

Abstract

The sesquiterpene synthase germacradiene-4-ol synthase (GdolS) from Streptomyces citricolor is one of only a few known high-fidelity terpene synthases that convert farnesyl diphosphate (FDP) into a single hydroxylated product. Crystals of unliganded GdolS-E248A diffracted to 1.50 Å and revealed a typical class 1 sesquiterpene synthase fold with the active site in an open conformation. The metal binding motifs were identified as D80DQFD and N218DVRSFAQE. Some bound water molecules were evident in the X-ray crystal structure, but none were obviously positioned to quench a putative final carbocation intermediate. Incubations in H218O generated labeled product, confirming that the alcohol functionality arises from nucleophilic capture of the final carbocation by water originating from solution. Site-directed mutagenesis of amino acid residues from both within the metal binding motifs and without identified by sequence alignment with aristolochene synthase from Aspergillus terreus generated mostly functional germacradien-4-ol synthases. Only GdolS-N218Q generated radically different products (∼50% germacrene A), but no direct evidence of the mechanism of incorporation of water into the active site was obtained. Fluorinated FDP analogues 2F-FDP and 15,15,15-F3-FDP were potent noncompetitive inhibitors of GdolS. 12,13-DiF-FDP generated 12,13-(E)-β-farnesene upon being incubated with GdolS, suggesting stepwise formation of the germacryl cation during the catalytic cycle. Incubation of GdolS with [1-2H2]FDP and (R)-[1-2H]FDP demonstrated that following germacryl cation formation a [1,3]-hydride shift generates the final carbocation prior to nucleophilic capture. The stereochemistry of this shift is not defined, and the deuteron in the final product was scrambled. Because no clear candidate residue for binding of a nucleophilic water molecule in the active site and no significant perturbation of product distribution from the replacement of active site residues were observed, the final carbocation may be captured by a water molecule from bulk solvent.
Terpenoids make up the largest family of natural products with many thousands of known compounds, spanning a wide range of biological activities, including pigments, semiochemicals, and medicinal compounds active against infectious diseases and many cancers.1−6 Typically, the biosynthetic generation of mature terpenoids occurs in two distinct phases. Mg2+-dependent cyclization of a linear isoprenyl diphosphate by a terpene synthase to generate a hydrocarbon product with a high degree of stereocomplexity is followed by an oxidative phase, whereby the cyclic hydrocarbon undergoes P450-dependent oxidations and other transformations to yield highly complex terpenoids with multiple stereocenters and a high degree of functionality.7,8
Class I sesquiterpene synthases share a common α-helical structure with the active site located in a hydrophobic cleft between two helices containing the highly conserved metal binding motifs DDXXD/E and NSE/DTE.9 Prior to initiation of the cyclization cascade through Mg2+-dependent diphosphate cleavage, the active site closes to protect the carbocationic intermediates from premature quenching by bulk solvent molecules.10,11 The enclosed active site volume of a terpenoid cyclase is typically just slightly larger than that of the substrate, which ensures a snug fit between the enzyme and the flexible isoprenoid substrate.12,13 However, X-ray crystallographic studies show that sometimes a water molecule can be trapped in the closed conformation of a terpenoid cyclase active site along with a bound substrate analogue or product.14,15 Notably, a limited number of the sesquiterpene synthases produce terpenoid alcohols and epoxides containing a single oxygen atom, presumably derived from a water molecule bound in the terpene cyclase active site along with the isoprenoid diphosphate substrate.16−19 The bacterial sesquiterpene synthase (−)-germacradien-4-ol synthase (GdolS) converts farnesyl diphosphate (FDP, 1) into the macrocyclic terpene alcohol (−)-germacradien-4-ol (2) (Figure 1). The enzyme exerts significant control over a catalytic water molecule, which quenches the final carbocation intermediate of the reaction cascade with high stereospecificity and regiospecificity.20
Figure 1.

Possible mechanism for the germacradien-4-ol synthase (GdolS)-catalyzed conversion of farnesyl diphosphate (1) to (−)-germaradien-4-ol (2).
According to a postulated mechanism for the GdolS-catalyzed conversion of 1 to 2 (Figure 1), the enzyme must first guide the ionized substrate through at least two carbocation intermediates before adding water at position 4 of the cyclic carbocation intermediate 4 and at the same time prevent the addition of water to 3 and the loss of a proton from 4 and 3. Here, we have used a combination of site-directed mutagenesis, incubation of GdolS with FDP analogues, kinetic measurements, and X-ray crystallography to investigate the mechanism of the GdolS-catalyzed conversion of FDP (1) to (−)-germacradien-4-ol (2). We demonstrate that the cyclization mechanism proceeds via ionization of FDP prior to cyclization, and it is proposed that the final quenching of the carbocation cascade is conducted by a molecule of water derived from bulk solvent. This water molecule could be trapped in the active site upon substrate binding, or it could access the active site at the end of the catalytic cycle if enabled by loop movements.
Materials and Methods
Protein Preparation and Purification
The expression vector containing the gene for GdolS (pET16b-SC1) was a generous gift from Y. Ohnishi (University of Tokyo, Tokyo, Japan). pET16b-SC1 contained the gene for GdolS with an in-frame sequence encoding an N-terminal decahistidine tag, permitting purification by nickel affinity chromatography.20Escherichia coli BL21(DE3) cells were transformed with pET16b-SC1, and a single transformed colony was used to inoculate 100 mL of LB medium containing ampicillin (100 mg L–1); this was grown overnight at 37 °C while being shaken. This starter culture was used to inoculate LB medium containing ampicillin (5 mL of culture per 500 mL of LB medium). The cultures were incubated at 37 °C until an OD600 of 0.6 was reached, at which time gene expression was induced with isopropyl β-d-1-thiogalactopyranoside (IPTG, 0.2 mM) and the culture allowed to grow for a further 3 h. Cells were harvested by centrifugation (3400g for 10 min), and the supernatant was decanted and discarded. Pellets were thawed and resuspended in 25 mL of cell lysis buffer [50 mM Tris, 100 mM NaCl, and 10 mM imidazole (pH 8.0)] and cells lysed by sonication (5 min, 40% amplitude, pulse 5 s on and 10 s off). The cell debris was discarded after centrifugation (38000g for 30 min) and the supernatant applied to a Ni-NTA affinity column (QIAGEN, 5 mL). Proteins were eluted with a 10 to 500 mM imidazole gradient in lysis buffer. Fractions containing GdolS [>95% pure as judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE)] were combined, dialyzed against 10 mM Tris (pH 8.0), and concentrated to 10 mL. Protein concentrations were measured using the method of Bradford21,22 and aliquots of GdolS stored at −20 °C.
