the enzyme onto phosphatidylcholine/cholesterol liposomes was also examined by fluorescence correlation spectroscopy (FCS). Lastly, mutations of residues homologous to active site residues in BcPLC were made to test the structural model generated for the recombinant LmPLC. The results show distinctive LmPLC enzymatic features that provide insights into the physiological function of this broad-range phospholipase in Listeria monocytogenes.
2. Materials & Methods
2.1 Chemicals
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dibutyryl-sn-glycero-3-phosphocholine (diC4PC), 1,2-dihexanoyl-sn-glycero-3-phosphocholine (diC6PC), 1,2-diheptanoyl-sn-glycero-3-phosphocholine (diC7PC) and 1,2-dioleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (sodium salt) (DOPG), 1,2-doleoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DOPS), bovine liver phosphatidylinositol (PI) and porcine brain sphingomyelin were obtained from Avanti Polar Lipids (Alabaster, AL). Alexa Fluor 488 C5 maleimide was obtained from Life Technologies (Eugene, OR). IPTG and ampicillin were obtained from American Bioanalytical (Natick, MA). The B. cereus PLC as well as the remaining chemicals were from Sigma-Aldrich (St. Louis, MO) unless stated otherwise.
2.2 Gene construction and protein purification
Plasmid pET29-plcB containing the gene plcB for mature LmPLC was provided by Dr. Helene Marquis, Cornell University. The plcB gene that codes mature enzyme LmPLC was amplified via polymerase chain reaction (PCR) using HotStartTaq DNA Polymerase (Qiagen, Inc., Valencia, CA) and primers 5 -GGTTGCTCTTCCAACTGGTCCG-3 (to introduce the SapI site) and 5 - GCTGCATATGTCAGTGGTGGTGG-3 (NdeI site). The amplified fragment was ligated into pTYB21 with T4 DNA ligase (both obtained from New England BioLabs, Ipswich, MA). The final construct was sequenced by Genewiz, Inc. (Cambridge, MA). The plasmid was transformed into E. coli expression strain BL21-AI (Life Technologies, Grand Island, NY). Protein expression was induced with both 2 mg/mL L-arabinose and 0.4 mM IPTG. Cells were shaken at 225 rpm and incubated at 20 °C for 6 h before harvesting. This system expresses recombinant LmPLC with an N-terminal intein tag containing a chitin-binding domain that allows isolation of PLC on a chitin resin. On-resin excision of the intein by incubation with dithiothreitol was performed following the manufacturer's (New England Biolabs) protocol. The resulting protein with N- and C-terminal residues equivalent to the mature native LmPLC, was precipitated with 40% ammonium sulfate. After gentle stirring for 1 h, the solution was centrifuged to generate a protein pellet that was then re-dissolved in storage buffer (20 mM HEPES, 1 M NaCl, 2 mM dithiothreitol, pH 8.5). The high salt and basic pH kept the protein from precipitating over time. Excess ammonium sulfate was removed by dialysis against fresh storage buffer. The purified recombinant protein exhibited a circular dichroism spectrum, shown in Figure 1 (recorded at 22 °C on an AVIV Model 420 spectrometer), consistent with a predominantly helical structure. Deconvolution of the spectrum with K2D2 [14] indicated 84 % α-helix The high helix content is consistent with the crystal structure of BcPLC [13]. Aliquots of purified protein were stored at 4 °C or frozen with liquid nitrogen in 20% glycerol and stored at -80 °C. Site-directed point mutations of PLC were made with the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Inc., Santa Clara, CA). Mutant proteins were expressed and purified following the same protocol as the recombinant wild type LmPLC. Recombinant LLO was expressed using a plasmid, pET29b-hly, obtained from Dr. Daniel A. Portnoy, University of California, Berkeley. LLO was expressed and purified as described previously [15], Protein concentrations of all samples used in this study were measured with a Pierce BCA protein assay kit (Pierce Biotechnology, Rockford, IL) [16].
Figure 1.
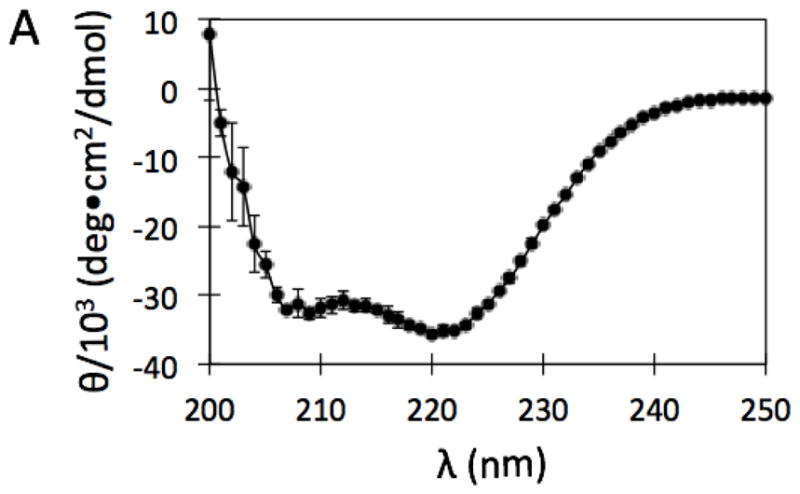
CD spectrum of purified recombinant LmPLC, in the absence of Zn2+, taken in storage buffer (20 mM HEPES, 1 M NaCl, 2 mM dithiothreitol, pH 8.5), at 22 °C. Data are shown as mean ± SD, n = 3 independent experiments.
2.3 Homology model of LmPLC
The online program SWISS-MODEL [17] was used to generate an LmPLC homology model structure based on the X-ray crystal structure of BcPLC [14].
2.4 Inductively coupled plasma mass spectrometry (ICP-MS)
The ICP-MS analysis was performed on a Perkin-Elmer NexION 300X ICP mass spectrometer (PerkinElmer, Waltham, MA) located at the University of Massachusetts Amherst, Department of Chemistry. Protein samples were subjected to acid digestion with a mixture of nitric acid and hydrogen peroxide. After digestion, samples were diluted to 10 mL. Triplicate measurements were taken for each sample. Both 63Zn and 65Zn were measured under kinetic energy discrimination (KED) mode. ICP-MS operating conditions were as follows: nebulizer flow rate: 0.95L/min; rf power: 1600 W; plasma Ar flow rate: 18 L/min; dwell time: 50 ms; KED cell gas: 4.6 mL/min.
