

Abstract
We describe a relatively convenient and reliable procedure for assessing the magnitude of free radical-mediated (chain) lipid peroxidation in biological systems. The approach is based on use of radiolabeled cholesterol ([14C]Ch) as a probe and determination of well-resolved oxidation intermediates/products ([14C]ChOX species), using high performance thin layer chromatography with phorphorimaging detection (HPTLC-PI). In a lipid hydroperoxide-primed liposomal test system treated with ascorbate and a lipophilic iron chelate, the following well-resolved [14C]ChOX are detected and quantified: 7α/7β-OOH, 7α/7β-OH, and 5,6-epoxide, their levels increasing with incubation time at 37 °C. [14C]Ch also serves as an excellent probe for lipid peroxidation in lipoproteins and plasma membranes of mammalian cells. Because this approach utilizes Ch as a natural in situ probe, it eliminates potential artifacts associated with artificial probes such as spin traps and fluorophores.
Keywords: Cholesterol, cholesterol oxides, free radicals, lipid peroxidation, thin layer chromatography, phosphorimaging
I. Introduction
Oxidative stress-induced peroxidation of unsaturated phospholipids and cholesterol in cell membranes and lipoproteins is of considerable biomedical interest because of its possible involvement in pathophysiological conditions such as chronic inflammation, ischemia-reperfusion injury, neurodegeneration, and atherogenesis [1–4]. Oxidative stress arising when pro-oxidant pressure exceeds antioxidant capacity is accompanied by formation of reactive oxygen species (ROS) such as superoxide (O2−·), hydrogen peroxide (H2O2), and hydroxyl radical (HO·), the latter being a strong indiscriminate oxidant that can initiate non-enzymatic free radical-mediated lipid peroxidation [3]. The initiating step might be abstraction of an allylic hydrogen from an unsaturated sn-2 fatty acyl group on a membrane phospholipid, e.g. 1 palmitoyl-2-linoleoyl-sn-glycero-phosphocholine (PLPC); see Eq. 1,
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
where LH denotes an unsaturated lipid. The resulting alkyl radical (L·) reacts rapidly with O2 to give a peroxyl radical (LOO·) (Eq. 2), which in turn can abstract an allylic hydrogen from another unsaturated lipid (L′H), giving a lipid hydroperoxide (LOOH) (Eq. 3). The accompanying L′· then begins the propagative phase of chain peroxidation (Eqs. 2 and 3). Meanwhile, if suitably ligated iron (or other redox metal ion) and a reductant are available, LOOH can undergo one-electron reduction to an oxyl radical (LO·), which rearranges and reacts with O2 to give an epoxyallylic peroxyl radical (OLOO·) (Eq. 4) [5]. The latter in turn can induce a new round of chain peroxidation via hydrogen abstraction from another proximal lipid (Eq. 5). The resulting hydroperoxide may also undergo iron-catalyzed one-electron reduction (Eq. 6), triggering a new chain via OLO·, which is reduced to an alcohol (Eq. 7). LOOHs can also arise in non-radical fashion, a key example being “ene” addition of singlet molecular oxygen (1O2), a photochemically-generated ROS, to an unsaturated lipid [4]. However, subsequent one-electron turnover of 1O2-derived LOOHs can induce chain peroxidation reactions (Eqs. 4–7) similar to those initiated by HO· [2–4].
Non-esterified cholesterol (Ch) is found in the outer layer of lipoproteins and in membranes of eukaryotic cells, most of it residing in the plasma membrane, where it comprises ~45 mol % of total lipid. As a monounsaturated lipid, Ch is susceptible to oxidative degradation, but less so than polyunsaturated fatty acyl groups in phospholipids. Oxidation occurring in the ring portion of Ch is predominantly non-enzymatic (free radical-mediated), whereas that occurring in the side-chain is usually enzymatic [6]. Free radical-mediated oxidation gives rise to a discrete number of cholesterol oxides (oxysterols), including hydroperoxides (ChOOHs), diols, epoxides, and ketone [6–8]. A lipid peroxyl radical can attack at the Ch double bond, giving epimeric 5,6-epoxides (5,6>O) (Eq. 8).
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
Alternatively, abstraction of a C-7 allylic hydrogen by a strong oxidant such as OLOO·, followed by O2 addition and hydrogen abstraction from another lipid gives the 7α- and 7β-hydroperoxides (7-OOHs) (Eqs. 9–11). As with LOOHs in general, iron-catalyzed one-electron reduction of either 7-OOH gives the corresponding diol (7-OH) (Eqs. 12 and 13). Alternatively, the oxyl radical intermediate may undergo β-hydrogen scission in the presence of a peroxyl radical, giving the 7-ketone (7=O) (Eq. 14). Structures of the Ch oxides (ChOX) referred to in Eqs. 8–14, and which are prominent in the analytical approach we describe, are shown in Fig. 1. It is important to note that whereas 7α- and 7β-OOH derive from free radical reactions, 5α-OOH and 6α/6β-OOH are generated exclusively by 1O2 attack on Ch and can serve as unambiguous reporters of 1O2 intermediacy in membrane oxidations [4,7,9].
Fig. 1.

Structures of cholesterol hydroperoxides and other oxides referred to in this article. Formal names of these species are provided in the list of abbreviations.
Ongoing interest in free radical-mediated lipid peroxidation has stimulated the development of highly sensitive and specific techniques for detecting and quantifying this process. These techniques typically involve measurement of reactive intermediates such as hydroperoxides or end-products such as hydroxides and aldehydes. They range from relatively simple “bulk-type” methods, e.g. the classic thiobarbituric acid and iodometric assays [9] to more sophisticated methods involving high performance liquid or gas chromatography with high sensitivity/specificity post-column detection of LOOHs and other characteristic species [10–12]. Perceiving the need for more convenience and less complexity in monitoring chain lipid peroxidation, the authors introduced a novel approach based on use of radiolabeled Ch as a natural probe for free radical lipid peroxidation in biological membranes and lipoproteins [13]. In this approach, [14C]Ch acts as a “sensor” of free radical activity in its surroundings. The intermediates and products of [14C]Ch oxidation, ([14C]ChOX species) serve as reporters of damaging peroxidation and are detected by high-performance thin layer chromatography with phosphorimaging (HPTLC-PI). We describe several different protocols that illustrate how this unique approach can be used to detect/quantify chain peroxidation in a variety of test systems ranging from relatively simple liposomes to mammalian cells. A simplified procedural flow diagram for determination of [14C]ChOX species in oxidatively stressed cells is shown in Scheme 1.
