

Abstract
RNA spatial dynamics play a crucial role in cell physiology and thus the ability to monitor RNA localization in live cells can provide insight into important biological problems. This article focuses on imaging RNAs using an “RNA mimic of GFP”. This approach relies on a RNA aptamer, called dimeric Broccoli, which binds to and switches on the fluorescence of DFHBI, a small molecule mimicking the fluorophore in GFP. Dimeric Broccoli is tagged to heterologously expressed RNAs and upon DFHBI binding the fluorescent signal of dimeric Broccoli reports the transcript’s localization in cells. This protocol describes the process of validating the fluorescence of dimeric Broccoli-labeled transcripts in vitro and in cells, flow cytometry analysis to determine overall fluorescence levels in cells, and fluorescence imaging in bacterial and mammalian cells. Overall, the current protocol should be useful for researchers seeking to image high abundance RNAs, such as transcribed off the T7 promoter in bacteria or off Pol III-dependent promoters in mammalian cells.
Keywords: RNA imaging, RNA aptamer, aptamer expression, fluorescence microscopy
INTRODUCTION
Imaging RNA in live cells can answer important questions about RNA synthesis, processing, transport, translation and degradation. While a number of methods, based on fluorescent proteins or fluorophore-labeled oligonucleotide probes have been developed (discussed in the Background part below), they all are complicated and require considerable effort to establish in a laboratory. Fluorophore-labeled oligonucleotides are complex or costly to synthesize and fluorescent protein-based imaging approaches require multiple plasmids for imaging. An ideal RNA imaging protocol would be simple, genetically encoded, minimally invasive and have little or no background fluorescence.
One strategy for RNA imaging is to append RNA of interest to an RNA mimic of green fluorescent protein (GFP) (Paige et al., 2011). The probe consists of an RNA aptamer, dubbed Spinach, and a small molecule fluorophore, DFHBI (3,5-difluoro-4-hydroxybenzylidene imidazolinone), which is very similar to that in GFP. DFHBI is readily cell-permeable and essentially non-fluorescent in solution, however, it becomes highly fluorescent upon Spinach binding. Thus, this system presents a simple and highly promising approach for live cell RNA imaging.
While bright in vitro and in bacteria, Spinach was dim in mammalian cells and thus improved versions of this system have been developed. Rational optimization of Spinach resulted in Spinach2 with increased folding and thermostability (Strack et al., 2013). However, both Spinach and Spinach2 were engineered in vitro and therefore had low cell compatibility, i.e. high dependence on non-physiological ion concentration or low resistance to cellular RNases. An alternative approach was to express aptamer libraries in live bacterial cells and use fluorescence-activated cell sorting to isolate the brightest and thus the most cell-compatible clones (Filonov et al., 2014). This allowed isolation of Broccoli and dimeric Broccoli (dBroccoli, discussed below) which display lower dependence on intracellular magnesium concentration and overall brighter fluorescent signal both in bacteria and mammalian cells compared to Spinach2 (Filonov et al., 2014).
Spinach, Spinach2 and Broccoli have been successfully used to image RNA both in bacterial and mammalian cells. Spinach and Broccoli were used to follow 5S relocalization in cells upon sucrose treatment while Spinach2 revealed the dynamic nature of toxic RNAs in cell nuclei (Filonov et al., 2014; Paige et al., 2011; Strack et al., 2013). Additionally, Spinach, Spinach2 and Broccoli have been fashioned into efficient small molecule and protein sensors for bacterial cells (Filonov et al., 2014; Kellenberger et al., 2015; Kellenberger et al., 2013; Paige et al., 2012; Song et al., 2013; You et al., 2015). Overall, RNA mimics of GFP have already proven themselves a potent approach for non-invasive RNA studies in a cell.
This article describes the process of using Broccoli for imaging of RNA in live bacterial and mammalian cells.
The first step (Basic Protocol 1) is used to detect expression of Broccoli-fused RNA in cells. Bacterial or mammalian cells are transformed or transfected, respectively, and upon expression of the RNA-Broccoli fusion the cells are lysed and total RNA is isolated. Total RNA is then separated using denaturing PAGE and Broccoli-containing bands are revealed with DFHBI staining. After that, total RNA is revealed using a non-selective nucleic acid fluorophore, such as SYBR Gold. DFHBI staining is very sensitive and allows detection of very small amounts of Broccoli-containing RNA. Additionally, this step ensures that the expressed transcript is not cleaved or processed in some other undesired way.
The second step (Basic Protocol 2) is to detect fluorescence in cells using flow cytometry. Flow cytometry is a very convenient and simple way to detect Broccoli fluorescence in cells. This experiment can give an indication as to whether fluorescence imaging on a microscope will be successful. Bacterial or mammalian cells are transformed or transfected, respectively, and Broccoli is expressed. Then the cells are incubated with DFHBI and analyzed on flow cytometer. Fluorescent cell detection ensures both successful Broccoli expression and folding. Finally, the last step (Basic Protocol 3) is the imaging of bacterial or mammalian cells.
Strategic planning
Selection of tags
Broccoli and Broccoli-containing tags are highly useful for tagging RNA due to their high brightness in mammalian and bacterial cells (Filonov et al., 2014). This increase in fluorescence relative to Spinach2 likely derives from improved folding and reduced dependence on free intracellular magnesium levels, which can be limiting in many cell types (Grubbs, 2002; Romani, 2013). One useful tag is dBroccoli, which is an aptamer containing two Broccoli units in one stem-loop with the total length of 92 nt vs. 49 nt in Broccoli (Filonov et al., 2014). dBroccoli is essentially twice as bright as a single Broccoli aptamer. dBroccoli is thus the brightest aptamer within the group of RNA mimics of GFP. Spinach and Spinach2, however, are more well-established platforms for sensor creation and their utilization should be considered when engineering sensors for novel molecules (Kellenberger et al., 2015; Paige et al., 2012; You et al., 2015).