2.5 Phosphate colorimetric assay
PLC activity towards monomeric synthetic short-chain phosphatidylcholine substrates (diC4PC, diC6PC, and diC7PC) and towards POPC or dioleoyl-phospholipids in Triton X-100 mixed micelles was measured with a phosphate colorimetric assay using slight modifications of a published protocol [18]. Specifically, reactions were carried out at 37 °C in 100 μL of 20 mM buffering agent (MES for pH 5.0, 5.5, and 6.0, MOPS for pH 6.5 and 7.0, HEPES for pH 7.5, 8.0, and 8.5), containing 150 mM NaCl, 0.1 mg/mL BSA, 50 μM ZnSO4 (unless otherwise specified) and the desired concentration of substrate. Buffers were pre-warmed for at least 20 min before the reaction was initiated by the addition of the PLC. At different time points, 15 μL of the reaction mixture was removed and quenched with 5 μL of 2 M Tris containing 0.4% SDS, pH 8.0 (the Tris cation is an inhibitor of BcPLC [19]). The sample was then boiled for 10 min. The resulting water-soluble glycerophosphoryl ester (glycerophosphocholine in the case of PC) was dephosphorylated by addition of 40 unit/ml of alkaline phosphatase. Inorganic phosphate was then quantified using a malachite green assay [20] with absorbance measured at 650 nm. Absorption measurements were carried out in triplicate in a SpectraMax M5 Microplate reader (Molecular Devices, Sunnyvale, CA).
2.6 Liposome preparation
Lipids were dissolved in chloroform and, as required, mixed 2:1 with cholesterol. After solvent removal with a rotary evaporator, the lipid film was further dried under vacuum overnight. Dry films were resuspended in the appropriate buffer for a particular experiment and extruded through polycarbonate membranes with 100 nm pores using an Avestin lipid extruder (Avestin, Ottawa, ON) to form large unilamellar vesicles (LUVs). Small unilamellar vesicles (SUVs) were prepared by ultrasonication of the multilamellar suspensions until solutions became nearly optically clear.
2.7 31P NMR assay of PLC enzymatic activity
The specific activity of PLC towards long-chain POPC in SUVs was measured using 31P NMR spectra acquired on an Agilent Direct Drive 600 MHz spectrometer (Santa Clara, CA). PLC was incubated with the vesicles for fixed times at 37 °C, then each sample was quenched by addition of 2 M Tris containing 0.4% SDS, pH 8.0 (1/3 of the reaction volume), followed by boiling the solution for 5 min. The unhydrolyzed POPC was solubilized in the SDS micelles, providing a sharp resonance for the PC that is well separated from that of the water-soluble glycerophosphocholine product. The concentration of product was determined by integrating the 31P resonances of PC and the glycerophosphocholine in spectra obtained with decoupling only during the acquisition. For all the 31P NMR assays, the reaction was not allowed to proceed to more than 10% hydrolysis of substrate.
2.8 Fluorescence correlation spectroscopy (FCS)
Use of FCS (and the specific instrument used to monitor protein binding to SUVs in this work) has been described in detail previously [21, 22]. Experiments were carried out at 20 °C with samples in chambered cover glass wells. Prior to use, the chambers were coated with 10 mg/mL of BSA and rinsed with reaction buffer containing 1 mg/mL of BSA to prevent protein adhesion to the sides of the wells. Protein samples (250 μL) were in MES-buffered saline (20 mM MES, 150 mM NaCl, pH 5.5) or HEPES-buffered saline (20 mM HEPES, 150 mM NaCl, pH 7.4) with 1 mg/mL of BSA. Vesicles were titrated into the sample containing a fixed concentration of fluorescent protein (10 nM) in the absence or presence of 50 μM Zn2+. The averaged diffusion coefficients, D, of fluorescent species were calculated using rhodamine 110 (D = 280 μm2/s) for calibration [23]. The protein used for these experiments, LmPLC variant C143S, has a single cysteine (Cys168) that was modified with a thiol-reactive Alexa Fluor 488 maleimide. The activity of the C143S mutant towards PC SUVs was comparable to that of the recombinant LmPLC, indicating that neither the mutation nor the labeling affects the overall behavior of the protein.
2.9 Hemolytic activity
Human red blood cells (obtained from Research Blood Components, Boston, MA) were washed three times in phosphate buffered saline (PBS, 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl), pH 7.4, and resuspended to 2% (v/v) in PBS at pH 7.4 or pH 5.5 (adjusted with HCl). Proteins were diluted to various concentrations in the appropriate pH buffer and mixed 1:1 (v/v) with the red blood cell solution. Three samples were prepared and monitored for each protein concentration. The resulting mixtures were gently shaken and incubated at 37 °C for 1 h, followed by centrifugation. Aliquots of the supernatant from each sample were transferred to a 96-well plate for absorbance measurement at 545 nm using a SpectraMax M5 Microplate reader (Molecular Devices, Sunnyvale, CA). The fractional hemolysis was calculated from the absorbance as (Aprotein - Abuffer)/(A+TX-100 - Abuffer). PBS buffer at pH 5.5 or pH 7.4 was mixed with cells (no LLO or other recombinant protein) to yield Abuffer, and complete hemolysis was achieved by the addition of 0.1 % Triton X-100 to the buffer.
3. Results
3.1 Protein characterization and Zn2+ content
The broad-range phospholipase C, encoded by the plcB gene, is synthesized and secreted as a proenzyme with a molecular weight of 33 kDa. Cleavage of the inactive proenzyme by a metalloprotease, generates an active, 29 kDa enzyme [24–27]. For heterologous protein expression in E. coli, an N-terminal intein fusion strategy was used, leading to expression and purification of LmPLC in a homogeneous and active form. Throughout the protein expression and purification procedures, LmPLC was in an environment without added Zn2+. The broad-range PLC displayed the same CD spectrum in the absence of added Zn2+ (Figure 1) and with 100 μM Zn2+ added.
To gauge the affinity of LmPLC for Zn2+, we set up a dialysis experiment with two different zinc concentrations. The protein was split into two portions. Each portion was dialyzed against 5 μM or 0 μM of Zn2+ in dialysis buffer (20 mM HEPES buffer, 150 mM NaCl, pH 8.0) overnight at 4 °C. The zinc content in both the post-dialysis buffer and the post-dialysis protein sample was measured by ICP-MS [28]. Zinc content measured in the post-dialysis buffers was referred to as “free Zn2+”. The difference between free Zn2+ and the total Zn2+ in the post-dialysis protein sample reflects the amount of PLC-associated Zn2+, from which the average zinc/protein ratio as, Zn2+/PLC, was calculated assuming different Kd values for each of the Zn2+ ions (Table 1). Each LmPLC bound an average of 0.42 zinc ions in buffer with no added Zn2+. Assuming that LmPLC, like BcPLC, binds three Zn2+, the dissociation constant of the first zinc ion from LmPLC can be estimated from Equation (1). A value of 54 nM is obtained for Kd1.