Scheme 1.

Flow diagram for labeling cells with [14C]Ch, applying an oxidative stress that induces free radical (chain) lipid peroxidation, and determining [14C]ChOX species by HPTLC-PI.
2. Materials
2.1. Chemicals and reagents
All chemicals and reagents should be of the highest purity available. Unlabeled Ch, 7α-OH, 7=O, Chelex-100, desferrioxamine, 8-hydroxyquinoline, ascorbic acid, fetal bovine serum, cell growth media, and media supplements are available from Sigma Chemical Co. (St. Louis, MO). [4-14C]Ch (~50 mCi/ml in toluene) is obtained from Amersham Life Sciences (Arlington Heights, IL). 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) are obtained from Avanti Polar Lipids (Birmingham, AL) and 5,6>O from Steraloids (Wilton, NH). Mobile phases for TLC separations are prepared using HPLC-grade solvents. 5α-OOH and 7α-OOH, which are not commercially available, are prepared by dye-sensitized photoperoxidation of Ch in pyridine or in liposomal form [9,12]. The hydroperoxides are isolated by reverse-phase HPLC, subsequent normal-phase HPLC being used to separate 7α-OOH from any 7β-OOH [11,12]. After confirmation of identity by proton NMR [14], 5α-OOH and 7α-OOH are stored in isopropanol at −20 °C. Immediately before experiments, peroxide levels of ChOOH preparations are determined by iodometric analysis [9]. Ultrapure Millipore quality water is used for all aqueous solutions. Before use, buffer solutions are treated with Chelex-100 to deplete redox metal ions such as Fe3+ and Cu2+.
2.2. Liposomes
Unilamellar liposomes suitable for initial experiments are prepared by an extrusion process using a device from Lipex Biomembranes (Vancouver, BC, Canada) with polycarbonate filters of requisite pore size [15]. Small vesicles (SUVs) or large vesicles (LUVs) of 50 nm or 100 nm average diameter, respectively, can be used. Before incorporation into liposomes, [14C]Ch (1–2 μCi) plus unlabeled carrier is separated from any preexisting oxides by TLC (see below). The major band of Ch is detected by radioscanning, recovered by scraping, eluted with chloroform/methanol (2/1 v/v), dried under N2, and stored at −20 °C. For a typical experiment in which chain-initiating potency of a ChOOH is to be evaluated, one can use LUVs consisting of 1.0 mM POPC, 0.75 mM [14C]Ch (0.5–1.0 μCi/ml), and 0.05 mM ChOOH (5α-OOH or 7α-OOH) in bulk aqueous phase. Initial ChOOH content of these LUVs (~3 mol %) models that of a natural plasma membrane subjected to some basal oxidative stress [13]. A suitable aqueous medium is phosphate buffered saline (pH 7.4 PBS) pre-treated with Chelex-100 to deplete redox metal ions; the latter might otherwise catalyze ChOOH decomposition. Liposomes are stored under argon at 4 °C and typically used for experiments within 48 h after preparation.
2.3. Low density lipoprotein
Whole plasma is prepared from freshly drawn blood provided by a local blood bank. The low density lipoprotein (LDL) fraction (density 1.019–1.063 g/mL) is isolated by flotation ultracentrifugation in KBr, as described [16]. All preparative solutions are dialyzed against Chelex-100 and sparged with argon before use. Immediately after harvesting, the LDL is dialyzed against argon-sparged PBS/DFO/EDTA, stored under argon at 4 °C, and used experimentally within one week. Preparations are standardized according to ApoB-100 protein content. In preparation for assessing endogenous chain peroxidation via [14C]ChOX formation, LDL is labeled with [14C]Ch using agarose-conjugated BSA as an easily removable vehicle [17,18]. A 1 mL aliquot of BSA solution (10 mg/mL in PBS/DFO) is added to 1 mL of PBS-washed Affi-Gel agarose beads (Bio-Rad, Richmond, CA). The slurry is gently mixed for 1 h at 37°C, after which the BSA-linked beads are washed with PBS to remove unbound protein and then sparged with argon. A 50 μl aliquot of [14C]Ch (~1 μCi) in acetone is added, followed by 4 h of slow mixing at 37 °C under argon. The [14C]Ch-charged BSA beads are then washed with and resuspended in PBS/DFO. To a 1 mL portion of the slurry is added 1 mL of stock LDL (2 mg protein/mL), followed by further incubation for 1 h under argon with gentle agitation. The [14C]Ch-labeling efficiency for LDL is typically >50%, as determined by scintillation counting.
2.4. Isolated erythrocyte membranes
Erythrocyte membranes (white ghosts) are prepared from freshly drawn human blood in 2 mM EDTA. Red blood cells are pelleted, washed several times with PBS, then lysed with at least 50 volumes of ice-cold 5 mM sodium phosphate/1 mM EDTA (pH 8.0). After pelleting and washing thoroughly with this buffer, the membranes are resuspended in PBS containing DFO and EDTA (100 μM each) and stored at 4 °C for no longer than one week. Membranes prepared in this way are sufficiently depleted of redox iron so as to minimize any LOOH formation/turnover during LOOH transfer to LDL [18]. Isolated ghost membranes are “primed” with ChOOHs and phospholipid counterparts (PLOOHs) by means of photosensitized oxidation, using a 1O2-generating sensitizer such as aluminum phthalocyanine disulfonate (AlPcS2) [9]. A typical reaction mixture in PBS/DFO consists of membranes (1.0 mg protein/ml or ~0.9 mg lipid/ml) and 25 μM AlPCS2 in a thermostatted beaker at 25 °C. The mixture is irradiated from above using a 90-W quartz-halogen source with slow magnetic stirring. A light fluence of 180 J/cm2 (~20 min of irradiation) produces ~45 μM total ChOOH and ~165 μM PLOOH in bulk suspension [18].