Scaffolds
dBroccoli performance in cells can be further enhanced by the use of a scaffold. A scaffold is a highly stable RNA structure which is fused to an aptamer of interest to force the correct folding (Ponchon and Dardel, 2007; Shu et al., 2014). Scaffolds solve one of the major problems with aptamer expression in cells, which is that aptamers fold poorly in cells (Filonov et al., 2015; Martell et al., 2002; Strack et al., 2013). Aptamers are prone to misfolding when surrounded by flanking sequences. Thus, the fluorescence signal of aptamers such as Spinach or Broccoli may be severely compromised. This is partially ameliorated in the case of Broccoli due to directed evolution, which exhibits a high degree of context-independent folding (Filonov et al., 2014). Nevertheless, to improve aptamer folding in vitro, and especially in vivo, we recommend using a scaffold. A good scaffold should not only improve aptamer folding but also be bioorthogonal. This means that it should not be recognized by the cellular RNA processing machinery and thus should not be cleaved or degraded.
We recently engineered F30 (Filonov et al., 2015), a scaffold based on the highly stable viral ϕ29 three-way junction RNA motif (Shu et al., 2014). F30 contains mutations that remove a cryptic mammalian transcription termination element (Filonov et al., 2015). F30 substantially improves Broccoli fluorescence and shows minimal or no cleavage in bacterial or mammalian cells (Filonov et al., 2015).
Combination of dBroccoli and the F30 scaffold result in a single tag that exhibits four-times the fluorescence of a single Broccoli. This tag comprises a single F30 scaffold, such that each of its two stem-loops contains a single dBroccoli. This results in F30-2xdBroccoli tag (Figure 1 A,B) (Filonov et al., 2015). F30-2xdBroccoli is highly useful for RNA imaging in live bacterial or mammalian cells. The protocol below utilizes this imaging tag.
Figure 1.

Sequence and schematic representation of the F30-2xdBroccoli tag. (A) Sequence of F30-2xdBroccoli is presented. Green indicates the dBroccoli units, orange indicates the F30 sequence. An RNA of interest can be appended on either 5′ or 3′ end, or F30-2xdBroccoli can be inserted inside of an RNA of interest. (B) Schematic drawing of F30-2xdBroccoli. Two Broccoli units (green) are inserted into each of two stem-loops of F30 (orange).
In vitro testing of RNA-Broccoli fluorescence
Before expressing an RNA tagged with F30-2xBroccoli in cells, we recommend testing its fluorescence in vitro. The F30-2xdBroccoli tag can be inserted at the 5′, 3′ or internal sites. In our experience, transcripts with RNA of interest appended to 5′ end of F30-2xdBroccol show higher fluorescent signal.
To confirm that the tag remains fluorescent after insertion into the target RNA, the tagged RNA can be generated by in vitro transcription. To do this, we recommend preparing the template for in vitro transcription by PCR. The PCR product encoding the F30-2xdBroccoli-tagged RNA should be prepared so that it contains a T7 promoter and can be transcribed using commercially available in vitro transcription kits (e.g., see the protocol for the AmpliScribe T7-Flash Transcription Kit, Epicenter). The RNA can be purified and then mixed with DFHBI to generate fluorescent signal. Comparing the fusion RNA fluorescent signal with that from F30-2xdBroccoli alone (positive control) and the fluorophore-only solution (negative control) allows determination of the folding efficiency of Broccoli within the fusion construct (use excess of fluorophore in the mixture, see Filonov et al., 2014, for further details on this protocol). An efficiency of 50% and higher indicates good folding of F30-2xdBroccoli appended to the RNA of interest and is sufficient for proceeding to the in-cell experiment.
In general, F30-2xdBroccoli remains fluorescent at most insertion points in a target RNA; however, the folding may be affected in regions with high G/C-content or sequence complementarity to F30 or dBroccoli.
Fluorophores
Currently there are two small-molecule fluorophores which can be used with F30-2xdBroccoli: DFHBI, mentioned earlier, and DFHBI-1T (3,5-difluoro-4-hydroxybenzylidene 1-trifluoroethyl-imidazolinone), which is a modified version of DFHBI (Song et al., 2014). Broccoli-DFHBI-1T is ~40% brighter that Broccoli-DFHBI due to higher extinction coefficient. Additionally DFHBI-1T shows lower fluorescent background, which further increases the signal-to-noise ratio during fluorescence imaging (Song et al., 2014). Finally, Broccoli-DFHBI-1T has excitation and emission maxima (472 nm and 507 nm, respectively) that are optimized for standard microscope FITC filter cubes, compared to Broccoli-DFHBI (447 nm and 501 nm). Overall, DFHBI-1T has diverse properties that enhance its use for imaging using Spinach or Broccoli. However, due to the higher cost of DFHBI-1T, compared to DFHBI, the latter can be preferred for in vitro and bacterial experiments, where the signal is already expected to be very bright due to high expression levels.
Promoter
Another consideration is the strength of a promoter used to express F30-2xdBroccoli-fused RNA in cells. The current protocol describes the expression of F30-2xdBroccoli or F30-2xdBroccoli-fused RNAs from very strong promoters, such as T7 (bacterial cells) or 5S and U6 (mammalian cells). While the high strength of fluorescent signal in bacterial cells allows using even significantly weaker promoters, the signal in mammalian cells is relatively dim and imaging of low abundance RNAs expressed from weaker promoters, e.g. mRNAs, has not yet been achieved. Engineering a cassette of multiple F30-2xdBroccoli units should provide an imaging tag bright enough for visualization of rare RNAs.