Table 1.
ICP-MS analysis of LmPLC bound Zn2+ content under different conditions a.
| sample | Free Zn2+ (μM) b | Total Zn2+ (μM) b | PLC-Zn2+ (μM)b | PLC (μM) c | Zn2+/PLC |
|---|---|---|---|---|---|
|
| |||||
| Non-Zn2+ buffer | 0.040±0.003 | ||||
| LmPLC in non-Zn2+ buffer | 0.040±0.003 | 1.30±0.02 | 1.26±0.02 | 2.97±0.15 | 0.42±0.02 |
| Zn2+-buffer | 5.04±0.06 | ||||
| LmPLC in Zn2+-buffer | 5.04±0.06 | 8.73±0.07 | 3.69±0.07 | 1.78±0.09 | 2.1±0.2 |
Data are shown as mean ± SD, n = 3 independent experiments.
Free Zn2+ is the zinc concentration measured in the post-dialysis buffer sample; total Zn2+ is the concentration measured in the post-dialysis protein sample; PLC-Zn2+ is total Zn2+ less free Zn2+.
LmPLC protein concentration was measured with BCA protein assay [17].
In buffer with 5 μM Zn2+ added, the dialysis yielded a ratio of 2.1 Zn2+ ions per protein. For this sample, a rough estimate of Kd3 using Equation 2 suggests a Kd3 ~ 60 μM. For comparison, when excess Zn2+ was removed from solutions with BcPLC, three metal ions were still bound to the enzyme [29]. Thus, LmPLC has a considerably weaker affinity for Zn2+ than the Bacillus enzyme.
| Eqn. (1) |
| Eqn. (2) |
3.2 Substrate head group and acyl chain length specificity
Recombinant LmPLC can catalyze the hydrolysis of a range of phospholipids with different head groups including DOPC, DOPE, DOPS, DOPG, and sphingomyelin. Of all the phospholipids tested (6 mM phospholipid in 20 mM Triton X-100), only phosphatidylinositol was not appreciably hydrolyzed under these conditions (Figure 2A). The dependence of specific activity on head group was DOPC > SM ~ DOPE ~ DOPS > DOPG, consistent with a previous report using LmPLC protein purified from L. monocytogenes [10] or culture supernatants overexpressing the enzyme [30]. Since PC was the best substrate for LmPLC, it was used as the substrate for more in-depth kinetic studies of the recombinant LmPLC.
Figure 2.

Effect of phospholipid polar headgroup and substrate aggregation state on LmPLC activity. (A) Relative activity of LmPLC towards different phospholipids (6 mM) dispersed in 20 mM Triton X-100, 20 mM HEPES, 150 mM NaCl pH 6, 37°C, compared to the % hydrolysis rate for PC (defined as 100). (B) Enzyme specific activity is shown as a function of diC7PC concentration in 20 mM MES, 150 mM NaCl, 50 μM Zn2+, 0.1 mg/mL BSA, pH 6. The black solid line is the fit for monomeric diC7PC as substrate; the dashed line is the fit for diC7PC micelles as substrates assuming a constant monomer activity and a sharp phase separation of monomers and micelles. Data are shown as mean ± SD, n = 3 independent experiments.
Most phospholipases, including BcPLC [31, 32], prefer substrate containing aggregates and micelles, e.g., synthetic short-chain phospholipids or long-chain phospholipids dispersed in detergents. Therefore, the effect of micelle formation on LmPLC activity was examined using diC7PC (Figure 2B). A clear, very steep transition in enzyme activity is observed around 1.8 mM diC7PC, a value very close to the published critical micelle concentration (CMC) of the pure phospholipid (1.5 mM [33]). Below this substrate concentration, there was an increase in activity that could be fit with a hyperbolic function to extract Km and kcat for monomeric diC7PC. Once micelles formed, above 1.8 mM diC7PC, LmPLC specific activity increased 3-fold compared to the activity towards monomeric substrate. As the catalytic mechanism is unaltered, the enhanced activity is likely due to processive catalysis and possibly facilitation of substrate binding and/or product dissociation by the micellar interface. The extent of the rate enhancement upon micelle formation is comparable to what has been observed for BcPLC activity towards a variety of short-chain PC and lyso-PC substrates [31, 34].
Pure diC6PC has a higher CMC, 14 mM [33], so it was used for further studies of the pH dependence of the LmPLC activity for monomeric substrates. The turnover number, kcat, exhibited a relatively flat profile between pH 5 and 8 (Figure 3A). In contrast, Km showed a reproducible minimum around pH 6 and 6.5 (Figure 3B). The variation in Km led to maximum of the overall catalytic efficiency kcat/Km around pH 6–6.5 (Figure 3C).
Figure 3.

Kinetic parameters for LmPLC catalyzed hydrolysis of diC6PC. The pH dependence of (A) kcat, (B) Km, and (C) kcat/Km are shown. All assays were conducted in the presence of 50 μM ZnSO4. (D) The variation in specific activity towards 5 mM diC6PC, pH 6.0, as a function of concentration of Zn2+ could be fit with an apparent KZn2+ = 1.9 ± 0.2 μM. Data are shown as mean ± SD, n = 3 independent experiments.
In order to compare different chain length short-chain PCs, we chose pH 6 where LmPLC has high catalytic efficiency towards monomeric substrate. A comparison of Km, kcat, and catalytic efficiency for diC4PC, diC6PC and diC7PC at this pH is shown in Table 2. For these three monomeric substrates, the Km values for LmPLC displayed a strong dependence on acyl chain length, while the kcat values were quite similar. This suggests that at least for monomeric substrates some degree of hydrophobicity is required for an isolated phospholipid to bind productively in the active site.
Table 2.
Kinetic parameters of LmPLC for substrates short-chain phosphatidylcholine substrates at pH 6 a.
| substrate | CMC (mM) b | Km (mM) | kcat (s−1) c | kcat/Km (mM−1 s−1) |
|---|---|---|---|---|
| diC4PC | 250 | 43.5±3.4 | 8.0±0.4 | 0.18±0.02 |
| diC6PC | 14 | 2.8±0.8 | 12.8±1.5 | 4.6±1.9 |
| diC7PC | 1.5 | 0.20±0.05 | 8.5±0.7 | 42±15 |
Data are shown as mean ± SD, n = 3 independent experiments.
CMC values for the short-chain PC molecules are from [33].
All assays were conducted in the presence of 50 μM ZnSO4.