2.5. Cultured mammalian cells
Experiments can be carried out with any cell line that is susceptible to deleterious free radical lipid peroxidation. COH-BR1 cells, an epithelial subline derived from a human breast tumor [19], is represented here as a special example. Wild type COH-BR1 cells are severely deficient in glutathione peroxidase type-4 (GPx4), the only enzyme known to catalyze two-electron reductive detoxification of ChOOHs as well as PLOOHs [20]. This deficiency is known to make these cells hypersensitive to damaging lipid peroxidation [21]. A transfectant COH-BR1 clone overexpressing mitochondrial GPx4 is also represented for comparing cell susceptibility to toxic peroxidation under oxidative stress. Cells are grown under standard culture conditions (95% air/5% CO2, 37 °C), using DME/F12 medium supplemented with 10% fetal bovine serum and antibiotics as described [21]. Experiments are carried out on cells at ~80% confluency in 10-cm culture dishes.
3. Protocols
3.1. Protocol 1: Chain peroxidation in liposomes with pre-existing LOOH (5α-OOH)
POPC/[14C]Ch/5α-OOH (1.0:0.75:0.05 by mol) LUVs (1.8 mM total lipid) in PBS are pre-incubated for ~5 min with a lipophilic iron chelate, ferric-8-hydroxyquinoline [Fe(HQ)3], typically at 1–2 μM. The reductant ascorbate (AH−) is then introduced at a starting concentration of 1 mM to initiate the iron-catalyzed one-electron reduction of the priming hydroperoxide. Ascorbate derives from a stock 100 mM ascorbic acid solution in PBS prepared immediately before the experiment. Reaction mixtures along with controls lacking Fe(HQ)3 or AH− are incubated at 37 °C with gentle stirring. At various time points, 50 μl samples are removed, mixed with 1 μl of 25 mM EDTA plus 0.2 ml of PBS and extracted with 0.4 ml of cold chloroform/methanol (2:1 v/v). After centrifugation, a 0.2 ml aliquot from each lower phase is recovered for [14C]ChOX analysis by HPTLC-PI.
High-performance silica gel-60 TLC plates (10 cm x 10 cm, 0.2 mm thickness) are obtained from EM Science (Gibbstown, NJ). Samples in a hexane/2-propanol mixture (97:3, v/v) are applied to a plate in a hairline nitrogen stream, using a programmable Linomat IV dispenser from Camag Scientific (Wilmington, NC). Chromatography is performed in a 9 cm x 20 cm x 24 cm glass chamber, using benzene/ethyl acetate (1:1, v/v) as the mobile phase. Analyte assignments are based on positions of 14C-labeled standards (e.g. [14C]7α/β-OOH, [14C]7α/β-OH) chromatographed alongside. The typical running time at room temperature is 8–10 min. After air-drying, plates are analyzed for [14C]ChOX using a storage phosphor system, e.g. the Storm-860 system with ImageQuant 4.2 software (Molecular Dynamics, Sunnyvale, CA) [13]. The development time for a phosphor screen is typically 48 h at room temperature. For the indicated specific radioactivity of liposomal [14C]Ch (see Sect. 2.2), the detection limit for [14C]7α/β-OH, for example, is ~5 pmol. Parent [14C]Ch, which is depleted by <5% in chain reactions and is well-resolved from [14C]ChOX species, can be used as a reliable internal standard to correct for any loading discrepancies [13].
3.2. Protocol 2: Chain peroxidation in photoperoxidized liposomes
Chain peroxidation can also be studied in liposomes that are being primed with LOOHs via photosensitized peroxidation of resident lipids. For example, POPC/[14C]Ch (1.0:0.8 by mol) LUVs in a thermostatted beaker at 37 °C are sensitized with AlPcS2 or protoporphyrin IX (PpIX) and irradiated for 45–60 min using a 90-W quartz halogen source positioned above the beaker. Irradiation is carried out in the presence of AH− (1 mM) and Fe(HQ)3 (1 μM) or absence of either as a control. The effect of a chain-breaking antioxidant such as nitric oxide (NO) can be examined by including a chemical NO donor such as spermine-NONOate (SPNO) before irradiation. Lipids in reaction and control samples are extracted and analyzed for [14C]ChOX levels as described in Sect. 3.1.
3.3. Protocol 3: Chain peroxidation in LDL with transfer-acquired LOOHs
Erythrocytes carry O2 in the blood circulation and are more susceptible to oxidative modification than most other cells. LOOHs generated in erythrocyte membranes can be translocated to acceptors such as LDL, where LOOH-initiated chain peroxidation may take place [18]. The resulting oxidized LDL could play a crucial role in formation of atherosclerotic plaques in the artery wall. Translocation of LOOHs from erythrocyte membranes to LDL is example of a general LOOH transfer phenomenon discovered in the authors’ laboratory [22,23]. One can examine the susceptibility of LDL to peroxidative damage from transfer-acquired erythrocyte LOOHs by incubating [14C]Ch-labeled LDL with photoperoxidized erythrocyte ghosts for 90 min in argon-sparged PBS at 37 °C, using gentle magnetic stirring throughout. LOOH levels in the peroxidized ghosts are determined by iodometric assay (total LOOH) or by HPLC-EC(Hg) for discrete LOOH classes [9]. Reaction mixture concentrations such as the following are appropriate: LDL (2–3 mM total lipid; ~1 μCi/ml); ghosts (50–60 μM PLOOH; 15–20 μM ChOOH). A control with non-peroxidized ghosts is run alongside. At various time intervals, samples are recovered, centrifuged to remove ghosts, and LDL in the supernatant fractions is incubated with pro-oxidant Cu2+ (e.g. 10–20 μM CuCl2) to evaluate enhanced sensitivity to chain peroxidation, as determined by [14C]ChOX analysis (see Sect. 3.1).