The QuikChange site-directed mutagenesis kit (Stratagene) was used to introduce the desired mutations following the manufacturer’s instructions using pairs of mutagenic primers (Table S1). Homology modeling of GdolS was conducted using the SWISS-MODEL workspace23−25 based on the crystal structure of a pentalenene synthase mutant [PS-N219L, Protein Data Bank (PDB) 1HM7].26
Steady-state kinetic parameters were measured at 30 °C using radiochemical assays modified slightly from those used to determine kinetic parameters in other sesquiterpene synthases.27−29 Reactions (final volume of 250 μL) were initiated by the addition of enzyme (30 nM) to assay buffer solutions [50 mM HEPES, 2.5 mM MgCl2, and 5 mM 2-mercaptoethanol (pH 8.0)] containing 0–10 μM [1-3H]FDP (24000 dpm μM–1) and overlaid with 1 mL of hexane. After incubation for 10 min, reactions were quenched by addition of EDTA (50 μL, 0.5 M) and mixtures vortexed for 30 s. The hexane was decanted and the sample extracted with hexane and Et2O (11:1) in the same way (2 × 750 μL). The pooled organic extracts were passed through a short column of silica (∼500 mg) into 15 mL of Ecoscint fluid (National Diagnostics), and the silica was then washed with a further portion of hexane and Et2O (750 μL) and analyzed by scintillation counting. Inhibition assays were performed in an identical fashion except for the addition of fluorinated FDPs at concentrations ranging from 0 to 10 μM.
Crystallization and Structure Determination
The gene encoding GdolS was cloned into a pET-28a vector, which improved the solution behavior of the expressed protein by decreasing the level of protein precipitation at the high concentrations required for crystallization. To facilitate crystallization by reducing protein flexibility, the E248A mutation was introduced on the basis of sequence analysis using the Surface Entropy Reduction Server (http://services.mbi.ucla.edu/SER/).30 E248 is a surface residue >14 Å from the nearest active site residue, so its mutation should not significantly affect enzyme activity (Figure 2). The E248A mutation was used exclusively for crystallization purposes. GdolS-E248A was expressed using the E. coli BL21-CodonPlus(DE3)-RIL strain (Novagen) as described above for wild-type GdolS, except that LB medium containing kanamycin (50 mg L–1) and chloramphenicol (34 mg L–1) was used. Expression was induced with 0.5 mM IPTG at 16 °C when an OD600 of 0.8–1.0 was reached. After expression for 18 h, cells were pelleted by centrifugation (3400g for 10 min).
Figure 2.
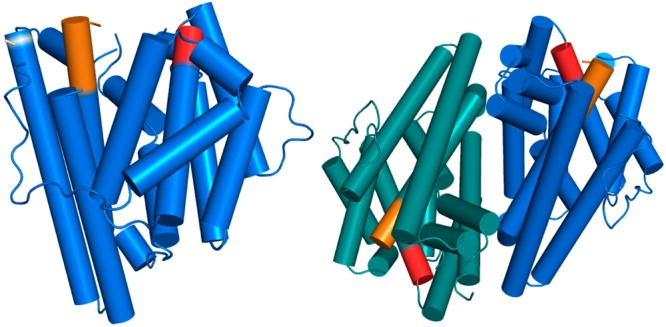
Structure of the GdolS-E248A monomer (left) and dimer (right). The aspartate-rich and NSE metal binding segments are colored red and orange, respectively. The position of the E248A substitution that permitted crystallization is indicated as a white band.
The cell pellet was resuspended in Ni-NTA (QIAGEN) wash buffer [50 mM K2HPO4, 300 mM NaCl, 10% (v/v) glycerol, and 3 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (pH 7.7)], and cells were lysed by sonication (10 min, 50% amplitude, pulse 1 s on and 2 s off). Cell debris was discarded after centrifugation (38000g for 30 min), and the supernatant solution was loaded on a Ni-NTA column (QIAGEN, 5 mL). Protein was eluted with a 0 to 400 mM imidazole gradient in Ni-NTA wash buffer. The GdolS fractions from the Ni-NTA column were combined, concentrated, and loaded onto a 10 mL HiTrap Q HP column (GE Healthcare) after exchange with Q wash buffer [10 mM Tris and 10 mM NaCl (pH 8.0)]. Elution was performed with a 0 to 500 mM NaCl gradient in Q wash buffer. Fractions containing GdolS (>95% pure as judged by SDS–PAGE) were loaded onto a HiLoad Superdex 200 PG column pre-equilibrated with 137 mM sodium chloride, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, and 5% (v/v) glycerol (pH 7.4). Again, fractions were analyzed by SDS–PAGE and the purest combined, concentrated to 5.5 mg/mL, and stored at −80 °C.
For crystallization, GdolS was treated with trypsin for 10 min at room temperature prior to setting up crystallization trays (1000:1 GdolS:trypsin molar ratio). Crystallization was achieved by the sitting-drop vapor diffusion method at room temperature. Typically, a 1 μL drop of protein solution [5 mg/mL GdolS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, and 5% (v/v) glycerol (pH 7.4)] was added to a 1 μL drop of precipitant solution [0.1 M Tris and 2.0 M ammonium sulfate (pH 8.5) (Hampton Research)] and equilibrated against an 80 μL reservoir of precipitant solution. Crystals generally formed within 1 week.
After growing to maximal dimensions, crystals of unliganded GdolS were transferred to a cryoprotectant solution (25% glycerol and 75% mother liquor) and flash-cooled in liquid nitrogen. X-ray diffraction data to 1.50 Å resolution were collected at beamline 4.2.2 of the Advanced Light Source (Lawrence Berkeley National Laboratory), indexed and integrated with Denzo in HKL2000, and scaled with SCALA.31,32 Crystals of the unliganded protein belonged to space group P21: unit cell parameters a = 51.4 Å, b = 74.1 Å, and c = 82.5 Å, with two monomers in the asymmetric unit and a Matthews coefficient (VM) of 2.1 Å3/Da (42% solvent content).
The crystal structure of unliganded GdolS was determined by molecular replacement using Phaser for Molecular Replacement in the CCP4 package33 using a monomer of pentalenene synthase (PDB entry 1PS1) as a search probe.34 Iterative cycles of refinement and manual adjustment of the model were performed with PHENIX and COOT, respectively.35,36 Water molecules were added to the model in the later stages of refinement. The following disordered segments were characterized by broken or missing electron density and accordingly were omitted from the final model: T158–Q161 and S227–V232 (chain A) and M1–T5, G160, Q161, and A224–D231 (chain B). Final refinement statistics are listed in Table 1.