The Zn2+ requirement of LmPLC was also examined using diC6PC as a substrate at pH 6. The increase in specific activity with added Zn2+ was hyperbolic and yielded an effective dissociation constant (Kd) for the weakest catalytically relevant zinc ion from LmPLC as 1.9 ± 0.2 μM (Fig. 3D). This value is significantly lower than what would be estimated from ICP-MS for occupying all the Zn2+ sites (Kd3 ~ 60 μM) at pH 8. Decreasing pH should decrease Zn2+ affinity, so the much lower kinetic estimate of Zn2+ binding needed for activity suggests that the third zinc ion binding to LmPLC is enhanced by the presence of substrate.
3.3 Mechanistic behavior of LmPLC compared to BcPLC with a monomeric substrate
Elegant work by Martin and coworkers with BcPLC [18, 29, 35, 36] provides a framework for analyzing the enzyme from L. monocytogenes. Variation of solvent viscosity was used to investigate the rate-limiting step of LmPLC. If the reaction is diffusion-controlled, such as by monomer substrate binding or product release, the enzymatic reaction will be affected by solvent viscosity since the diffusion rate of small molecules is inversely proportional to the microviscosity of the solvent. However, the activity of LmPLC was independent of solvent viscosity (Figure 4A). For comparison, the dashed line shows the expected relationship of enzyme efficiency for a diffusion-controlled reaction. Similar to BcPLC, product release is not the rate-limiting step in the enzymatic hydrolysis of the water-soluble substrate.
Figure 4.

Effect of solution viscosity and mole fraction D2O on the enzyme efficiency, Eff = (kcat/Km), for LmPLC hydrolysis of 5 mM diC6PC, pH 6.0. (A) Relative enzyme efficiency, Effo/Eff = (kcat/Km)o / (kcat/Km), where the subscript o denotes H2O buffer in the absence of sucrose, is shown as a function of solution viscosity (varied by the addition of sucrose). η is the viscosity of the sucrose containing solutions and ηo is the solution viscosity in the absence of sucrose. (kcat/Km)o is the LmPLC efficiency towards diC6PC in the absence of sucrose. (B) Variation of relative enzyme efficiency with mole fraction of D2O (XD2O). Effo = (kcat/Km)o is the enzymatic efficiency in the absence of D2O. Data are shown as mean ± SD, n = 3 independent experiments.
Solvent isotope effects on LmPLC activity were also examined. If proton transfer is involved in the rate-limiting step of the reaction with soluble substrate, varying the D2O content of the solvent should affect the reaction rate. Indeed, when D2O was added to the reaction buffer, the activity decreased linearly with a slope of −0.47 (R2=0.90). The linear fit also indicates a single proton is transferred during the rate-determining step for the LmPLC as was observed for BcPLC [36]. However, there is a significant difference in the behavior of the two enzymes with the same monomeric substrate. In examining the enzyme efficiency kcat/Km, we see that LmPLC has an acidic pH optimum generated by an unusual Km profile that has a minimum around pH 6–7. In contrast, BcPLC kcat/Km is characterized by an increase from ~30 mM−1 s−1 at pH 5 to a plateau of ~300 mM−1 s−1 between pH 6.5 and 8 [37].
3.4 pH profile of LmPLC towards aggregated substrates
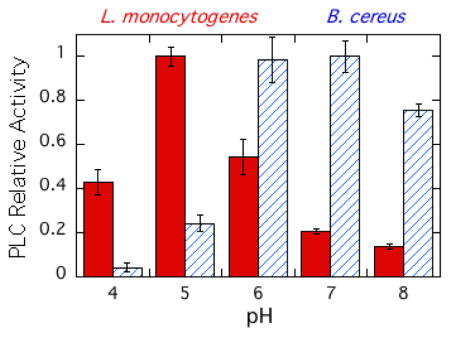
Expression, activation, and release of the virulence factor LmPLC is pH-sensitive [38, 39] making it important to characterize the enzyme's pH dependence towards substrates in an aggregate (micelles and bilayer vesicles) as well as short-chain PC monomers. Recombinant LmPLC specific activity towards POPC (6 mM) in 20 mM Triton X-100 micelles was maximal at pH 5 (21 μmol min−1 mg−1), more acidic conditions than observed for the monomer substrate diC6PC (Figure 5A). For comparison, with the same assay and substrates, a commercially available BcPLC yielded the highest activity at pH 7 (Figure 5A). Reports for recombinant BcPLC activity for POPC/Triton X-100 micelles are typically 200–500 μmol min−1 mg−1 depending on the ratio of detergent to PC [40]. The pH profile for recombinant LmPLC differs from what was reported by Goldfine et al. [10] for LmPLC isolated from Listeria culture supernatant. Those researchers found a broad pH profile, from pH 5.5 to 8.0. There are several differences in assay conditions used for the recombinant enzyme. Here a higher substrate concentration, 6 mM, was used (1.85 mM previously), with a lower ratio of Triton X-100 to PC (3.3:1 compared to 8:1). All our assays showed less than 10% substrate hydrolysis and specific activities were reproducible for different preparations of the recombinant protein.
Figure 5.
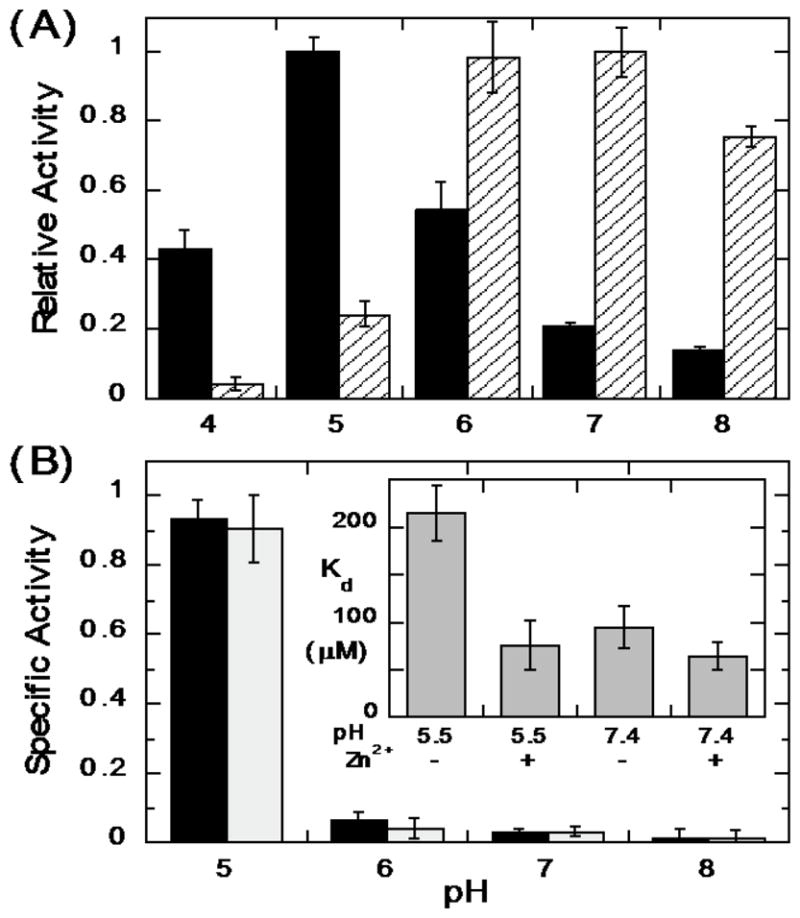
The pH dependence of LmPLC activity towards POPC presented in micelles and vesicles. (A) Enzyme activities relative to the enzyme maximal activity towards POPC (6 mM) dispersed in 20 mM Triton X-100 for LmPLC (black) and BcPLC (hatched). (B) Specific activity (μmol min−1 mg−1) of LmPLC towards 4 mM POPC (black bar) or POPC /cholesterol (4 mM /2 mM) (grey bar) SUVs as a function of pH. The inset in (B) shows the apparent dissociation constant (Kd) of LmPLC (10 nM) binding to POPC/POPG (0.95/0.05) SUVs at pH 5.5 (20 mM MES) or pH 7.4 (20 mM HEPES), with 150 mM NaCl, 50 μM Zn2+, and 1 mg/mL BSA. Data are shown as mean ± SD, n = 3 independent experiments.