3.4. Protocol 4: Chain peroxidation in mammalian cells
Translocated or photogenerated LOOHs, even at relatively low starting levels, can also induce toxic bursts of chain peroxidation in living cells and these can be attenuated by chain-breaking antioxidants, including NO, which can intercept oxyl and peroxyl radicals [24,25]. One can demonstrate this by first labeling cells (e.g. sub-confluent COH-BR1 cells in 10-cm dishes) with [14C]Ch via incubation with a [14C]Ch/serum albumin complex (1:15 mol/mol) as described [18], followed by washing to remove loosely associated sterol. Most of the bound sterol resides in the plasma membrane, allowing chain peroxidation to be probed with high sensitivity and selectivity in this compartment. Cells may then be metabolically photosensitized with PpIX via dark incubation with its precursor, 5-aminolevulinic acid (ALA), using a procedure that allows most of the sensitizer to diffuse to plasma membrane [26]. Following this, cells are treated with Fe(HQ)3 (1–2 μM), then irradiated in the absence or presence of SPNO (0.2–0.4 mM), added immediately before irradiation. After 1 h of dark incubation, lipids are extracted and analyzed for [14C]ChOX levels (see Sect. 3.1). Effects of SPNO-derived NO on ChOX accumulation are evaluated.
One can also use [14C]ChOX analysis to determine ability of a mammalian enzymatic antioxidant, viz. selenoperoxidase GPx4, to protect cells against peroxidative toxicity. A vector control of wild type (GPx4-deficient) COH-BR1 cells and a mitochondrial GPx4 transfectant clone (6G4) exhibiting ~160-times greater activity are compared for ability to defend against an SUV-ChOOH challenge. After plasma membrane labeling with [14C]Ch, both cell types are incubated in the presence of DMPC/Ch/7α-OOH (1.0:0.5:0.3 by mol) SUVs, DMPC/Ch (1.0:0.8 by mol) SUVs serving as a control. After various incubation times, cells are washed, harvested, and extracted, the lipid fractions being analyzed for [14C]ChOX by HPTLC-PI.
4. Expected Results
4.1. Application 1: Chain peroxidation in 5α-OOH-primed liposomes
Results of a relatively simple experiment illustrating LOOH-stimulated ChOX formation in liposomal membranes are shown in Fig. 2 [25]. In this example, POPC LUVs were labeled with [14C]Ch and “primed” with 5α-OOH. At time zero, >99% of the radioactivity resided in the Ch band, which migrated well ahead of any oxides. The LUVs underwent chain peroxidation during incubation with Fe(HQ)3 and AH−, as indicated by the time-dependent accumulation of radiolabeled 7α/7β-OOH, 7α/β-OH, and 5,6>O over at least 20 min (Fig. 2A). 7=O is not seen because it co-migrates with more abundant 5,6>O. Whereas the 7α/7β-OOH levels peaked at ~20 min, the 7α/7β-OH, and 5,6>O levels continued to rise through at least 45 min (Fig. 2B). Like 5α-OOH, 7α/7β-OOH are susceptible to one-electron reductive turnover, which explains their rise to an apparent steady state maximum near 20 min, followed by a decline. The higher level of 7β-OOH than 7α-OOH throughout probably reflects the greater thermodynamic stability of 7β-OOH [27].
Fig. 2.

Time courses for [14C]ChOX accumulation in liposomes as determined by HPTLC-PI. POPC/[14C]Ch/5α-OOH (1.0:0.75:0.05 by mol) LUVs (1.8 mM total lipid in bulk phase PBS) were incubated at 37 °C in the presence of 1 μM Fe(HQ)3 and 1 mM AH−. At the indicated times, lipids were extracted and analyzed by HPTLC-PI. (A) Chromatographic profiles of [14C]ChOX species. Identities were based on co-migration of authentic standards (not shown). (B) Integrated band intensities: 7β-OOH (●), 7α-OOH (○), 7α-OH (▲), 7β-OH (△), 5,6>O (□). A control without AH−, but with Fe(HQ)3 is also represented (+). Reproduced from Ref. 25 with permission.
It is important to point out that some ChOOHs have only a limited ability to initiate chain peroxidation. This was discovered when free radical-generated 7α-OOH was compared with 1O2-generated 5α-OOH and 6β-OOH [28]. All three ChOOHs in liposomal form were found to decay at the same rate when incubated with Fe(HQ)3 and AH−. Moreover, 5α-OOH and 7α-OOH produced [14C]ChOX at an equal rate. However, under the same reaction conditions (including starting hydroperoxide concentration), 6β-OOH produced <5% of the [14C]ChOX generated by 5α-OOH or 7α-OOH [28]. Correspondingly, 5α-OOH and 7α-OOH were each ~50-times more toxic to leukemia cells than 6β-OOH. Since all three ChOOHs appeared to undergo one-electron reduction to oxyl radicals at equal rates, it was deduced that (a) the 6β-oxyl radical might be poorly oxidizing compared with 5α-oxyl or 7α-oxyl radical, i.e. the redox potential of 6β-oxyl radical might be significantly lower, or (b) 6β-oxyl radical might be kinetically restrained from initiating chains due to some structural disorientation in the membrane [28]. Which of these two alternatives may be correct remains to be established.
4.2. Application 2: LOOH free radical turnover in photoperoxidized liposomes
A somewhat more complex scenario involves [14C]Ch-containing LUVs in which LOOHs are generated de novo by PpIX-sensitized photooxidation and undergo immediate one-electron reduction in the presence of Fe(HQ)3 and AH− [29]. As shown in Fig. 3A, several [14C]ChOX appear under these conditions including ChOOHs (5α-OOH, 6α/β-OOH,7α/β-OOH – some only partially resolved) along with well-resolved 5α-OH, 7α-OH, 7β-OH, and 6β-OH. 7α-OH and 7β-OH levels were substantially diminished when photooxidation was carried out in the presence of SPNO (Fig. 3A,C), consistent with NO scavenging of free radical intermediates [24,25,29]. However, 5α-OH and 6β-OH levels were unchanged (Fig. 3A,B), indicating that neither rapid photogeneration of primary ChOOHs nor iron reduction had been affected by NO at the level generated in this system.