Table 1. Data Collection and Refinement Statistics of Unliganded GdolS.
| resolution limits (Å) | 55.1–1.50 |
| space group | P21 |
| unit cell parameters | |
| a, b, c (Å) | 51.4, 74.1, 82.5 |
| α, β, γ (deg) | 90.0, 91.7, 90.0 |
| total no. of reflections | 346240 |
| no. of unique reflections | 98277 |
| redundancya | 3.5 (3.2) |
| Rmergea,b | 0.070 (0.978) |
| Rpima,c | 0.044 (0.629) |
| CC1/2a,d | 0.998 (0.588) |
| I/σ(I)a | 11.6 (1.2) |
| completeness (%)a | 99.4 (99.8) |
| no. of reflections used in work set/test set | 98143/1994 |
| Rworka,e | 0.157 (0.269) |
| Rfreea,e | 0.200 (0.305) |
| no. of protein atomsf | 4977 |
| no. of solvent atomsf | 658 |
| no. of sulfate ionsf | 10 |
| root-mean-square deviation | |
| bonds (Å) | 0.015 |
| angles (deg) | 1.3 |
| average B factor (Å2) | |
| main chain | 21 |
| side chain | 24 |
| solvent | 38 |
| SO4– anion | 76 |
| Ramachandran plot (%) | |
| allowed | 96.6 |
| additionally allowed | 3.4 |
| generously allowed | 0.0 |
| disallowed | 0.0 |
Numbers in parentheses refer to the highest-resolution shell of data.
Rmerge for replicate reflections. R = ∑|Ih – ⟨Ih⟩|/∑⟨Ih⟩, where Ih is the intensity measure for reflection h and ⟨Ih⟩ is the average intensity for reflection h calculated from replicate data.
Rpim = ∑[1/(n – 1)]1/2|Ih – ⟨Ih⟩|/∑⟨Ih⟩, where n is the number of observations (redundancy).
CC1/2 = στ2/(στ2 + σε2), where στ2 is the true measurement error variance and σε2 is the independent measurement error variance.
Rwork = ∑||F0| – |Fc||/∑|F0| for reflections contained in the working set. Rfree = ∑||F0| – |Fc||/∑|F0| for reflections contained in the test set held aside during refinement (5% of total). |F0| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Per asymmetric unit.
Molecular Modeling
The substrate molecule (3R)-nerolidyl diphosphate (8) was geometry-optimized in its chair conformation, followed by a single-point calculation at the Hartree–Fock level of theory with a standard 6-31G basis set, and subsequent population analysis. The LUMO molecular orbital was selected for the sake of illustrating the mechanism of allylic bond reshuffling as it displays the expected alternation of orbital coefficients after ring closure. The diphosphate group was replaced by OH for the sake of convenience of calculation. The calculations were performed using GAUSSIAN09.37
Results
Structure of GdolS
Unliganded GdolS-E248A adopts the α-helical fold characteristic of a class I terpenoid synthase, as first observed in the crystal structure of avian farnesyl diphosphate synthase.38 GdolS crystallizes as an isologous dimer in the asymmetric unit through the interactions of helices E, F, H, and I. As often observed for dimeric terpenoid cyclases,39 the active sites of the GdolS dimer are oriented in antiparallel fashion. The H-1α loop, the loop following the NSE motif, and the F–G loop are not resolved because of the high mobility of these regions (Figure 2).
Sequence alignments with other terpene synthases show that two metal binding motifs D80DQFD and N218DVRSFAQE are conserved (boldface indicates presumptive metal binding ligands). The crystal structure of GdolS shows that these segments flank the mouth of the active site on helices D and H, respectively. While the penultimate residue in the second metal binding motif is usually an arginine or a lysine residue, GdolS has a glutamine at this position. The function of this residue is to hydrogen bond with the substrate diphosphate group, so it is interesting to note the substitution of the shorter, uncharged side chain for the longer, positively charged side chain that is normally conserved for this function in other sesquiterpene synthases.40 An omit electron density map showing the metal binding motifs in unliganded GdolS is shown in Figure 3.
Figure 3.

Simulated annealing omit map calculated for the metal binding segments D80DQFD and N218DVRSFAQE in GdolS (wall-eyed stereoview contoured at 2.5σ).
The size of the GdolS active site, approximately 15 Å deep and 10 Å wide, is comparable to that of other sesquiterpene synthases. The unliganded enzyme crystallized in the open conformation as had also been observed in the crystal structure of unliganded aristolochene synthase from Penicilium roqueforti (PR-AS)41 and several monomers of aristolochene synthase from Aspergillus terreus (AT-AS).10 The active site is predominantly nonpolar with a contour defined by aliphatic and aromatic side chains. Curiously, although GdolS utilizes a water molecule to quench the final carbocation intermediate in the cyclization cascade, the active site does not contain any polar side chains that could readily serve as a general base to facilitate the reactivity of a trapped water molecule. In the structure of the open active site conformation presented here, a number of ordered solvent molecules are observed in the GdolS active site. However, we cannot conclude that any of these water molecules play specific structural or catalytic roles.
Metal Binding Residues
To probe individual amino acid residue contributions to the GdolS-catalyzed conversion of FDP to (−)-germacradien-4-ol (2), various residues were replaced using site-directed mutagenesis and product distributions were analyzed by gas chromatography and mass spectrometry (GC–MS) of organic extractable products. Steady-state kinetics were measured for all functional mutants.
Single-amino acid replacements in the aspartate-rich region, specifically D80, D81, and D84, with glutamate created functional GdolS variants with only minor effects on the product distribution (Table 2). Each of these enzymes produced minor amounts of germacrene A (5) and D (6) (total <2%) in the pentane extractable products, and while functional, the measurement of the steady-state kinetic parameters showed that these GdolS variants were significantly less active than the wild-type enzyme. GdolS-D80E was too slow to determine steady-state kinetic values. The GdolS-D81E- and GdolS-D84E-catalyzed reactions had turnover numbers, kcat, approximately 10-fold lower than that of the wild-type enzyme. Each mutation had little effect on the binding of FDP to the enzymes (KM values of 0.82 ± 0.11 and 0.92 ± 0.09 μM, respectively).