The specific activity of LmPLC was also examined towards 4 mM POPC SUVs in the absence or presence of cholesterol (4 mM POPC ± 2 mM cholesterol). LmPLC, similar to BcPLC and most other bacterial phospholipase enzymes, has considerably lower activity towards POPC SUVs relative to micelles (Figure 5B). For PC presented in a vesicle, LmPLC exhibits dramatically higher specific activity at pH 5 compared to pH 7. Incorporating cholesterol into the vesicles had no specific effect on enzyme activity. In contrast, BcPLC exhibits lower activity at pH 5 compared to pH 7 [41].
3.5 Binding of PLC to vesicles
For monomer substrates, Km dictates the catalytic efficiency across the pH range from 5 to 8.5 (Figure 3). Therefore, the affinity of LmPLC for phospholipid vesicles was measured using FCS to see if increased vesicle affinity is correlated with the higher activity observed at acidic pH. Under the experimental conditions used (10 nM LmPLC purified without added Zn2+ and POPC SUVs at 20 °C), the specific activity of LmPLC towards the vesicles is sufficiently low so that very little PC would be hydrolyzed during sample preparation and FCS measurements (typically under 30 min). Even with added Zn2+, product formation from POPC in the SUVs would be minimal. This allowed us to measure the partitioning of LmPLC onto the POPC vesicles.
The protein used for FCS was LmPLC C143S, which has a single cysteine (Cys168) available for conjugation with a fluorescent dye. Nanomolar concentrations of labeled protein in solution were titrated with POPC/POPG (0.95/0.05 mole fraction) SUVs. The small amount of negatively charged POPG was added to prevent fusion of the vesicles. The apparent dissociation constant (Kd) describing protein binding to these small vesicles is shown in the inset in Figure 5B. Two parameters were varied: zinc concentration and pH. The apparent Kd at pH 7.4 was 65–85 μM with no significant change when excess Zn2+ was added. The same apparent Kd was observed at pH 5.5 when 50 μM Zn2+ was present. However, at the acidic pH without Zn2+ added, the apparent Kd of the protein for the SUV was about 3-fold higher. The similar partitioning of the protein onto SUVs at pH 5.5 and 7.4 with an apparent Kd below 0.1 mM under the assay conditions (i.e., with excess Zn2+) indicates that bulk partitioning of the protein on vesicles is not responsible for the lower enzymatic activity observed at neutral compared to acidic pH.
3.6 Predicted model for LmPLC and mutation of active site residues
The high sequence homology between LmPLC and BcPLC allowed us to use the BcPLC crystal structure [13, 19, 42] to model LmPLC (Fig. 6A). The modeled structure, not surprisingly, is very similar to that for BcPLC showing a helical single domain protein. Work by Martin and coworkers showed that residues Asp55, Tyr56, and Phe66 are part of the active site and important for enzymatic activity [29, 43]. LmPLC conserves two of these three residues with the exception of His56, which corresponds to a Tyr in BcPLC.
Figure 6.

Predicted structure of L. monocytogenes broad-range PLC showing the location of the mutated residues and the effects on enzymatic activity. (A) The model for LmPLC was based on the crystal structure of BcPLC (PDB ID: 1AH7) [13]; the colored residues (with the exception of the two Cys residues) were mutated based on their sequence homology to key active site residues in BcPLC. (B) Specific activity of LmPLC variants towards 5 mM diC6PC in 20 mM MES, 150 mM NaCl, 50 μM Zn2+, 0.1 mg/mL BSA, pH 6. Data are shown as mean ± SD, n = 3 independent experiments.
In the BcPLC studies, Asp55 was suggested as the putative general base that aids in activating the nucleophilic water for attack on the phosphodiester since alterations at this position reduced enzymatic activity 104~106-fold. For LmPLC, replacement of Asp55 with asparagine also reduced the enzyme activity towards diC6PC monitored at pH 6 (Fig. 6B). This suggests that Asp55 likely has a similar role in LmPLC catalysis where it helps to polarize the water molecule that will attack the phosphorus (aided and abetted by other side chains). In the structure of a BcPLC-inhibitor complex [42] the choline moiety of the analogous inhibitor could form a cationic complex with the π-system of one or both Tyr56 and Phe66. Mutation of these two adjacent aromatic residues affect BcPLC substrate recognition and enzyme specificity [43]. In LmPLC, the activity of mutant H56Y was comparable to wild type enzyme as seen for assays using culture supernatant expressing the mutant protein LmPLC H56Y [44]. However, F66Y, a mutation that might be expected to be conservative, caused a dramatic loss of activity. It is possible that in LmPLC F66Y the added hydroxyl group might form hydrogen bonds with other active site residues so that this residue is no longer aligned for cation-π interactions with the substrate.
A unique feature of LmPLC compared to BcPLC is that it has two cysteine residues, Cys143 and Cys168, which are absent in the BcPLC sequence. Both cysteine residues, on the surface of the protein, were individually mutated to serine to see if there was any effect on LmPLC activity. C143S actually exhibited slightly higher diC6PC hydrolysis activity than the wild type enzyme, while the activity of C168S was reduced (Figure 6). These results indicate that the two cysteine residues are not essential for catalysis. However, these residues could allow the enzyme to be sensitive to the redox environment.