Fig. 3.

[14C]ChOX accumulation during photoperoxidation of liposomes: inhibitory effects of NO. (A) POPC/[14C]Ch/PpIX (100:80:0.2 by mol) LUVs were irradiated for 45 min (~120 J/cm2 light fluence) in the presence of 1 μM Fe(HQ)3 and 1 mM AH− (lane 3) or 1 μM Fe(HQ)3, 1 mM AH−, and 0.2 mM SPNO (lane 4). Also represented is a dark control with Fe(HQ)3/AH− (lane 1) and a light control without Fe(HQ)3/AH−/SPNO (lane 2). (B) 5α-OH (○,●) and 6β-OH (□,■) buildup with increasing light fluence. (C) 7α-OH (△,▲) and 7β-OH (▽,▼) buildup with increasing light fluence. Irradiation was carried out in the absence (open symbols) or presence (closed symbols) of 0.2 mM SPNO. Plotted data are means ± deviation of values from duplicate experiments. Reproduced from Ref. 28 with permission.
4.3. Application 3: Free radical turnover of transfer-acquired LOOHs in LDL
A third illustration involves priming an acceptor system, viz. [14C]Ch-labeled LDL, via spontaneous LOOH translocation from a donor system, viz. photooxidized erythrocyte ghost membranes [18]. HPTLC-PI profiles depicting [14C]ChOX accumulation in the LDL after 0, 1, 1.5, and 4 h of incubation with Cu2+ are shown in Fig. 4A. System a represents control LDL (pre-incubated with non-photooxidized ghosts) and System b experimental LDL (pre-incubated with photooxidized ghosts). Initial total LOOH (predominantly PLOOH plus ChOOH) in the experimental LDL was ~4.6 μM in bulk suspension, whereas LOOH in control LDL was <25 nM. Five [14C]ChOX species are identified which intensify over time: 7α-OH, 7β-OH, 5,6>O, and partially resolved 7α- and 7β-OOH. Importantly, the ChOX appeared earlier in System b and System a, suggesting more rapid and greater free radical activity in the former [18]. ChOX kinetic plots B (Fig. 4B) confirm this point, showing a 30-min longer lag period in System a than in System b, whereas post-lag accumulation rates were the same.
Fig. 4.

[14C]ChOX accumulation in transfer LOOH-enriched LDL. [14C]Ch-labeled LDL (2.5 mM lipid; ~0.5 μCi/ml) was incubated with photoperoxidized erythrocyte ghosts (0.25 mM lipid; ~75 μM PLOOH plus ChOOH) for 1.5 h in argon-sparged PBS at 37 °C. After centrifugation, LDL was recovered, diluted ~4-fold in PBS, and incubated in the presence of 20 μM CuCl2. A control that had been treated with non-photoperoxidized ghosts was incubated alongside. Samples were recovered at various times up to 4 h, extracted, and lipid fractions subjected to HPTLC-PI analysis. (A) [14C]ChOX profiles as a function of incubation time for System a (control LDL) and System b (LOOH-exposed LDL). Sample load: ~0.1 μg lipid per lane. (B) Time courses for total ChOX accumulation: (△) System a; (○) System b. Data are from the same experiment as in panel (A) with additional time points represented. The plots show ChOX concentrations in bulk reaction mixtures. Reproduced from Ref. 18 with permission.
4.4. Application 4: Lipid chain peroxidation in mammalian cells: inhibition by NO
[14C]Ch can also be used to monitor free radical peroxidation in plasma membranes of living cells. Figure 5 shows results of such an experiment in which [14C]Ch-labeled COH-BR1 breast tumor cells were subjected to a specific type PpIX-sensitized (1O2-mediated) photooxidation that resulted in primary LOOH generation mainly in the plasma membrane [26], where most of the [14C]Ch was located. This generation strongly stimulated [14C]ChOX formation and this was further enhanced by Fe(HQ)3. On the other hand, SPNO added immediately before irradiation reduced all [14C]ChOX levels (Fig. 5A,B), consistent with OLOO· /OLO· interception by NO [29,30].
Fig. 5.

Effects of NO on ChOX accumulation in cells subjected to a photodynamic challenge. [14C]Ch-labeled COH-BR1 cells were treated with ALA alone or ALA plus Fe(HQ)3, then irradiated for 30 min in the absence or presence of 0.4 mM SPNO (added immediately before irradiation). After 1 h of dark incubation, lipids were extracted and analyzed by HPTLC-PI. (A) [14C]ChOX profiles showing origin (O), 7α-OH (1), 7β-OH (2), 5α-OH (3), 5,6>O/6-OOH (4), 7-OOH/5α-OOH (5), Ch (6), and cholesteryl esters (7), and solvent front (F). Each lane represents lipid from ~5 x 106 cells. Lane a: ALA/ Fe(HQ)3 dark control; lane b: ALA/light; lane c: ALA/SPNO/light; lane d: ALA/Fe(HQ)3/light; lane e: ALA/Fe(HQ)3/SPNO/light. (B) ChOX yields as percentages of total radioactivity in each sample lane; values were background-corrected. ChOOH denotes unresolved 7α/7β-OOH and 5α-OOH. Means ± deviation of values from duplicate experiments are plotted. *P<0.005 vs. ALA/light; **P<0.001 vs. ALA/ Fe(HQ)3/light. Reproduced from Ref. 29 with permission.