Photostability
The final important point is that all DFHBI (or DFHBI-1T)-binding aptamers engineered so far show low photostability when imaged under strong light illumination (Han et al., 2013). DFHBI undergoes a light-induced isomerization which renders the RNA-fluorophore complex non-fluorescent. While this process is fast and substantially decreases the fluorescent signal level, the bleaching is reversible since the isomerized fluorophore can come off the RNA and be replaced with fresh fluorophore from solution. Overall it is recommended to minimize RNA-fluorophore complex exposure to bright light. If, however the bleaching has occurred, it is recommended to allow the fluorophore to exchange and thus the signal to restore by letting the cells (or solution) sit in the dark for 1–2 minutes. Another strategy is to utilize pulse illumination, which was shown to increase bacterial cell brightness and improve imaging (Han et al., 2013). However, we have not thoroughly tested this approach and thus it is not included in this protocol.
BASIC PROTOCOL 1: In-gel staining of Broccoli-tagged RNA to assess expression level and integrity of Broccoli-tagged RNA
When a Broccoli-tagged RNA is expressed in cells, it is important to ensure that the RNA is expressed and that it is primarily present in cells as a full-length product and not a truncated or degraded transcript. Typically, the most desirable RNA is one where the full-length product is tagged with Broccoli, and none of abundant breakdown products contain Broccoli. This way, fluorescence signals in cells clearly derive from the full-length RNA. To establish the integrity and quantity of the Broccoli-tagged RNA, a simple and fast in-gel staining protocol, presented below, is typically used. In this procedure F30-2xdBroccoli is expressed in bacterial cells and 5S-F30-2xdBroccoli and U6+27-F30-2xdBroccol are expressed in mammalian cells. The latter construct is expressed off the strong U6 promoter and has the +27 leader sequence of U6 on the 5′ end (Paul et al., 2003). Then, total cellular RNA is isolated, separated based on size using denaturing PAGE, and visualized by soaking the gel in a DFHBI-1T staining solution. When the gel is imaged on a gel-imaging device, Broccoli-tagged RNAs are readily detectable. The gel can be subsequently stained with a non-selective RNA fluorophore, such as SYBR Gold. This method was presented and discussed in detail in a previous paper (Filonov et al., 2015).
Materials
Bacterial expression
pET-28c-F30-dBroccoli plasmid (available at Addgene). The plasmid is generated by inserting dBroccoli with a part of T7 promoter into pET-28c-Broccoli (Filonov et al., 2014) cut with BglII and NheI enzymes.
pET28c (Novagen)
LB media with kanamycin (see recipe)
LB/agar dishes with kanamycin (see recipe)
Super Optimum Culture Media (SOC, Life Technologies, cat. no. 15544)
Chemically competent Escherichia coli BL21 Star (DE3) cells (Invitrogen, cat. no. C6010-03)
1 M IPTG solution (see recipe)
1.5 ml microcentrifuge tubes (USA Scientific, cat. no. 1615–5500)
Petri dishes (VWR International, cat. no. 25384–094)
Culture tubes (VWR International, cat. no. 60818–667)
25-ml Erlenmeyer flasks with baffles (Corning, cat. no. 355115)
Parafilm
42 °C water bath or heat block
37 °C air incubator
Orbital shaker
Cuvettes for NanoDrop (VWR International, cat. no. 470138–468)
Microcentrifuge (Eppendorf, cat. no. 5424)
NanoDrop (Thermo Scientific) or similar instrument
Mammalian cell expression
pAVU6+27 plasmid (Paul et al., 2003), available by request from the corresponding author of the reference publication.
pAV5S plasmid (Paul et al., 2003), available by request from the corresponding author of the reference publication.
pAVU6+27-F30-2xdBroccoli plasmid (available at Addgene) (Filonov et al., 2015).
pAV5S-F30-2xdBroccoli plasmid (available at Addgene). The plasmid is generated by inserting dBroccoli into pAV5S plasmid cut with SalI and XbaI restriction enzymes.
HEK293T/17 cells (ATCC, cat. no. CRL-11268)
Mammalian cell culture media (see recipe)
1x PBS solution (see recipe)
TrypLE (Life technologies, cat. no. 12604021)
FuGENE HD (Promega, cat. no. E2311)
Pasteur pipettes
12-well plate (Costar, cat. no. 3513))
Cell culture incubator
Cell culture hood
RNA isolation and gel imaging
DNase, RNase-free water (Gibco, cat. no. 10977)
TRIzol LS (Life Technologies, cat. no. 10296028)
Chloroform (Fisher Scientific, cat. no. C607-4)
Isopropanol (Thermo Scientific, cat. no. BP26181)
Ethanol, 70% (see recipe) (Koptec, cat. no. V1016)
Glycogen, 20 mg/ml (Thermo Scientific, cat. no. R0551))
TE buffer (see recipe)
RiboRuler Low Range RNA Ladder (Life Technologies, cat. no. SM1831)
DFHBI-1T staining solution (see recipe)
SYBR Gold (Life Technologies, S-11494)
1x TBE buffer (see recipe)
40 mM stock of DFHBI-1T (see recipe)
1.5 ml microcentrifuge tubes (USA Scientific, cat. no. 1615–5500)
Novex TBE-Urea Sample Buffer (2X) (Life Technologies, cat. no. LC6876)
Precast Novex 10% TBE-Urea Gel (Life Technologies, cat. no. EC6875BOX)
1.5 ml microcentrifuge tubes (USA Scientific, cat. no. 1615–5500)
XCell SureLock Mini-Cell Electrophoresis System (Life technologies, cat. no. EI0001)
Power supply (Bio-Rad, cat. no. 1645050 or similar))
Microcentrifuge (Eppendorf, cat. no. 5424)
NanoDrop (Thermo Scientific) or similar instrument
ChemiDoc MP imager (Bio-Rad)
Bacteria transformation
-
1
Thaw 2 tubes of competent E. coli BL21 Star (DE3) cells on ice.