3.7 Do LmPLC and LLO act synergistically?
Both LLO and LmPLC are L. monocytogenes virulence factors that contribute to phagosomal membrane disruption. It is tempting to speculate that they could act synergistically. With this in mind, we examined (i) the effect of the recombinant LmPLC on LLO-mediated hemolysis of red blood cells and (ii) the effect of LLO pore-formation on the LmPLC-catalyzed hydrolysis of POPC/cholesterol (2:1) LUVs.
In the hemolytic assay, the presence of LmPLC (up to a maximum concentration of 20 nM, which on its own is not lytic) had little to no statistically significant impact on hemolysis caused by LLO (Figure 7A). This is presumably due to the fact that LLO itself is a very potent cytolysin that disrupts the target membrane before significant lipid hydrolysis by LmPLC.
Figure 7.

LmPLC and LLO work independently. (A) Adding increasing amounts of LmPLC (0.0 (filled circle), 0.20 (empty circle), 2.0 (filled box), and 20 (empty box) nM), had little significant effect on LLO hemolysis of human erythrocytes (in PBS, pH 5.5). (B) Time-course of LmPLC (10 nM) hydrolysis of 5 mM POPC/cholesterol (2:1) LUVs in 20 mM MES buffer, 1 mM NaCl, pH 5.5, 50 μM Zn2+, 0.1 mg/mL BSA. LLO concentrations in μM were: 0 (filled circle), 0.05 (empty circle) 0.1 (filled box), 1 (empty box), and 4 (filled triangle). In both (A) and (B) the data is from one experiment that is representative of three independent experiments.
POPC/cholesterol LUVs were used to assess the effect(s) of LLO on LmPLC activity. As seen in Fig. 7B, large vesicles are not good substrates for LmPLC (the specific activity of the enzyme toward SUVs is roughly 10-fold higher). The kinetics exhibit a lag phase as observed for many other phospholipase enzymes. This lag is often attributed to a requirement for significant product build-up leading to formation of defects that can promote critical interactions between the membrane and the protein [45]. Adding LLO to LUVs to disrupt the large vesicle structures (which presumably generates defects), did have an effect on LmPLC activity. The difference was most pronounced at 30 min where it appears that LLO modification of the vesicles increases PC hydrolysis in an LLO-dependent manner. The maximum amount of LLO added, 4 μM, was more than enough to disrupt the integrity of the LUVs producing a variety of much smaller bilayer structures. However, stimulation of LmPLC activity was relatively small, two-fold at 30-min time point. Thus, in these in vitro experiments, there was no effect of the LmPLC on the LLO–induced hemolysis; there was a small effect of LLO treatment of LUVs that shortened the lag phase for LmPLC -catalyzed hydrolysis of PC.
4. Discussion
The broad-range LmPLC, while similar in many respects to the PLC from B. cereus, has distinctive enzymatic features that are likely to be important for its role in L. monocytogenes pathogenicity. These are likely to be important since as noted previously [10,44], LmPLC exhibits much lower (~10-fold) activity for PC in Triton X-100 micelles. The significantly lower specific activity for LmPLC is also seen for short-chain PC monomers and micelles as well as vesicles. However, the most striking difference is the preference of recombinant LmPLC for acidic pH for optimal enzyme efficiency regardless of substrate physical state. For monomeric diC6PC this shifted pH optimum of enzyme efficiency is due to the pH dependence of Km, not kcat. Towards interfacial substrates, LmPLC activity is much lower as the pH increases. Under our assay conditions, the well-folded protein is essentially all partitioned onto vesicles. However, bulk binding of the protein to a vesicle may occur independently of a phospholipid occupying the active site. Either directly or indirectly through a conformational change, the protonated group must control substrate binding in the active site. When it is deprotonated, substrate access is more difficult.
Further insight into what contributes to substrate binding in the active site is provided by measurements of LmPLC enzyme kinetics with monomeric short-chain PC substrates. Even though the hydrolysis reaction catalyzed by LmPLC occurs at the polar head group of a phospholipid, there is a significant variation in Km, but not in the turnover rate (kcat), with acyl chain length. The decrease in CMC tracks the decrease in the Km for the substrate (Table 2). For example, the 18-fold decrease in CMC from diC4PC to diC6PC leads to a 16-fold decrease in Km. These results indicate that hydrophobic interactions aid in substrate docking into the active site of LmPLC. However, the nature of these interactions is harder to pin down. Although the substrates on their own are monomeric significantly below the CMC (and around the value measured as the Km), it is possible that the protein binds several phospholipids weakly and could thus nucleate a micro- or mini-micelle. The resulting cluster of lipids with shielded acyl chains could facilitate diffusion of a substrate molecule into and product out of the active site. The longer the monomer chain length, the easier it would be for several of these molecules to interact with the protein providing a reservoir of hydrophobic acyl chains. Such transient structures could optimize entry and release from the active site and thereby enhance enzymatic efficiency. There are several instances in the recent literature of phospholipids at monomer concentrations interacting with proteins and forming much larger aggregates [45, 46]. This phenomenon may be a relatively common property of amphitropic proteins.
As a virulence factor, LmPLC is expressed, secreted, and proteolytically activated with acidification of the environment [7, 27, 37, 38]. It has therefore been implicated in assisting Listeria escape from acidic phagosomes/endosomes [5, 7–9]. Similarly, LLO, the essential factor in L. monocytogenes phagosomal escape, exhibits an acidic pH optimum for pore formation [4, 16]. This compartmentalization of LLO toxicity is critical for the intracellular life of L. monocytogenes. Substituting a cholesterol-dependent cytolysin that does not exhibit an acidic pH requirement for pore formation leads to cytotoxicity in host cells which would prevent bacterial growth [16]. A similar pH regulation of LmPLC activity would maximize lipolytic activity inside the vacuoles for efficient bacterial escape and inhibit phospholipid hydrolysis in the more basic conditions of the cytosol thereby minimizing damage to host cell membranes and reducing LmPLC toxicity.
5. Conclusions
Nonspecific phospholipase C from Listeria monocytogenes is an important virulence factor of that bacterial pathogen [5–9] whose role in pathogenicity has not been clearly defined. This work provides a systematic examination of multiple factors that affect LmPLC catalysis and lays a solid foundation for a better understanding of LmPLC s biological role. Kinetics with monomer substrates suggest hydrophobicity of the substrate aids in binding in the active site, and that, similar to the homologous PLC from B. cereus, proton transfer is rate limiting. However, LmPLC has an unusual pH profile that is acidic compared to BcPLC. Bulk binding of the protein to bilayers is not responsible for the higher activity at acidic pH. The lower specific activity of this PLC compared to that for secreted PLCs also contributes to its activity in contributing to vacuolar escape upon acidification and quick inhibition once in the cytosol.