Table 2. Steady-State Kinetic Parameters and Product Distributions (%) for the Conversion of FDP.
| product
distribution |
|||||
|---|---|---|---|---|---|
| enzyme | kcat (s–1) | KM (μM) | germacradien-4-ol (2) | germacrene A (5) | germacrene D (6) |
| GdolS-WT | 0.079 ± 0.003 | 1.07 ± 0.13 | 100 | – | – |
| GdolS-D80E | ndaa | ndaa | >98 | <1 | <1 |
| GdolS-D80N | n/ab | n/ab | n/ab | n/ab | n/ab |
| GdolS-D81E | 0.010 ± 0.0006 | 0.82 ± 0.11 | >99 | <1 | <1 |
| GdolS-D81N | ndaa | ndaa | >99 | <1 | – |
| GdolS-D84E | 0.009 ± 0.0003 | 0.92 ± 0.09 | >98 | <1 | <1 |
| GdolS-D84N | n/ab | n/ab | n/ab | n/ab | n/ab |
| GdolS-E226D | ndaa | ndaa | >99 | <1 | – |
| GdolS-S222A | ndaa | ndaa | >98 | – | <2 |
| GdolS-N218Q | ndaa | ndaa | >47 | >50 | <2 |
| GdolS-N218L | ndaa | ndaa | >99 | <1 | – |
| GdolS-N218T | n/ab | n/ab | n/ab | n/ab | n/ab |
| GdolS-N218E | n/ab | n/ab | n/ab | n/ab | n/ab |
No detectable activity; activity too low for kinetic parameters to be determined.
Not applicable.
Replacement of D80, D81, and D84 with asparagine led to inactive enzymes with only GdolS-D81N showing small amounts of activity. Products could be observed in the pentane extracts, but activity was too low to allow the measurement of kinetic parameters. These results are consistent with previously reported effects of amino acid replacements in the metal binding domains, with replacement of the first aspartate in the DDXXD motif (D80 in GdolS) having a much greater effect on catalytic activity than replacement of the others.26,42,43 This is also consistent with molecular modeling results reported by van der Kamp et al.,11 who predicted that upon formation of the Michaelis complex two of the requisite Mg2+ ions are coordinated to this first aspartate of AT-AS.
Replacement of N218 of the NSE motif of GdolS with glutamine generated a functional enzyme that converted FDP to almost identical quantities of germacradien-4-ol (2) (47.6%) and germacrene A (5) (50.7%), in addition to a small amount of germacrene D (6) (1.7%). However, this substitution had a detrimental impact on the catalytic efficiency and the steady-state kinetic parameters could not be measured. Because the amino acids of the NSE motif chelate one of the three Mg2+ ions required for catalysis, it is likely that the longer ligand side chain perturbs ideal metal coordination geometry and thus perturbs the catalytic function of the Mg2+3 cluster. In an attempt to further shift the product distribution toward germacrene A (5), N218 was also replaced with leucine, a residue with similar shape and volume but with no metal coordination or hydrogen bonding capability. With the loss of a critical metal coordination interaction in the NSE motif, as well as the loss of possible hydrogen bond interactions with the N218 side chain, coupled with a corresponding increase in hydrophobicity through the N218L substitution, it was proposed that this might prevent ingress of water into the active site and further reduce the proportion of germacradien-4-ol produced. GdolS-N218L was an inefficient enzyme that produced >98% germacradien-4-ol and only traces of germacrene A (5), so water still had ready access to the active site in this mutant. However, the catalytic activity of GdolS-N218L is severely compromised. Other replacements of N218 with amino acids of varying degrees of hydrogen bond donors and acceptors (threonine and glutamate) yielded inactive proteins.
Origin of the Hydroxyl Group of 2 and Potential Water-Directing Residues
To trace the origin of the oxygen atom of 2, incubations were conducted in buffer containing 50% (v/v) H218O and the pentane extractable products were analyzed by GC–MS. The mass spectrum of isolated germacradien-4-ol showed incorporation of an 18O atom with a 1:0.65 16O:18O ratio (Figure S8), indicating that the hydroxyl group of 2 stems from bulk solvent through quenching of the final carbocation in the reaction sequence. A lack of obvious candidates that might act as water binding residues to hydrogen bond with and/or activate H2O as a nucleophile led us to examine the structures of other terpene synthases to gain further insight. A crystallographic study of AT-AS revealed a trapped water molecule in the upper active site hydrogen bonding with N213, N299, and S303 (Figure 4).15 Sequence alignment of AT-AS with GdolS showed that these three residues correspond to N218, Y303, and E307 in GdolS. With N218 having been identified as influencing the incorporation of water into the GdolS product, the function of Y303 and E307 was also investigated by site-directed mutagenesis.
Figure 4.
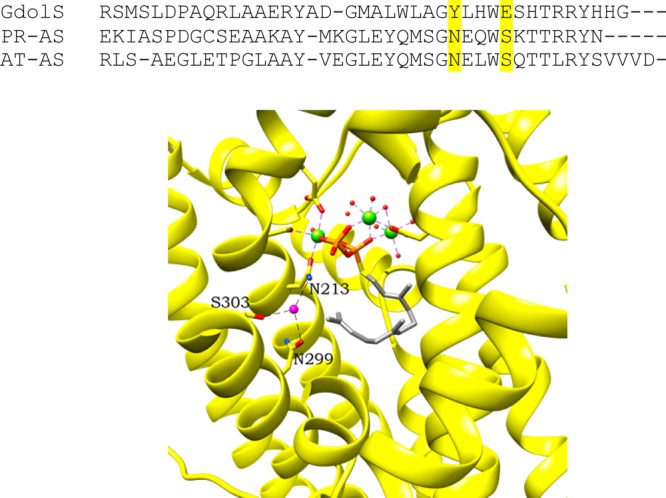
Sequence alignment of the terminal helix and loop of GdolS with AT-AS and PR-AS. N299 and S303 in AT-AS and the aligning residues in GdolS and PR-AS are highlighted in yellow. AT-AS bound to FSDP (farnesyl S-thiolodiphosphate) and [Mg2+]3-PPi,15 with the active site water colored magenta and interactions with N213, N299, and S303 shown as black dashes (PDB entry 4KUX, chain A). Residues are shown with carbons in the respective ribbon color, oxygens in red, nitrogens in blue, and hydrogens in white. Phosphorus atoms are colored orange and sulfur atoms yellow; magnesium is shown as green spheres and water as small red spheres, with the exception of the highlighted magenta water.