We use the BL21 Star (DE3) strain, which shows very high expression level of F30-2xdBroccoli. However, any strain can potentially be used for expression. If using pET28c-based plasmids, as in the current protocol, verify that the cells produce T7 polymerase, necessary for expression off the T7 promoter, by checking the genetic background of the strain used: conventionally, these strains are designated (DE3). -
2
Add 40 ng of pET28c (negative control) to the first tube. To the second tube, add 40 ng of pET-28c-F30-dBroccoli. Mix the plasmids with the cells by gently flicking the tubes and incubate the mixtures on ice for 20 min.
-
3
Heat shock cells in a 42°C water bath for 30 s. Place tubes back on ice immediately after and chill them for 2 minutes.
-
4
Add 300 μl SOC medium to each tube and incubate the tubes at 37 °C with rotation or shaking for 45 minutes.
-
5
Plate 50 μl of each cell mixture onto a petri dish containing LB medium supplemented with 50 μg/μl kanamycin. Incubate plates at 37 °C overnight to grow individual colonies.
Plates containing transformed E. coli cells can be sealed with parafilm and stored at 4°C for up to four weeks.
Aptamer expression in bacteria
-
6
Inoculate a single colony from Step 5 into a sterile culture tube containing 5 ml LB medium with 50 μg/μl kanamycin. Grow cultures at 37 °C with shaking overnight.
-
7
Measure the optical density (OD) at 600 nm for a 1:10 dilution (with PBS) of each overnight culture.
-
8
Start fresh cultures by diluting cells to a starting concentration of 0.05 OD600 units/ml in a 25-ml baffled Erlenmeyer flask containing 10 ml of LB medium with 50 μg/μl kanamycin.
-
9
Grow cells at 37 °C with shaking to OD600 = 0.4 (~2 h).
-
10
Add IPTG to 1 mM final concentration. Continue to grow cells at 37 °C with shaking for another 2–4 h.
RNA isolation
-
11
Pellet bacterial cells by centrifugation at 2,500 x g for 5 min. Aspirate the media with vacuum aspirator.
-
12
Resuspend bacterial cells in 1 ml of PBS.
-
13
Measure the optical density (OD) at 600 nm for a 1:10 dilution (with PBS) of bacteria.
-
14
Adjust the OD600 of the non-diluted cells to achieve a final OD600 of 1.
-
15
Centrifuge 1 ml of bacterial suspension from the previous step at 2,500 x g for 5 min. Use 1 ml per 1.5 ml microcentrifuge tube.
-
16
Aspirate the media, and resuspend the pellet in 250 μl of PBS.
-
17
Add 750 μl of TRIzol LS, mix by inverting up and down for 10 s or until no cell suspension is seen.
TRIzol contains phenol, an irritating and toxic chemical, and should be handled with gloves while wearing eye protection in a chemical hood. Close the tube caps tightly to avoid solution seeping through. All phenol-contaminated plasticware should be disposed according to the regulations of your institution. -
18
Add 200 μl of chloroform per 750 μl of TRIzol LS Reagent used. Cap the tubes securely.
Handle chloroform in a chemical hood. Protect hands with gloves and avoid breathing fumes. -
19
Shake the tubes vigorously by hand for 15 seconds. Incubate for 2–15 minutes at room temperature.
-
20
Centrifuge the samples at 12,000×g for 15 minutes at 4°C.
-
21
Remove the upper aqueous phase of the sample by angling the tube at 45° and pipetting the solution out (~500 μl). Avoid drawing any of the interphase or organic layer into the pipette when removing the aqueous phase. Use gloves from this point onwards to avoid RNA degradation.
Follow the general rules of working with RNA (Nielsen, 2011). In general, use gloves, change them frequently. Use standard approaches for avoiding RNase contamination. -
22
Transfer the aqueous phase (~0.5 ml) into a new tube, add 5–10 μg (0.25–0.5 μl) of RNase-free glycogen as a carrier to the aqueous phase.
The phenol-chloroform phase should be disposed according to the regulation of your institution. -
23
Add 0.5 mL of 100% isopropanol to the aqueous phase, vortex for 5 s, incubate for 10 minutes at RT.
-
24
Centrifuge at 12,000 × g for 10 minutes at 4°C. After centrifugation a small white pellet should be observed at the bottom of the tube. Remove the supernatant from the tube by vacuum aspiration, leaving only the RNA pellet. Be careful while removing the liquid as the pellet is easy to dislodge and can be sucked into the aspirator.
-
25
To wash the RNA pellet, add 0.7 ml of 75% ethanol. Wash the pellet by inverting the tube 10–15 times. The pellet may dislodge during this step.
-
26
Centrifuge the sample at 12,000 × g for 5 minutes at 4°C, and discard the supernatant by vacuum aspiration. Be careful not to suck out the pellet.
-
27
Do a quick spin to collect all the liquid on the bottom. Gently aspirate the liquid using a pipette with a 20 μl tip.
-
28
Leave the tubes on the bench for 5–10 minutes, upright, to air dry the RNA pellet. Dry pellet should look nearly transparent. No ethanol droplets should be seen in the tubes.