4.5. Application 5: Enzymatic inhibition of lipid chain peroxidation in cells
Another example of how [14C]ChOX can accurately report on cellular chain peroxidation is represented in Fig. 6. This experiment tested the ability of GPx4, which catalyzes the GSH-dependent detoxification of PLOOHs and ChOOHs, to curtail damaging chain peroxidation induced by 7-OOH-bearing SUVs. As shown in Fig. 6A,B, transfectant clone 6G4 of COH-BR1 cells (which overexpressed mitochondrial GPx4) accumulated [14C]ChOX significantly slower than a vector control (VC), which, like wild type cells (not represented), expressed almost negligible GPx4 [21]. Other experiments with 6G4 vs. VC COH-BR1 cells showed that the former, in addition to being more resistant to chain peroxidation, were more resistant to a loss of mitochondrial membrane potential (Δψm) and to cell death via intrinsic apoptosis [21]. Thus, GPx4 inhibited damaging chain lipid peroxidation, but unlike NO or other chain-breaking antioxidants, did so by inactivating chain-initiating LOOHs [20,21].
Fig. 6.
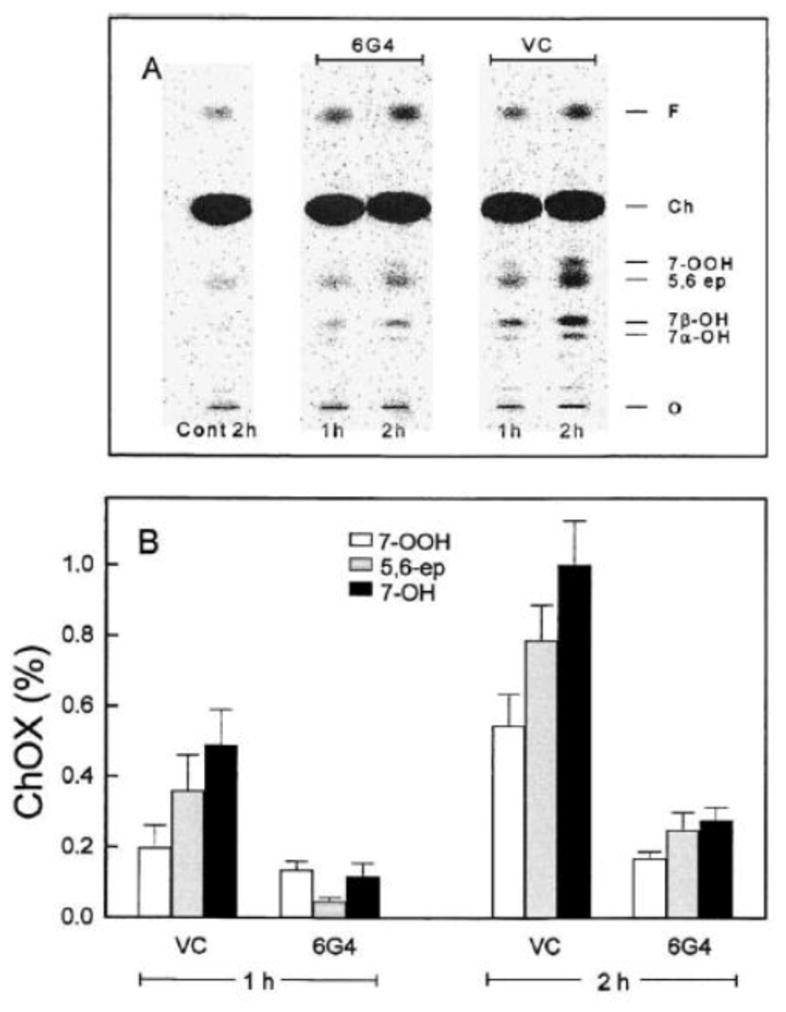
Effect of GPx4 overexpression on ChOOH-induced chain peroxidation in tumor cells. A GPx4-overexpressing clone (6G4) of COH-BR1 cells and a vector control (VC) were labeled with [14C]Ch by incubating with a [14C]Ch-BSA complex (1:15 mol/mol). The labeling strength for both cell types was approximately the same. Labeled cells were incubated for 1 h or 2 h with non-peroxidized (control) or photoperoxidized DMPC/Ch (1.0/0.8 by mol) SUVs. The initial 7-OOH concentration in the latter system was ~150 μM. After incubation, cells were washed, harvested, and extracted; lipid fractions were analyzed by HPTLC-PI. (A) [14C]ChOX profiles. (B) Percent yields of various [14C]ChOX species. Means ± deviation of values from duplicate experiments are plotted. Reproduced from Ref. 21 with permission.
5. Notes and Precautions
Protocols 1–4 involve the use of cholesterol radiolabeled with Carbon-14, which is a weak β-particle emitter. As true for all radioisotopes, precautions should be observed when purifying [14C]Cholesterol and inserting it into liposomal and cellular membranes and also when isolating [14C]ChOX for HPTLC-PI analysis. Thus, protective gloves should be worn for these procedures in order to avoid skin contact with radioactive material. The light sources and photosensitizers used in Protocols 2 and 4 involve no obvious hazards when used as described.