Y303 was replaced sequentially with F, I, and T. GdolS-Y303F was generated to test whether the phenolic OH was responsible for hydrogen bonding with the nucleophilic water molecule, while GdolS-Y303I removes hydrogen bonding capability altogether. The side chain of T is an alternative hydrogen bond donor/acceptor equivalent. However, both F and I replacements of Y303 yielded functional enzymes with only minor effects on product distribution while GdolS-Y303T was inactive. Indeed, it appears that Y303 has a greater role in the stabilization of carbocation intermediates given the dramatic reduction of catalytic activity observed for GdolS-Y303I.
Replacement of E307 with Q produced an enzyme that was an effective GdolS, and only traces of germacrene A (5) and germacrene D (6) side products were observed. GdolS-E307Q was approximately 10 times less efficient than the wild-type enzyme (Table 3). Replacement of the same residue with methionine had little effect on product distribution, albeit with a dramatic loss of catalytic efficiency.
Table 3. Steady-State Kinetic Parameters and Product Distribution (%) of Wild-Type GdolS and Y303 Mutants.
| product
distribution |
|||||
|---|---|---|---|---|---|
| enzyme | kcat (s–1) | KM (μM) | germacradien-4-ol (2) | germacrene A (5) | germacrene D (6) |
| GdolS-WT | 0.079 ± 0.003 | 1.07 ± 0.13 | 100 | – | – |
| GdolS-Y303I | ndaa | ndaa | >99 | trace | – |
| GdolS-Y303F | 0.014 ± 0.006 | 0.66 ± 0.12 | >96 | >2 | <1 |
| GdolS-Y303T | n/ab | n/ab | n/ab | n/ab | n/ab |
| GdolS-E307Q | 0.013 ± 0.0004 | 2.24 ± 0.16 | >98 | <1 | <1 |
| GdolS-E307M | ndaa | ndaa | >95 | >4 | <1 |
No detectable activity; activity too low for kinetic parameters to be determined.
Not applicable.
Incubations of GdolS with FDP Analogues
To investigate the catalytic mechanism of GdolS with regard to the diphosphate ionization and subsequent carbocationic reaction cascade, the wild-type enzyme was incubated with various FDP analogues. The nature of the initial carbocation was explored with the fluorinated FDP analogues 2-F-FDP (1b), 15,15,15-F3-FDP (1c), and 12,13-F2-FDP (1d), which might intercept putative ring closure steps through the destabilization of carbocation intermediates. Incubation of 1b and 1c with GdolS generated no detectable products in the pentane extracts as judged by GC–MS analysis, even under extended incubation times. The effect of these FDP analogues as inhibitors was investigated by measuring steady-state kinetic parameters in the presence of varying amounts of fluorinated FDP. Both 2-F-FDP and 15,15,15-F3-FDP were found to be potent inhibitors of GdolS with measured Ki values of 1.1 ± 0.56 and 3.9 ± 0.2 μM, respectively. Double-reciprocal plots indicated that the mode of inhibition was largely noncompetitive, suggesting that they may only partially occupy the active site upon binding. The binding interactions of prenyl diphosphates with a terpene synthase are most likely dominated by the interaction of the negatively charged diphosphate group with the positively charged magnesium ions, and it is not surprising that the Ki values are comparable to substrate KM values. These results may suggest that the initial formation of germacryl cation from FDP proceeds in a stepwise fashion through linear farnesyl cation prior to 1,10-ring closure (Figure 5) because the electron-withdrawing fluorine atoms on C2 and C15 increase the energy of the allylic farnesyl cation and thereby prevent its formation. In previous work, we observed that the terpene cyclases PR-AS44 and δ-cadinene synthase from Gossypium arboreum(45) are capable of converting 2F-FDP to cyclic products, indicating that an SN2-like synchronous cyclization can take place for this substrate; this result strongly suggests that farnesyl cation formation is required for GdolS.
Figure 5.

Reaction outcomes catalyzed by GdolS in this study. The wild-type reaction is colored red. Abortive, potential products, not generated, are colored gray. (a) Reaction catalyzed by wild-type GdolS from FDP (1a). (b) Alternative (minor) pathway observed for some GdolS mutants resulting in germacrene D. (c) Alternative (minor) pathway observed for some GdolS mutants resulting in germacrene A. Panel A shows products arising from incubation of GdolS with deuterated FDP analogues.
To further probe this, GdolS was incubated with 12,13-F2-FDP (1d), a substrate analogue that should not prevent farnesyl cation formation because the fluorine atoms are located on the distal isopropylidene group but will prevent 1,10-ring closure because the π-electron density on C10 and C11 is reduced, thereby lowering the double bond nucleophilicity and increasing the energy of a putative cation with the charge on C11.46 Incubation of 1d with GdolS generated a single pentane extractable product, 12,13-difluoro-(E)-β-farnesene (7), as judged by GC–MS. 7 was identified by comparison with the product generated upon incubation of 1d with (E)-β-farnesene synthase. This result lends strong support to the proposal of a stepwise formation of germacryl cation because this must arise from loss of diphosphate from the abortive substrate and subsequent loss of a proton from C15 to generate the farnesene product.
Incubation of GdolS with nerolidyl diphosphate (NDP, 8), often proposed as a reactive intermediate in sesquiterpene synthase-catalyzed reactions,45,47 produced germacradien-4-ol as a single product. Measurement of the steady-state kinetic parameters with racemic [3H]NDP48 gave values for kcat and KM of 0.076 ± 0.003 s–1 and 1.3 ± 0.03 μM, respectively, remarkably similar to those measured for FDP (Table 4). Use of enantiopure (3R)-[3H]NDP49 for the measurement of kinetic parameters showed a 3-fold increase in kcat paired with a 2-fold increase in KM (0.24 ± 0.007 s–1 and 2.6 ± 0.18 μM, respectively), leading to a modest improvement in catalytic efficiency over that of the substrate FDP (1).