-
29
Dissolve the pellet in 10–20 μl of TE.
-
30
Measure RNA concentration using a NanoDrop or similar instrument that enables nucleic acid quantification in small volumes.
You should expect to have 10–20 μl of total RNA with the concentration of 100–500 ng/μl. A much lower concentration may indicate protocol failure.
RNA PAGE
-
31
Assemble the electrophoresis chamber with a 8% or 10% TBE-Urea gel. Fill the chamber with 1x TBE and pre-run the gel at 300 V for 30–60 min at room temperature.
Choose the gel percentage based on the expected size of your transcript. For the transcripts with the size as in this protocol, both options are acceptable. A 10% gel is better for resolving individual bands and bands in the low-molecular weight range (e.g. 50–500 nt) but requires longer runs. -
32
While the gel is running, prepare RNA samples. Use water to adjust the volume of the RNA samples from step 30 so each sample has 200 ng of bacterial total RNA in a final volume of 5–10 μl. Also prepare 1 μl of the RNA ladder mixed with 4 μl of water.
-
33
Add equal amount of 2xSample Buffer (1 μl per μl of sample) to all samples and the RNA ladder. Heat the mixture with a heat block at 75°C for 5 min, then chill on ice.
-
34
Load samples and the ladder on a gel and run at 270 V for 30–60 min depending on the expected size of the tagged RNA.
For the RNA used in this protocol we routinely run until the xylene cyanol fluorophore band, which is slowest out of two fluorophores present in the Sample Buffer, runs off the end of the gel (~1 h). Adjust the running time depending on the length of your particular transcript. However, if F30-2xdBroccoli (234 nt) is used for your RNA labelling, the time recommendation above will be appropriate.
Gel staining
-
35
When electrophoresis is completed, disassemble the cassette, remove the gel and wash it 3×5 min with ~15 ml of water in a small container.
We routinely use lids from pipette tip boxes as containers for gel staining. -
36
Incubate the gel in ~15 ml of DFHBI-1T staining solution for 15–20 min at room temperature.
The staining solution can be used for more than one staining, however for the best performance we recommend changing the solution after ~5 stainings. DFHBI, which is less expensive, compared to DFHBI-1T, can be used for the staining solution preparation with similar efficiency. -
37
Image the gel using a ChemiDoc MP imager. Broccoli fluorescence is detected using a channel with 470±15 nm excitation and 532±14 nm emission (preset Alexa488 imaging protocol). Observe specific staining of the Broccoli-containing bands on a gel (Fig. 2A). Ensure that your RNA is of expected size. F30-2xdBroccoli in bacteria is expected to be 284 nt long, which is what is observed on the gel. This size includes the length of the tag itself plus the transcription terminator sequence at the 3′ end.
The gel imaging protocol is highly sensitive. As little as ~1 fmole (~200 pg) Broccoli can be detected, so RNA present in cells at a concentration range of 0.1 μM and higher can be visualized (Filonov et al., 2015). -
38
Wash out DFHBI-1T with water, three times 5 min each at room temperature on an orbital shaker.
-
39
Stain with the RNA fluorophore of your choice. We use SYBR Gold diluted 1/10000 in ~15 ml of TBE and incubate for 30 min at RT on an orbital shaker.
Never use the same container to stain gels with DFHBI-1T and SYBR Gold. The residual amount of SYBR Gold staying or sticking on the plastic of the container is enough to stain RNA in the gel non-specifically and contaminate a DFHBI-1T staining solution. -
40
Image the gel using a ChemiDoc MP imager (Bio-Rad) with the preset SYBR Gold protocol (302 nm excitation and 590±55 nm emission). The SYBR Gold staining should reveal all the RNA fragments in the sample (Fig. 2A).
A successful gel image should show crisp bacterial total RNA bands indicating that the samples were prepared correctly and no degradation of the RNA material has occurred. The RNA from the pET28c-F30-2xdBroccoli-transformed cells in the Fig. 2A is an example of the proper sample preparation. The pET28c-transformed cell RNA (negative control) though shows a noticeable smear which may indicate partial degradation. While the major bands of the total RNA, such as 5S, are still clearly seen, the overall negative control sample preparation is suboptimal.The band amounts for the RNA ladder used in this protocol (RiboRuler Low Range RNA Ladder) are known. This makes it easy to quantify the amount of Broccoli-containing RNA in a SYBR Gold-stained band which in turn will allow back calculation of that RNA concentration in cells. However, this may not be possible if the SYBR-stained Broccoli band is obscured by an abundant endogenous RNA band, such as 5S. In that case another option is to calculate the amount of the RNA of interest based on the Broccoli fluorescence. For that it is necessary to generate RNA standards by in vitro transcribing the same transcript and loading known amounts on the same gel as serial dilutions.
Figure 2.

Broccoli expression is readily detected using the in-gel staining protocol. (A) Bacterial cells were transformed with either pET28c plasmid (negative control) or with pET28c-F30-2xdBroccoli and the RNA was expressed for four hours. Then total RNA was isolated, separated using denaturing PAGE and stained with DFHBI-1T and then with SYBR Gold, as described the Basic Protocol 1. DFHBI-1T specifically reveals Broccoli-containing bands while SYBR Gold detects total cellular RNA. (B) Mammalian (HEK293T) cells were either untransfected or transfected with pAV5S or pAVU6+27 (negative controls) or with pAV5S-F30-2xdBroccoli or pAVU6+27-F30-2xdBroccoli. After 72 h expression total RNA was isolated, separated using denaturing PAGE and stained with DFHBI-1T and then with SYBR Gold, as described in the Alternate Protocol 1. Broccoli-containing bands are clearly detectable even in such a complex mixture as total mammalian cell RNA.