6. Summary and Conclusions
We have described a straightforward new technique for monitoring free radical-mediated lipid peroxidation in cell membranes and lipoproteins whereby radiolabeled cholesterol is exploited as a natural lipid probe. Among the many attractive features of this approach are its relative simplicity and convenience, which should make it useful to investigators seeking better ways to monitor oxidative stress-induced lipid peroxidation. One can use this approach to accurately assess the chain-initiating potency of pre-existing or primary PLOOHs and ChOOHs. [14C]Ch incorporated into a membrane or lipoprotein gives rise to [14C]ChOX species, which are readily monitored by HPTLC-PI. These species, which include redox-active intermediates (e.g. 7α/β-OOH) as well as redox-inactive products (7α/β-OH, 7=O, 5,6>O), report on chain lipid peroxidation taking place in the vicinity of a primary hydroperoxide, and by implication, the oxidizability of surrounding lipids and availability of catalytic metal ions and reductants. There are several advantages of using [14C]Ch as a probe lipid. First, Ch autoxidation has been studied extensively over the last 50 years and most of its important oxidation intermediates/products have been well characterized [7,8,31]. To a great extent, this reflects the fact that Ch, unlike natural phospholipids, exists as a single molecular species, thus facilitating individual ChOX isolation and identification. Second, because our approach exploits Ch as a natural in situ probe, it avoids potential artifacts associated with use of exogenous probes such as spin traps [32] or fluorophores such as DPPP and C11-BODIPY [33,34]. Upon binding to a membrane or lipoprotein, such agents might alter responses by perturbing existing structure, thus potentially interfering with chain propagation. Of course, these concerns are at least partially offset by the advantage of directly analyzing intact cells rather than extracts of cellular lipids. Third, since most of the Ch in mature mammalian cells (>80%) is located in the plasma membrane, [14C]Ch would be an excellent probe for free radical activity occurring in this compartment, e.g. activity initiated by LOOHs from external 1O2 attack or LOOHs translocated from other cells under greater oxidative pressure, e.g. red blood cells. Fourth, Ch is capable of spontaneous and reasonably rapid translocation from a donor membrane, e.g. unilamellar liposome, to an acceptor membrane or lipoprotein [23]. This makes [14C]Ch labeling of acceptors for chain peroxidation assessment relatively convenient. Finally, [14C]Ch gives rise to a relatively small number of [14C]ChOX species, which are well resolved by HPTLC-PI and can be individually quantified, as described. In addition to assessing chain-initiating potency of a primary ChOOH or PLOOH by this approach, one can easily assess chain length by integrating [14C]ChOX levels for any given reaction time, thus providing information that is not available when spin trap or fluorophore probes are used.
Highlights.
[14C]Cholesterol can probe chain lipid peroxidation in membranes and lipoproteins.
Resulting [14C]ChOX can be analyzed by HPTLC with phosphor-imaging.
[14C]ChOX analysis is widely applicable for assessing lipid peroxidation.
Artifacts from exogenous probes (spin traps, fluorophores) are avoided.
Acknowledgments
Research support from the following agencies is gratefully acknowledged: (i) USPHS Grants CA70823, CA72630, TW001386, and HL85677 (to AWG); (ii) NSF Grant MCB-9106117 (to AWG); (iii) Polish National Science Center Grant NZ3/00833 (to WK).
Abbreviations
- AH−
ascorbate
- ALA
5-aminolevulinic acid
- Ch
cholesterol [(3β)-cholest-5-en-3-ol)]
- ChOOH
cholesterol hydroperoxide
- ChOX
cholesterol oxide(s)
- DFO
desferrioxamine
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- Fe(HQ)3
ferric 8-hydroxyquinoline
- GPx4
type-4 glutathione peroxidase
- HPLC-EC(Hg)
high-performance liquid chromatography with mercury cathode electrochemical detection
- HPTLC-PI
high-performance thin layer chromatography with phosphorimaging detection
- LDL
low density lipoprotein
- LH
unsaturated lipid
- LOOH
lipid hydroperxide
- PLOOH
phospholipid hydroperoxide
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SPNO
spermine NONOate
- SUV/LUV
small and large unilamellar vesicles (liposomes)
- 5α-OOH
3β-hydroxy-5α-cholest-6-ene-5-hydroperoxide
- 5α-OH
5α-cholest-6-ene-3β,5-diol
- 5,6>O
cholestan-5,6-epoxy-3β-ol
- 6α/6β-OOH
3β-hydroxycholest-4-ene-6α- and 6β-hydroperoxide
- 7α/7β-OOH
3β-hydroxycholest-5-ene-7α- and 7β-hydroperoxide
- 7α/7β-OH
cholest-5-ene-3β,7α- and 7β-diol
- 7,O
3β-hydroxycholest-5-ene-7-one
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sies H. Biochemistry of oxidative stress. Angew Chem Int Ed Engl. 1986;25:1058–1071. [Google Scholar]
- 2.Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- 3.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Clarendon Press; Oxford, UK: 1999. [Google Scholar]
- 4.Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res. 1998;39:1529–1542. [PubMed] [Google Scholar]
- 5.Wilcox AL, Marnett LJ. Polyunsaturated fatty acid alkoxyl radicals exist as carbon-centered epoxyallylic radicals: a key step in hydroperoxide-amplified lipid peroxidation. Chem Res Toxicol. 1993;6:413–416. doi: 10.1021/tx00034a003. [DOI] [PubMed] [Google Scholar]
- 6.Brown AJ, Jessup W. Oxysterols: sources, cellular storage and metabolism, and new insights into their roles in cholesterol homeostasis. Mol Aspects Med. 2009;30:111–122. doi: 10.1016/j.mam.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Girotti AW. Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J Photochem Photobiol B. 2001;63:103–113. doi: 10.1016/s1011-1344(01)00207-x. [DOI] [PubMed] [Google Scholar]