Table 4. Comparison of Steady-State Kinetic Parameters for the GdolS-Catalyzed Turnover of FDP, Racemic [3H]NDP, and (3R)-[3H]NDP.
| 1-3H-labeled substrate | kcat (s–1) | KM (μM) | kcat/KM (s–1 μM–1) |
|---|---|---|---|
| FDP | 0.079 ± 0.003 | 1.07 ± 0.13 | 73.8 × 10–3 |
| racemic [3H]NDP | 0.076 ± 0.003 | 1.30 ± 0.03 | 58.5 × 10–3 |
| (3R)-[3H]NDP | 0.240 ± 0.007 | 2.60 ± 0.18 | 92.3 × 10–3 |
To probe the nature and stereochemistry of the hydride shift following germacrenyl cation formation, GdolS was incubated with [1-2Hn]FDP analogues. GdolS was incubated with [1-2H2]FDP [1e (Figure 5)], and the pentane extractable products were analyzed by GC–MS. The mass spectrum of the resulting [2H2]germacradien-4-ol (Figure S23) contained a molecular ion at m/z 224 consistent with the incorporation of both deuterons. The dehydrated molecular ion fragment [M – H2O]+ showed an ion at m/z 206, indicating that it is also doubly deuterated. The fragment derived from the loss of the isopropyl group shows an ion at m/z 162, indicating that a single deuterium atom has been lost and only one remains on the remaining ring fragment, hence supporting a [1,3]-hydride shift mechanism (Figures 1 and 5); consecutive [1,2]-hydride shifts, as observed for (R)-germacrene D synthase,50 would leave both deuterons on this fragment (m/z 163). The stereochemistry of the 1,3-hydride shift was also investigated through the incubation of GdolS with (R)-[1-2H]FDP (1f). Interestingly, analysis of the mass spectrum of germacradien-4-ol arising from this showed fragments at both m/z 161 and 162, with no clear enrichment in either, e.g., as is evident in the same experiment with δ-cadinene synthase.45 This indicates that the hydride shift is not stereospecific possibly due to a small degree of rotation around the C10, C11 bond that would allow a shift of either hydride. Alternatively, there may be scrambling of the stereochemical information through equilibration of the stabilized allylic carbocation and the germacryl cation, which would lead to no overall isotopic enrichment of either species upon fragmentation of the isopropyl group.
Discussion
GdolS, first discovered through genome mining of Streptomyces citricolor,20 provides an exciting case study for examining how sesquiterpene synthases generate alcohol products through an exquisitely selective carbocationic reaction cascade during which the high-energy and reactive intermediates must be sheltered from the aqueous medium yet subsequently generate the product through nucleophilic capture of the final carbocation with a molecule of water.
Attempts to form cocrystals of GdolS with diphosphate, Mg2+, and farnesyl diphosphate analogues were frustratingly unproductive in this study, but diffracting crystals of unliganded apo-GdolS were obtained. The 1.50 Å resolution structure revealed that GdolS folds as a classic class 1 sesquiterpene synthase with the metal binding motifs identified as D80DQFD and N218DVRSFAQE. Some ordered water molecules were observed in the active site of this structure, but with the active site in an open conformation and disorder preventing observation of the complete H-1α and F–G loops, it is not clear whether these waters play a role in catalysis. There are no obvious active site residues that could hydrogen bond with or activate a nucleophilic water molecule. Site-directed mutagenesis of the metal binding residues in the DDXXD and NSE motifs resulted in either inactive enzymes or functional mutants with little change in product distribution. Only GdolS-N218Q showed a significant perturbation of the product distribution with 51% germacrene A (5) formed alongside 48% germacradien-4-ol (2) and some germacrene D (6). Changes in the Mg2+B coordination polyhedron presumably caused by substitution of the larger glutamine residue for the asparagine ligand to Mg2+B in wild-type GdolS most likely reorient the diphosphate group in the active site so that instead of playing a spectator role, it may prematurely quench the germacryl cation by acting as general base for removal of a proton from C12 or C13 to generate germacrene A (5) (Figure 5). Incubation of wild-type GdolS with FDP in the presence of labeled water confirmed the incorporation of a solvent water molecule (Figure S8). There is a precedent for active site-trapped water molecules in bornyl diphosphate and aristolochene synthases,14,15 and this was believed to be the most likely source of the incorporation of water by GdolS. However, we have found no evidence of any residues involved in the direct incorporation of an active site water molecule. An alternative hypothesis is that the final carbocation is simply quenched by a molecule of water from the bulk solvent. With the carbocation still bound in the active site, specific movements of the H-1α and E–F loops restrict water molecules to approach from the re face of the final carbocation, resulting in the stereo- and regiospecific formation of (−)-germacradien-4-ol (2).
Incubation of GdolS with the fluorinated FDP analogues 2-F-FDP (1b) and 15,15,15-F3-FDP (1c) yielded no detectable products, and both compounds proved to be potent noncompetitive inhibitors. Incubation of 12,13-F2-FDP (1d) with GdolS produced exclusively 12,13-difluoro-(E)-β-farnesene (5), suggesting a stepwise formation of germacryl cation via an intermediate farnesyl cation (Figure 5). Incubation of GdolS with deuterated analogues [1-2H2]FDP (1e) and (R)-[1-2H]FDP (1f) demonstrated that the [1,3]-hydride shift in the next step of the catalytic cycle occurs with scrambling of the stereochemistry at C1.
The results from incubation of GdolS with NPD (8) are intriguing, because (3R)-NDP appears to be a more efficient substrate than FDP itself. While this may suggest that the catalytic mechanism of GdolS proceeds through isomerization of FDP (1) to NDP prior to 1,10-ring closure, this isomerization seems unnecessary. Molecular modeling of the LUMO of (3R)-nerolidol (nerolidol was used rather than NDP to simplify the modeling) constrained to the presumed GdolS active site volume shows that in this conformation the orbitals are aligned to productively drive ring closure and the allylic elimination of the diphosphate upon promotion of electrons from the HOMO (Figure 6). While product release is generally rate-limiting for the overall conversion of FDP by terpene synthases, the initial ionization of FDP is widely accepted as the rate-determining chemical step.51,52 This configuration of NDP is optimal for directly generating the transoid-germacryl cation in a concerted manner.
Figure 6.

Proposed concerted ring closure and allylic elimination of diphosphate in (3R)-NDP. Calculated LUMO of (3R)-nerolidol constrained to the active site volume.
Taken together, the results reported here suggest that after the formation of the Michaelis complex the GdolS-catalyzed conversion of FDP (1) to (−)-germacradiene-4-ol (2) proceeds through farnesyl cation and rapid 1,10-ring closure to germacrenyl cation. The subsequent rapid 1,3-hydride shift yields a stabilized allylic carbocation, which is then quenched by a water molecule just before product release. The mechanism of water capture is unclear, but in the absence of evidence of an active involvement of residues from GdolS, enzyme loop movements may be required to facilitate ingress of water to quench the final carbocation.
Acknowledgments
We thank Prof. Yasuo Ohnishi (University of Tokyo) for plasmid pET16b-SC1 harboring the cDNA of GdolS, Dr. Juan A. Faraldos for helpful discussions, and Dr. Rob Jenkins, Robin Hicks, Simon Waller, and Thomas Williams (Cardiff University) for assistance with mass spectrometry and NMR. We thank Jay Nix for help with data collection at beamline 4.2.2 at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00115.