ALTERNATE PROTOCOL 1
This alternate protocol describes mammalian cell RNA isolation, separation and staining. The major difference between this protocol and the Basic Protocol 1 is RNA expression in a cell, which is described below in details. Protocols for RNA isolation, gel PAGE and staining are essentially the same and differ only in details, which are listed.
Cell transfection and aptamer expression
-
1
Start with a 10 cm dish of nearly confluent HEK293T cells. Aspirate the media with a Pasteur pipette connected to a vacuum line, add 10 ml of PBS, gently rinse the cells by rocking the dish, aspirate PBS.
For this protocol, we recommend using HEK293T cells, which in our experience are the best for producing large amounts of exogenous RNA. While other cell lines can potentially be used, we advise to transfect HEK293T cells in a parallel experiment and compare the transcript level in HEK293T and the cell line of your choice later on a gel. HEK293T cells are very easy to detach. Use caution when adding liquid to or aspirating media from the cell bed. Also, change liquid quickly to avoid cell death from overdrying. All manipulations with cells should be performed in a sterile tissue culture hood. -
2
Add 1.5 ml of TrypLE, incubate for 5 min at room temperature.
-
3
Add 1.5 ml of media, resuspend cells with 1 ml pipette by aspirating up and down.
-
4
Count cells using hemocytometer and calculate cell concentration.
-
5
Prepare a master mix of cells and media in a 15 ml or 50 ml conical tube. Have 2.5×105 cells per well in 2 ml of media per well.
For the current experiment we need 5 wells (4 plasmids and one untransfected well), so we use 15 ml conical tube and mix 1.25×106 cells in a media volume adjusted to 10 ml. -
6
Seed HEK293T cells into five wells of 12-well plate. Grow overnight in the cell culture incubator.
Make sure that you will have enough of plasmid material for the transfection the next day. See the step 9 for the plasmid amount needed. -
7
The next day, make sure that the confluence is around 80%. Change the media on cells to pre-warmed fresh one, 2 ml per well.
-
8
Pre-warm FuGENE HD reagent at RT.
-
9
Prepare solution of 1.1 μg of the pAV5S, pAVU6+27 (negative controls), pAVU6+27-F30-2xdBroccoli or pAV5S-F30-2xdBroccoli plasmid in 52 μl of water.
Consider co-transfecting a plasmid expressing a red fluorescent protein to have a transfection efficiency control. We routinely use the pSuper-mCherry plasmid (Belly et al., 2010) for that purpose. The ratio of the pSuper-mCherry plasmid to a pAV-based plasmid is 1:19–1:39. Control cells should also be transfected with pSuper-mCherry to establish bleed-through fluorescence levels during imaging using the FITC filter cube. -
10
Add 3.3 μl of FuGENE HD, mix carefully by pipetting up and down (15 times) or by vortexing briefly. Incubate for 5–10 minutes at room temperature.
Refer to the manufacturer’s manual for more detailed information on transfection and its optimization. -
11
Add 50 μl of mixture dropwise to each well and mix thoroughly by rocking the plate sideways.
-
12
Incubate cells in the tissue culture incubator for 48–72 hours at 37 °C.
Check the cells daily. Change the culturing media to a fresh one if it turns yellow,
RNA isolation
-
13
48–72 hours after the transfection aspirate media from cells.
-
14
Add ~1 ml of PBS to each well, gently wash and aspirate.
-
15
Add 200 μl TrypLE, incubate 5 min at RT.
-
16
Add 800 μl of PBS, resuspend cells and transfer to a microcentrifuge tube.
-
17
Centrifuge at 100 x g for 5 min at RT.
-
18
Resuspend cells in 250 μl of PBS.
-
19
Follow the steps 17–30 in the Basic Protocol 1.
After RNA pellet resuspension in 15 μl of TE expect to have concentration in a range of 0.5–5 μg/μl.
RNA PAGE and staining
-
20
Follow steps 31–40 in the Basic Protocol 1. Use 5 μg of total RNA from mammalian cell to load into one well, instead of 200 ng. After staining completion, ensure that your RNA is of expected size. 5S-F30-2xdBroccoli is expected to be 385 nt long (including 5S and transcription terminator) and U6+27-F30-2xdBroccoli is expected to be 293 nt long (including +27 sequence and transcription terminator) which is what is observed on the gel.
The amount of exogenous transcript expressed in mammalian cells is much lower than that in bacteria and thus your specific band will likely be co-migrating with some endogenous RNA. If the amount of Broccoli RNA in cells needs to be calculated, we recommend generating transcript standards by doing in vitro transcription, as mentioned earlier. Running known amounts of these standards along with the actual sample and then staining with DFHBI-1T will allow precise calculation of the RNA amount in your band of interest.We recommend including U6+27-F30-2xdBroccoli-transfected cell total RNA in all your subsequent experiments. This total RNA always shows readily detectable Broccoli-containing band and will serve as a positive control which gives you a reference on how much of expressed RNA you should expect.
BASIC PROTOCOL 2: Flow cytometry analysis of transformed bacterial cells
Before proceeding with fluorescence imaging, it is useful to make sure that the dBroccoli-tagged RNA is expressed well in mammalian or bacterial cells. The simplest approach is to use flow cytometry to assess cellular brightness on the whole population level. The protocols below employ delivering of the genetic material into cells, expressing RNA, pretreating cells with DFHBI-1T, and finally flow cytometry and data processing. This protocol uses F30-2xdBroccoli expressed in bacterial cells and 5S-F30-2xdBroccoli and U6+27-F30-2xdBroccolin in mammalian cells. The cells are incubated with DFHBI-1T and analyzed on a cell sorter to detect the fluorescent population.