Highlights.
The broad-range phospholipase C (LmPLC) from Listeria monocytogenes was purified and its detailed enzymatic features in dependent of Zn2+, substrate, and active site residues were characterized.
The distinct acidic pH enzymatic optimum was shown for LmPLC, which suits its role as the virulence factor of the intracellular pathogen.
The interdependence of LmPLC with another major Listeria virulence factor LLO was evaluated in in vitro systems.
Acknowledgments
We are grateful to Professor Helene Marquis for providing us with the gene for the L. monocytogenes PLC. We also acknowledge Professor Richard W. Vachet and Gokhan Elci at the University of Massachusetts, Amherst, for help with the ICP-MS analysis of the LmPLC samples. These studies were supported by National Institute of Health grant R01 GM60418 (to Mary F. Roberts).
Footnotes
Abbreviations: BSA, bovine serum albumin; CMC, critical micelle concentration; FCS, fluorescence correlation spectroscopy; ICP-MS, inductively coupled plasma mass spectrometry; LLO, listeriolysin O; LUV, large unilamellar vesicle; PC, phosphatidylcholine; diC4PC, 1,2-dibutyryl-PC; diC6PC, 1,2-dihexanoyl-PC; diC7PC, 1,2-diheptanoyl-PC; DOPC, 1,2-dioleoyl-PC; DOPG, 1,2-dioleoyl-sn-glycero-3-phospho-(1'-rac-glycerol); DOPS, 1,2-doleoyl-sn-glycero-3-phospho-L-serine; PI, phosphatidylinositol; PLC, phospholipase C; LmPLC, Listeria monocytogenes PLC; BcPLC, Bacillus cereus PLC; SUV, small unilamellar vesicle;.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pizarro-Cerdá J, Kühbacher A, Cossart P. Entry of Listeria monocytogenes in mammalian epithelial ells: an updated view. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a010009. pii: a010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cossart P, Lebreton A. A trip in the New Microbiology with the bacterial pathogen Listeria monocytogenes. FEBS Lett. 2014;588:2437–2445. doi: 10.1016/j.febslet.2014.05.051. [DOI] [PubMed] [Google Scholar]
- 3.Seveau S. Multifaceted activity of listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Subcell Biochem. 2014;89:161–195. doi: 10.1007/978-94-017-8881-6_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith GA, Marquis HH, Jones S, Johnston NC, Portnoy DA, Goldfine H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun. 1995;63:4231–4237. doi: 10.1128/iai.63.11.4231-4237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camilli A, Goldfine H, Portnoy DA. Listeria monocytogenes mutants lacking phosphatidylinositol-specific phospholipase C are avirulent. J Exp Med. 1991;173:751–754. doi: 10.1084/jem.173.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marquis H, Doshi VV, Portnoy DA. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect Immun. 1995;63:4531–4534. doi: 10.1128/iai.63.11.4531-4534.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gründling A, Gonzalez MD, Higgins DE. Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J Bacteriol. 2003;185:6295–6307. doi: 10.1128/JB.185.21.6295-6307.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alberti-Segui C, Goeden KR, Higgins DE. Differential function of Listeria monocytogenes listeriolysin O and phospholipases C in vacuolar dissolution following cell-to-cell spread. Cell Microbiol. 2007;9:179–195. doi: 10.1111/j.1462-5822.2006.00780.x. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell G, Ge L, Huang Q, Chen C, Kianian S, Roberts MF, Schekman R, Portnoy DA. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun. 2015;83:2175–2184. doi: 10.1128/IAI.00110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldfine H, Johnston NC, Knob C. Nonspecific phospholipase C of Listeria monocytogenes: activity on phospholipids in Triton X-100-mixed micelles and in biological membranes. J Bacteriol. 1993;175:4298–4306. doi: 10.1128/jb.175.14.4298-4306.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hergenrother PJ, Martin SF. Phosphatidylcholine-preferring phospholipase C from B. cereus. function, structure, and mechanism. Top Curr Chem. 2001;211:131–167. [Google Scholar]
- 12.Titball RW, Naylor CE, Basak AK. The Clostridium perfringens α-toxin. Anaerobe. 1999;5:51–64. doi: 10.1006/anae.1999.0191. [DOI] [PubMed] [Google Scholar]
- 13.Hough E, Hansen LK, Birknes B, Jynge K, Hansen S, Hordvik A, Little C, Dodson E, Derewenda Z. High-resolution (1.5 Å) crystal structure of phospholipase C from Bacillus cereus. Nature. 1989;338:357–360. doi: 10.1038/338357a0. [DOI] [PubMed] [Google Scholar]
- 14.Perez-Iratxeta C, Andrade-Navarro MA. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol. 2008;8:25. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J Cell Biol. 2002;156:1029–1038. doi: 10.1083/jcb.200201081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith PK, Krohn RL, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 17.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 18.Hergenrother PJ, Martin SF. Determination of the kinetic parameters for phospholipase C (Bacillus cereus) on different phospholipid substrates using a chromogenic assay based on the quantitation of inorganic phosphate. Anal Biochem. 1997;251:45–49. doi: 10.1006/abio.1997.2251. [DOI] [PubMed] [Google Scholar]
- 19.Hansen S, Hansen LK, Hough E. The crystal structure of tris-inhibited phospholipase C from Bacillus cereus at 1.9 Å resolution; nature of the metal ion in site 2. J Mol Biol. 1993;231:870–876. doi: 10.1006/jmbi.1993.1333. [DOI] [PubMed] [Google Scholar]