Synthesis of FDP analogues, protein characterization, table of mutagenic primers, gas chromatograms from enzymatic incubations, mass spectra, and kinetic data (PDF)
Accession Codes
The atomic coordinates and crystallographic structure factors of GdolS-E248A have been deposited in the Protein Data Bank as entry 5I1U.
This work was supported by the U.K.’s Biotechnology and Biological Sciences Research Council (BBSRC) through Grants BB/H01683X/1 and BB/G003572/1, the U.K.’s Biotechnology and Biological Sciences Research Council (BBSRC), the U.K.’s Engineering and Physical Sciences Research Council through Grant EP/L027240/1, the Cardiff Synthetic Biology Initiative, Cardiff University, and National Institutes of Health Grant GM56838.
The authors declare no competing financial interest.
Supplementary Material
References
- Gershenzon J.; Dudareva N. (2007) The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414. 10.1038/nchembio.2007.5. [DOI] [PubMed] [Google Scholar]
- Stevens R. (1992) in Dictionary of terpenoids (Connolly J. D., and Hill R. A., Eds.) Chapman & Hall, London. [Google Scholar]
- Hick A. J.; Luszniak M. C.; Pickett J. A. (1999) Volatile isoprenoids that control insect behaviour and development. Nat. Prod. Rep. 16, 39–54. 10.1039/a705984a. [DOI] [Google Scholar]
- Pickett J. A.; Allemann R. K.; Birkett M. A. (2013) The semiochemistry of aphids. Nat. Prod. Rep. 30, 1277–1283. 10.1039/c3np70036d. [DOI] [PubMed] [Google Scholar]
- Manfredi J. J.; Horwitz S. B. (1984) Taxol: an antimitotic agent with a new mechanism of action. Pharmacol. Ther. 25, 83–125. 10.1016/0163-7258(84)90025-1. [DOI] [PubMed] [Google Scholar]
- White N. J. (2008) Qinghaosu (artemisinin): the price of success. Science 320, 330–334. 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- Tetzlaff C. N.; You Z.; Cane D. E.; Takamatsu S.; Omura S.; Ikeda H. (2006) A gene cluster for biosynthesis of the sesquiterpenoid antibiotic pentalenolactone in Streptomyces avermitilis. Biochemistry 45, 6179–6186. 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.; Cane D. E. (2009) Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor. Mechanism and stereochemistry of the enzymatic formation of epi-isozizaene. J. Am. Chem. Soc. 131, 6332–6333. 10.1021/ja901313v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson D. W. (2006) Structural biology and chemistry of the terpenoid cyclases. Chem. Rev. 106, 3412–3442. 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- Shishova E.; Di Costanzo L.; Cane D. E.; Christianson D. W. (2007) X-ray crystal structure of aristolochene synthase from Aspergillus terreus and evolution of templates for the cyclization of farnesyl diphosphate. Biochemistry 46, 1941–1951. 10.1021/bi0622524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kamp M. W.; Sirirak J.; Zurek J.; Allemann R. K.; Mulholland A. J. (2013) Conformations change and ligand Binding in the aristolochene synthase catalytic cycle. Biochemistry 52, 8094–8105. 10.1021/bi400898k. [DOI] [PubMed] [Google Scholar]
- Rynkiewicz M. J.; Cane D. E.; Christianson D. W. (2002) X-ray crystal structures of D100E trichodiene synthase and its pyrophosphate complex reveal the basis for terpene product diversity. Biochemistry 41, 1732–1741. 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- Köksal M.; Jin Y.; Coates R. M.; Croteau R.; Christianson D. W. (2011) Taxadiene synthase structure and evolution of modular architecture in terpene biosynthesis. Nature 469, 116–120. 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington D. A.; Wise M. L.; Urbansky M.; Coates R. M.; Croteau R. B.; Christianson D. W. (2002) Bornyl Diphosphate Synthase: Structure and Strategy for Carbocation Manipulation by a Terpenoid Cyclase. Proc. Natl. Acad. Sci. U. S. A. 99, 15375–15380. 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Al-Lami N.; Janvier M.; D’Antonio E. L.; Faraldos J. A.; Cane D. E.; Allemann R. K.; Christianson D. W. (2013) Mechanistic insights from the binding of substrate and carbocation intermediate analogues to aristolochene synthase. Biochemistry 52, 5441–5453. 10.1021/bi400691v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane D.; Tandon M.; Prabhakaran P. C. (1993) Epicubenol synthase and the enzymic cyclization of farnesyl diphosphate. J. Am. Chem. Soc. 115, 8103–8106. 10.1021/ja00071a023. [DOI] [Google Scholar]
- Faraldos J. A.; Wu S.; Chappell J.; Coates R. M. (2010) Doubly deuterium-labeled patchouli alcohol from cyclization of singly labeled [2-2H1]-farnesyl diphosphate catalyzed by recombinant patchoulol synthase. J. Am. Chem. Soc. 132, 2998–3008. 10.1021/ja909251r. [DOI] [PubMed] [Google Scholar]
- Nakano C.; Tezuka T.; Horinouchi S.; Ohnishi Y. (2012) Identification of the SGR6065 gene product as a sesquiterpene cyclase involved in (+)-epicubenol biosynthesis in Streptomyces griseus. J. Antibiot. 65, 551–558. 10.1038/ja.2012.68. [DOI] [PubMed] [Google Scholar]
- Jiang J. Y.; He X. F.; Cane D. E. (2006) BIOL 223 - Geosmin biosynthesis. Streptomyces coelicolor germacradienol/germacrene D synthase converts farnesyl diphosphate to geosmin. Abstr. Pap. Am. Chem. Soc. 232, 944–944. [DOI] [PubMed] [Google Scholar]
- Nakano C.; Kudo F.; Eguchi T.; Ohnishi Y. (2011) Genome mining reveals two novel bacterial sesquiterpene cyclases: (−)-Germacradien-4-ol and (−)-epi-α-Bisabolol Synthases from Streptomyces citricolor. ChemBioChem 12, 2271–2275. 10.1002/cbic.201100418. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Zor T.; Selinger Z. (1996) Linearization of the Bradford protein assay increases its sensitivity: Theoretical and experimental studies. Anal. Biochem. 236, 302–308. 10.1006/abio.1996.0171. [DOI] [PubMed] [Google Scholar]