Materials
Chemically competent Escherichia coli BL21 Star (DE3) cells (Invitrogen, cat. no. C6010-03)
HEK293T/17 cells (ATCC, cat. no. CRL-11268)
Cell culture hood
1x PBS solution (see recipe)
TrypLE (Life technologies, cat. no. 12604021)
PBS with 4% FBS (see recipe)
40 mM stock of DFHBI-1T (see recipe)
Round-bottom tubes with cell strainer cap (VWR, cat. no. 21008–948)
FACSaria II (BD biosciences)
Transform bacteria and express F30-2xdBroccoli as described in the Basic Protocol 1, steps 1-10.
-
Inoculate 20 μl of crude bacterial culture into 1 ml PBS with 40 μM DFHBI-1T, incubate 10 min at room temperature (RT).
Consider preparing bacterial solution in PBS without DFHBI-1T as an additional control. Transfer bacterial suspension into round-bottom tubes with cell strainer cap. Squirt the suspension into the cap to strain cell clumps. Tap the tube against the bench to facilitate the suspension passage through the strainer.
-
Analyze bacterial suspension using FACSaria II or similar instrument. Aim to detect events in the standard FITC channel (488 nm excitation and 530/30 emission filter). Alternatively, for better signal detection, consider using a broader emission collection filter (e.g. 525/50 nm).
FACSaria II is a cell sorter, which we also conventionally used for cell analysis. However, if a flow cytometer (cell analyzer) is available in your laboratory or core facility, then it is more practical to use it for this protocol. Run the negative control (pET28c-transformed cells with DFHBI-1T). Then run F30-2xdBroccoli-expressing cells. You will observe events that are significantly brighter than the negative control.
Analyze data using the FlowJo software (TreeStar) or other program designed to process flow cytometry data. Successful expression of dBroccoli aptamer in bacterial cells results in a very bright population of cells (Fig. 3A).
Figure 3.

Flow cytometry analysis reveals bright fluorescent cell population. (A) Flow cytometry analysis of the pET28c- or pET28c-F30-2xdBroccoli-transformed bacteria. The RNA was expressed for 4 h and then the cells were treated with 40 μM DFHBI-1T and analyzed using a FACSAria II instrument as described in the Basic Protocol 2. Fluorescent events were detected in the modified FITC channel (488 nm excitation and 530±25 emission filter). The data is presented as an overlay of the histograms for the pET28c- and pET28c-F30-2xdBroccoli-transformed bacteria. Broccoli-expressing cells are more than 100 fold brighter than the negative cell population. (B) Flow cytometry analysis of the untransfected mammalian cells or the cells transfected with pAV5S or pAVU6+27 (negative controls) or with pAV5S-F30-2xdBroccoli or pAVU6+27-F30-2xdBroccoli. mCherry-expressing plasmid was co-transfected as a transfection efficiency control. The cells were harvested 72 h after transfection and treated with 40 μM DFHBI-1T as described in the Alternate Protocol 2. The data is presented as a two-parameter dot plot with both FITC (488 nm excitation and 530±25 emission filter) and PE-Texas Red (561 nm excitation and 610±10 nm emission) channels presented. Broccoli-expressing cells show high transfection efficiency and robust fluorescent signal in both channels, compared to the controls.
ALTERNATE PROTOCOL 2
This alternate protocol describes mammalian cell FACS analysis. Again, the major difference between this protocol and the Basic Protocol 2 is RNA expression in a cell. Also, the procedure of cell preparation for analysis follows a different protocol, which is described below. Finally, we recommend utilization of the second fluorophore during the analysis. We use mCherry expressed in cells, which serves as a convenient transfection marker.
-
1
Transfect cells and express F30-2xdBroccoli-labelled RNAs in cells as described in the Alternate Protocol 1, steps 1–12. Generate all the controls described and consider co-transfecting red fluorescent protein plasmid.
-
2
48–72 hours after the transfection aspirate media from cells.
-
3
Add ~1 ml of PBS to each well, gently wash and aspirate.
-
4
Add 200 μl TrypLE, incubate 5 min at RT.
-
5
Add 800 μl PBS with 4% FBS containing 50 μM DFHBI-1T (final concentration of DFHBI-1T is 40 μM).
-
6
Resuspend cells and transfer to round-bottom tubes with cell strainer cap. Push the cell suspension through the strainer. Tap tubes against the bench to facilitate the solution passage. Keep the tubes on ice from that point.
Cells are exposed to DFHBI-1T for 10–15 min during resuspension and cell straining, so additional incubation with DFHBI-1T is not needed.If needed, cells can be divided into two parts before DFHBI-1T addition. One part will not be treated with the fluorophore and used as an additional control. -
7
Analyze the mammalian cell suspension using FACSAria II (BD biosciences) or similar instrument. Aim to detect Broccoli-specific events in the standard FITC channel (488 nm excitation and 530±15 emission filter). Alternatively, for better signal detection, consider using broader emission collection filter (e.g. 525±25 nm). If co-transfecting mCherry, as in this protocol, also monitor events in the PE-Texas Red channel (561 nm excitation and 610±10 nm emission).
-
8
Run negative controls (pAV5S- and pAVU6+27-transfected cells with DFHBI-1T). Then run F30-2xdBroccoli-expressing cells. You will observe mCherry fluorescence only in transfected cells. You will see green fluorescent events in the Broccoli-expressing cells only.