- 8.Murphy RC, Johnson KM. Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J Biol Chem. 2008;283:15521–15525. doi: 10.1074/jbc.R700049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girotti AW, Korytowski W. Cholesterol as a singlet oxygen detector in biological systems. Methods Enzymol. 2000;319:85–100. doi: 10.1016/s0076-6879(00)19011-1. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto Y, Frei B, Ames BN. Assay of lipid hydroperoxides using high-performance liquid chromatography with isoluminal chemiluminescence detection. Methods Enzymol. 1990;186:371–380. doi: 10.1016/0076-6879(90)86130-n. [DOI] [PubMed] [Google Scholar]
- 11.Korytowski W, Geiger PG, Girotti AW. High-performance liquid chromatography with mercury cathode electrochemical detection: application to lipid hydroperoxide analysis. J Chromatogr B. 1995;670:189–197. doi: 10.1016/0378-4347(95)00182-4. [DOI] [PubMed] [Google Scholar]
- 12.Korytowski W, Geiger PG, Girotti AW. Lipid hydroperoxide analysis by high-performance liquid chromatography with mercury cathode electrochemical detection. Methods Enzymol. 1999;300:23–33. doi: 10.1016/s0076-6879(99)00109-3. [DOI] [PubMed] [Google Scholar]
- 13.Korytowski W, Wrona M, Girotti AW. Radiolabeled cholesterol as a reporter for assessing one-electron turnover of lipid hydroperoxides. Anal Biochem. 1999;270:123–132. doi: 10.1006/abio.1999.4070. [DOI] [PubMed] [Google Scholar]
- 14.Korytowski W, Bachowski GJ, Girotti AW. Chromatographic separation and electrochemical determination of cholesterol hydroperoxides generated by photodynamic action. Anal Biochem. 1991;197:149–156. doi: 10.1016/0003-2697(91)90371-y. [DOI] [PubMed] [Google Scholar]
- 15.Mayer LD, Hope MJ, Cullis PR. Vesicles of variable size produced by a rapid extrusion procedure. Biochim Biophys Acta. 1986;858:161–168. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- 16.Thomas JP, Kalyanaraman B, Girotti AW. Involvement of preexisting lipid hydroperoxides in Cu(2+)-stimulated oxidation of low-density lipoprotein. Arch Biochem Biophys. 1994;315:244–254. doi: 10.1006/abbi.1994.1496. [DOI] [PubMed] [Google Scholar]
- 17.Miida T, Fielding CJ, Fielding PE. Mechanism of transfer of LDL-derived free cholesterol to HDL subfractions in human plasma. Biochemistry. 1990;29:10469–10474. doi: 10.1021/bi00498a007. [DOI] [PubMed] [Google Scholar]
- 18.Vila A, Korytowski W, Girotti AW. Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low-density lipoprotein: assessment of reaction kinetics and prooxidant effects. Biochemistry. 2002;41:13705–13716. doi: 10.1021/bi026467z. [DOI] [PubMed] [Google Scholar]
- 19.Esworthy RS, Baker MA, Chu FF. Expression of selenium-dependent glutathione peroxidase in human breast tumor cell lines. Cancer Res. 1995;55:957–962. [PubMed] [Google Scholar]
- 20.Thomas JP, Maiorino M, Ursini F, Girotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J Biol Chem. 1990;265:454–461. [PubMed] [Google Scholar]
- 21.Hurst R, Korytowski W, Kriska T, Esworthy RS, Chu FF, Girotti AW. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free Radic Biol Med. 2001;31:1051–1065. doi: 10.1016/s0891-5849(01)00685-2. [DOI] [PubMed] [Google Scholar]
- 22.Vila A, Korytowski W, Girotti AW. Dissemination of peroxidative stress via intermembrane transfer of lipid hydroperoxides: model studies with cholesterol hydroperoxides. Arch Biochem Biophys. 2000;380:208–218. doi: 10.1006/abbi.2000.1928. [DOI] [PubMed] [Google Scholar]
- 23.Girotti AW. Translocation as a means of disseminating lipid hydroperoxide-induced oxidative damage and effector action. Free Radic Biol Med. 2008;44:956–968. doi: 10.1016/j.freeradbiomed.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korytowski W, Zareba M, Girotti AW. Nitric oxide inhibition of free radical-mediated cholesterol peroxidation in liposomal membranes. Biochemistry. 2000;39:6918–6928. doi: 10.1021/bi000393e. [DOI] [PubMed] [Google Scholar]
- 25.Korytowski W, Zareba M, Girotti AW. Inhibition of free radical-mediated cholesterol peroxidation by diazeniumdiolate-derived nitric oxide: effect of release rate on mechanism of action in a membrane system. Chem Res Toxicol. 2000;13:1265–1274. doi: 10.1021/tx000160o. [DOI] [PubMed] [Google Scholar]
- 26.Kriska T, Korytowski W, Girotti AW. Hyperresistance to photosensitized lipid peroxidation and apoptotic killing in 5-aminolevulinate-treated tumor cells overexpressing mitochondrial GPX4. Free Radic Biol Med. 2002;33:1389–1402. doi: 10.1016/s0891-5849(02)01078-x. [DOI] [PubMed] [Google Scholar]
- 27.Teng JI, Kulig MJ, Smith LL, Kan G, Van Lier JE. Sterol metabolism. XX. Cholesterol 7β-hydroperoxide. J Org Chem. 1973;38:119–123. doi: 10.1021/jo00941a024. [DOI] [PubMed] [Google Scholar]
- 28.Korytowski W, Schmitt JC, Girotti AW. Surprising inability of singlet oxygen-generated 6-hydroperoxycholesterol to induce damaging free radical lipid peroxidation in cell membranes. Photochem Photobiol. 2010;86:747–751. doi: 10.1111/j.1751-1097.2010.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niziolek M, Kowytowski W, Girotti AW. Nitric oxide inhibition of free radical-mediated lipid peroxidation in photodynamically treated membranes and cells. Free Radic Biol Med. 2003;34:997–1005. doi: 10.1016/s0891-5849(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 30.Niziolek M, Korytowski W, Girotti AW. Chain-breaking antioxidant and cytoprotective action of nitric oxide on photodynamically stressed tumor cells. Photochem Photobiol. 78:262–270. doi: 10.1562/0031-8655(2003)078<0262:caacao>2.0.co;2. 200. [DOI] [PubMed] [Google Scholar]
- 31.Smith LL. Cholesterol Autoxidation. Plenum Press; New York: 1981. [Google Scholar]
- 32.Wagner BA, Buettner GR, Burns CP. Free radical-mediated lipid peroxidation in cells: oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry. 1994;33:4449–4453. doi: 10.1021/bi00181a003. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi M, Shibata M, Niki E. Estimation of lipid peroxidation of live cells using a fluorescent probe, diphenyl-1-pyrenylphosphine. Free Radic Biol Med. 2001;31:164–174. doi: 10.1016/s0891-5849(01)00575-5. [DOI] [PubMed] [Google Scholar]
- 34.Drummen GP, van Liebergen LC, Op den Kamp JA, Post JA. C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic Biol Med. 2002;33:473–490. doi: 10.1016/s0891-5849(02)00848-1. [DOI] [PubMed] [Google Scholar]