- 20.Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. A malachite green colorimetric assay for protein phosphatase activity. Anal Biochem. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- 21.Pu M, Roberts MF, Gershenson A. Fluorescence correlation spectroscopy of phosphatidylinositol-specific phospholipase C monitors the interplay of substrate and activator binding sites. Biochemistry. 2009;48:6835–6845. doi: 10.1021/bi900633p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng J, Karri S, Grauffel C, Wang F, Reuter N, Roberts MF, Wintrode PL, Gershenson A. Does changing the predicted dynamics of a phospholipase C alter activity and membrane binding? Biophys J. 2012;104:185–195. doi: 10.1016/j.bpj.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magde D, Elson EL, Webb WW. Fluorescence correlation spectroscopy II. An experimental realization. Biopolymers. 1974;13:29–61. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 24.Geoffroy C, Raveneau J, Beretti J, Lecroisey A, Vazquez-Boland J, Alouf JE, Berche P. Purification and characterization of an extracellular 29-kilodalton phospholipase C from Listeria monocytogenes, Infect Immun. 1991;59:2382–2388. doi: 10.1128/iai.59.7.2382-2388.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raveneau J, Geoffroy C, Beretti JL, Gaillard JL, Alouf JE, Berche P. Reduced virulence of a Listeria monocytogenes phospholipase deficient mutant obtained by transposon insertion into the zinc metalloprotease gene. Infect Immun. 1992;60:916–921. doi: 10.1128/iai.60.3.916-921.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marquis H, Goldfine H, Portnoy DA. Proteolytic pathways of activation and degradation of a bacterial phospholipase C during intracellular infection by Listeria monocytogenes. J Cell Biol. 1997;137:1381–1392. doi: 10.1083/jcb.137.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Neil HS, Forster BM, Roberts KL, Chambers AJ, Bitar AP, Marquis H. The propeptide of the metalloprotease of Listeria monocytogenes controls compartmentalization of the zymogen during intracellular infection. J Bacteriol. 2009;191:3594–3603. doi: 10.1128/JB.01168-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whittal RM, Ball HL, Cohen RE, Burlingame AL, Prusiner SB, Baldwin MA. Copper binding to octarepeat peptides of the prion protein monitored by mass spectrometry. Prot Sci. 2000;9:332–343. doi: 10.1110/ps.9.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin SF, Hergenrother PJ. General base catalysis by the phosphatidylcholine-preferring phospholipase C from Bacillus cereus: the role of Glu4 and Asp55. Biochemistry. 1998;37:5755–5760. doi: 10.1021/bi972948k. [DOI] [PubMed] [Google Scholar]
- 30.Zuckert WR, Marquis H, Goldfine H. Modulation of enzymatic activity and biological function of Listeria monocytogenes broad-range phospholipase C by amino acid substitutions and by replacement with the Bacillus cereus ortholog. Infect Immun. 1998;66:4823–4831. doi: 10.1128/iai.66.10.4823-4831.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Sayed MY, DeBose CD, Coury LA, Roberts MF. Sensitivity of phospholipase C (Bacillus cereus) activity to phosphatidylcholine structural modifications. Biochim Biophys Acta. 1985;837:325–335. doi: 10.1016/0005-2760(85)90056-6. [DOI] [PubMed] [Google Scholar]
- 32.Little C. Phospholipase C from Bacillus cereus. Action on some artificial lecithins. Acta Chem Scand. 1977;B31:267–272. doi: 10.3891/acta.chem.scand.31b-0267. [DOI] [PubMed] [Google Scholar]
- 33.Bian J, Roberts MF. Thermodynamic comparison of lyso- and diacylphosphatidyl-cholines. J Colloid Interface Sci. 1992;153:420–428. [Google Scholar]
- 34.El-Sayed MY, Roberts MF. Charged detergents enhance the activity of phospholipase C (Bacillus cereus) towards micellar short-chain phosphatidylcholine. Biochim Biophys Acta. 1985;831:133–141. doi: 10.1016/0167-4838(85)90160-8. [DOI] [PubMed] [Google Scholar]
- 35.Hergenrother PJ, Martin SF. Determination of the kinetic parameters for phospholipase C (Bacillus cereus) on different phospholipid substrates using a chromogenic assay based on the quantitation of inorganic phosphate. Anal Biochem. 1997;251:45–49. doi: 10.1006/abio.1997.2251. [DOI] [PubMed] [Google Scholar]
- 36.Martin SF, Hergenrother PJ. Catalytic cycle of the phosphatidylcholine-preferring phospholipase C from Bacillus cereus. Solvent viscosity, deuterium isotope effects, and proton inventory studies. Biochemistry. 1999;38:4403–4408. doi: 10.1021/bi9821216. [DOI] [PubMed] [Google Scholar]
- 37.Martin SF, Spaller MR, Hergenrother PJ. Expression and site-directed mutagenesis of the phosphatidylcholine-preferring phospholipase C of Bacillus cereus: probing the role of the active site Glu146. Biochemistry. 1996;35:12970–12977. doi: 10.1021/bi961316f. [DOI] [PubMed] [Google Scholar]
- 38.Marquis H, Hager EJ. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol Microbiol. 2000;35:289–298. doi: 10.1046/j.1365-2958.2000.01708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeung PS, Zagorski N, Marquis H. The metalloprotease of Listeria monocytogenes controls cell wall translocation of the broad-range phospholipase C. J Bacteriol. 2005;187:2601–2608. doi: 10.1128/JB.187.8.2601-2608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan CA, Hehir MJ, Roberts MF. Cloning, overexpression, refolding, and purification of the nonspecific phospholipase C from Bacilus cereus. Protein Expr Purif. 1997;10:365–372. doi: 10.1006/prep.1997.0756. [DOI] [PubMed] [Google Scholar]
- 41.Shimanouchi T, Kawasaki H, Fuse M, Umakoshi H, Kuboi R. Membrane fusion mediated by phospholipase C under endosomal pH conditions. Colloids Surf B Biointerfaces. 2013;103:75–83. doi: 10.1016/j.colsurfb.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 42.Hansen S, Hough E, Svensson LA, Wong YL, Martin SF. Crystal structure of phospholipase C from Bacillus cereus complexed with a substrate analog. J Mol Biol. 1993;234:179–187. doi: 10.1006/jmbi.1993.1572. [DOI] [PubMed] [Google Scholar]
- 43.Martin SF, Follows BC, Hergenrother PJ, Trotter BK. The choline binding site of phospholipase C (Bacillus cereus): insights into substrate specificity. Biochemistry. 2000;39:3410–3415. doi: 10.1021/bi9919798. [DOI] [PubMed] [Google Scholar]
- 44.Zuckert WR, Marquis H, Goldfine H. Modulation of enzymatic activity and biological function of Listeria monocytogenes broad-range phospholipase C by amino acid substitutions and by replacement with the Bacillus cereus ortholog. Infect Immun. 1998;66:4823–4831. doi: 10.1128/iai.66.10.4823-4831.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gradziel CS, Wang Y, Stec B, Redfield AG, Roberts MF. Cytotoxic amphiphiles and phosphoinositides bind to two discrete sites on the Akt1 PH domain. Biochemistry. 2014;53:462–272. doi: 10.1021/bi401720v. [DOI] [PubMed] [Google Scholar]
- 46.Wei Y, Stec B, Redfield AG, AG, Weerapana E, Roberts MF. Phospholipid-binding sites of phosphatase and tensin homolog (PTEN): exploring the mechanism of phosphatidylinositol-4,5-bisphosphate. J Biol Chem. 2015;290:1592–1606. doi: 10.1074/jbc.M114.588590. [DOI] [PMC free article] [PubMed] [Google Scholar]