- Biasini M.; Bienert S.; Waterhouse A.; Arnold K.; Studer G.; Schmidt T.; Kiefer F.; Cassarino T. G.; Bertoni M.; Bordoli L.; Schwede T. (2014) SWISS-MODEL: modeling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–258. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modeling. Bioinformatics 22, 195–201. 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Bordoli L.; Kiefer F.; Arnold K.; Benkert P.; Battey J.; Schwede T. (2008) Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13. 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- Seemann M.; Zhai G.; de Kraker J.-W.; Paschall C. M.; Christianson D. W.; Cane D. E. (2002) Pentalenene synthase. Analysis of active site residues by site-directed mutagenesis. J. Am. Chem. Soc. 124, 7681–7689. 10.1021/ja026058q. [DOI] [PubMed] [Google Scholar]
- Cane D. E.; Watt R. M. (2003) Expression and mechanistic analysis of a germacradienol synthase from Streptomyces coelicolor implicated in geosmin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 100, 1547–51. 10.1073/pnas.0337625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert M. J.; Ashton P. R.; Allemann R. K. (2002) Germacrene A is a product of the aristolochene synthase-mediated conversion of farnesyl pyrophosphate to aristolochene. J. Am. Chem. Soc. 124, 11636–11641. 10.1021/ja020762p. [DOI] [PubMed] [Google Scholar]
- Gennadios H. A.; Gonzalez V.; Di Costanzo L.; Li A.; Yu F. L.; Miller D. J.; Allemann R. K.; Christianson D. W. (2009) Crystal structure of (+)-δ-cadinene synthase from Gossypium arboreum and evolutionary divergence of metal binding motifs for catalysis. Biochemistry 48, 6175–6183. 10.1021/bi900483b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L.; Cooper D. R.; Derewenda Z. S.; Eisenberg D. (2007) Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 16, 1569–1576. 10.1110/ps.072914007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesburg C. A.; Zhai G.; Cane D. E.; Christianson D. W. (1997) Crystal structure of pentalenene synthase: Mechanistic insights on terpenoid cyclization reactions in biology. Science 277, 1820–1824. 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) COOT: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., and Fox D. J. (2010) Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT. [Google Scholar]
- Tarshis L. C.; Yan M.; Poulter C. D.; Sacchettini J. C. (1994) Crystal structure of recombinant farnesyl diphosphate synthase at 2.6 Å resolution. Biochemistry 33, 10871–10877. 10.1021/bi00202a004. [DOI] [PubMed] [Google Scholar]
- Rynkiewicz M. J.; Cane D. E.; Christianson D. W. (2001) Structure of trichodiene synthase from Fusarium sporotrichioides provides mechanistic inferences on the terpene cyclization cascade. Proc. Natl. Acad. Sci. U. S. A. 98, 13543–13548. 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.; Cane D. E.; Ikeda H. (2012) Diversity and analysis of bacterial terpene synthases. Methods Enzymol. 515, 123–162. 10.1016/B978-0-12-394290-6.00007-0. [DOI] [PubMed] [Google Scholar]
- Caruthers J. M.; Kang I.; Rynkiewicz M. J.; Cane D. E.; Christianson D. W. (2000) Crystal structure determination of aristolochene synthase from the blue cheese mold Penicillium roqueforti. J. Biol. Chem. 275, 25533–25539. 10.1074/jbc.M000433200. [DOI] [PubMed] [Google Scholar]
- Cane D. E.; Xue Q.; Fitzsimons B. C. (1996) Trichodiene synthase. Probing the role of the highly conserved aspartate-rich region by site-directed mutagenesis. Biochemistry 35, 12369–12376. 10.1021/bi961344y. [DOI] [PubMed] [Google Scholar]
- Felicetti B.; Cane D. (2004) Aristolochene synthase: mechanistic analysis of active site residues by site-directed mutagenesis. J. Am. Chem. Soc. 126, 7212–7221. 10.1021/ja0499593. [DOI] [PubMed] [Google Scholar]
- Miller D. J.; Yu F.; Allemann R. K. (2007) Aristolochene Synthase-Catalyzed XCyclization of 2-Fluorofarnesyl-Diphosphate to 2-Fluorogermacrene A. ChemBioChem 8, 1819–1825. 10.1002/cbic.200700219. [DOI] [PubMed] [Google Scholar]
- Faraldos J. A.; Miller D. J.; Gonzalez V.; Yoosuf-Aly Z.; Cascon O.; Li A.; Allemann R. K. (2012) A 1,6-ring closure mechanism for (+)-δ-cadinene synthase?. J. Am. Chem. Soc. 134, 5900–5908. 10.1021/ja211820p. [DOI] [PubMed] [Google Scholar]
- Yu F.; Miller D. J.; Allemann R. K. (2007) Probing the reaction mechanism of aristolochene synthase with 12,13-difluorofarnesyl diphosphate. Chem. Commun. 4155–4157. 10.1039/b709562g. [DOI] [PubMed] [Google Scholar]
- Picaud S.; Olofsson L.; Brodelius M.; Brodelius P. E. (2005) Expression, purification, and characterization of recombinant amorpha-4,11-diene synthase from Artemisia annua L. Arch. Biochem. Biophys. 436, 215–226. 10.1016/j.abb.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Karp F.; Zhao Y.; Santhamma B.; Assink B.; Coates R. M.; Croteau R. B. (2007) Inhibition of monoterpene cyclases by inert analogues of geranyl diphosphate and linalyl diphosphate. Arch. Biochem. Biophys. 468, 140–146. 10.1016/j.abb.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane D. E.; Ha H. J. (1988) Trichodiene biosynthesis and the role of nerolidyl pyrophosphate in the enzymatic cyclization of farnesyl pyrophosphate. J. Am. Chem. Soc. 110, 6865–6870. 10.1021/ja00228a041. [DOI] [Google Scholar]
- Schmidt C. O.; Bouwmeester H. J.; Franke S.; König W. A. (1999) Mechanisms of the biosynthesis of sesquiterpene enantiomers (+)- and (−)-germacrene D in Solidago canadensis. Chirality 11, 353–362. 10.1002/(SICI)1520-636X(1999)11:5/6<353::AID-CHIR2>3.0.CO;2-L. [DOI] [Google Scholar]
- Cane D. E.; Chiu H. T.; Liang P. H.; Anderson K. S. (1997) Pre-steady-state kinetic analysis of the trichodiene synthase reaction pathway. Biochemistry 36, 8332–8339. 10.1021/bi963018o. [DOI] [PubMed] [Google Scholar]
- Mathis J. R.; Back K.; Starks C.; Noel J.; Poulter C. D.; Chappell J. (1997) Pre-steady-state study of recombinant sesquiterpene cyclases. Biochemistry 36, 8340–8348. 10.1021/bi963019g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.