-
9
Analyze data using the FlowJo software (TreeStar). Successful expression of dBroccoli aptamer in bacterial cells results in a substantially brighter, compared to negative, population of cells observed in the FITC channel (Fig. 3B).
To clearly see the positive population, overlay the dot plot of the F30-2xdBroccoli-expressing cells with the negative controls (5S- or U6+27-expressing ones and untransfected ones), as shown in Figure 3B. Successful transfection and expression should result in at least ~30% positive cells, from the total population, as on the figure.
BASIC PROTOCOL 3: dBroccoli fluorescence imaging in live cells
Once you confirm that you can detect fluorescence of your dBroccoli-fused RNA in cells using flow cytometry and that your construct is intact using in-gel staining, you can proceed to imaging. This portion of the protocol will describe how to image F30-2xdBroccoli in live bacterial cells and 5S-F30-2xdBroccoli and U6+27-F30-2xdBroccolin live mammalian cells. Again, dBroccoli-fused RNAs are expressed in either bacterial or mammalian cells and then the cells are pre-incubated with DFHBI-1T and imaged using fluorescent microscopy.
Materials
Chemically competent Escherichia coli BL21 Star (DE3) cells (Invitrogen, cat. no. C6010-03)
HEK293T/17 cells (ATCC, cat. no. CRL-11268)
1x PBS solution (see recipe)
TrypLE (Life Technologies, cat. no. 12604021)
40 mM stock of DFHBI-1T (see recipe)
Clear mammalian cell culture media (see recipe)
Mouse laminin (Fisher Scientific, cat. no. 50948048)
DNase, RNase-free water (Gibco, cat. no. 10977)
Hoechst 33342 (Life Technologies, cat. no. H3570)
Poly-D lysine-coated 3.5 cm glass-bottom plates (Mattek Corporation, cat. no. P35GC-1.5-14-C)
Cell culture incubator
Cell culture hood
Inverted widefield fluorescent microscope (Nikon, TE2000)
Motorized stage (Ludl, BioPrecision Motorized Flat Top Stage) or similar
High NA fluorescence objective (Nikon, Plan Apo, X60/ 1.4, oil)
CoolSnap HQ2 CCD camera (Photometrics) or similar
Filter cube suitable for green fluorescence imaging such as FITC or GFP filter sets (e.g., with a sputter coated excitation filter 470±20, dichroic mirror 495 (long pass), and emission filter 525±25 (Chroma Technology))
Filter cubes suitable for imaging of auxiliary fluorescent signals, such as Hoechst 33342-stained nuclei and mCherry-filled cells. We use the Texas Red filter cube with 560±20 nm excitation filter, 585 nm (long pass) dichroic mirror and 630±37.5 nm emission filter for imaging mCherry (Chroma Technology) and the DAPI filter cube with 350±25 nm excitation filter, 400 nm (long pass) dichroic mirror and 460±25 nm emission filter (Chroma technology) for imaging Hoechst 33342 staining.
Temperature-controlled environmental chamber for microscope. Our microscope is housed in a custom-built environmental chamber that maintains a temperature of 35–37°C and CO2 level of 5%.
NIS Elements (Nikon) or similar image acquisition software
Fiji (http://fiji.sc/Fiji), ImageJ, or similar image analysis software
Preparing bacterial cells for imaging
-
1
Transform cells and express aptamers following steps 1–10 in the basic protocol 1. Again, have pET28c-transformed cells as negative control and pET28c-F30-2xdBroccoli- transformed cells for dBroccoli imaging.
-
2
Turn on the environmental chamber to pre-warm the microscope to 37 °C for imaging.
If the environmental chamber is not available, then a heated stage is also an option, especially for bacteria, which do not require CO2 level to be controlled. Conceivably, the imaging can even be done at room temperature if temperature is not a critical parameter for your experiment. -
3
Take a 200 μl aliquot from each flask and transfer each to a fresh microcentrifuge tube. Centrifuge 2 min at 5,000 x g at room temperature. Remove media from pellet by vacuum aspiration. Be careful to not suck out the cells.
-
4
Resuspend each pellet in 1 ml PBS, use pipetting or vortexing. Make sure no cell clumps are seen.
-
5
Remove two 3.5 cm glass-bottom dishes from its packaging.
-
6
Add 200 μl of each cell suspension sample to a separate glass-bottom dish. Incubate dishes at 37 °C for 45 min to allow E. coli cells to adhere to the glass bottom.
-
7
Wash wells twice with 500 μl PBS media to remove unattached cells. Be extra careful not to wash away loosely attached cells. Also, be careful not to suck out the liquid completely since the cells exposed to air will die.
-
8
Replace PBS media with 200 μl PBS media supplemented with 40 μM DFHBI-1T and incubate cells at 37 °C for 30 min.
Bacteria imaging
-
9
Bring the dishes to the Inverted widefield fluorescent microscope (such as Nikon TE2000, or similar). Focus on adhered E. coli using a 60X oil objective under bright light illumination.
-
10
Switch to fluorescence acquisition with a FITC filter set. Optimize the exposure time, start with 200 ms.
-
11
Take DIC and fluorescent images.
DIC gives sharper images, however phase contrast can also be used if DIC setup is not available.dBroccoli, similarly to Broccoli and Spinach, bleaches quickly under direct illumination, and this can be noticed when imaging bacterial cells. However, the fluorescent signal of dBroccoli in E. coli is bright enough that even bleached dBroccoli can still be detected. If a higher signal is desired, allow the dBroccoli fluorescence to recover in the dark for 30–60 s and then take an image again. -
12
Image the negative control cells under the same conditions and using the same settings.
-
13
Process images in ImageJ or other software of your choice. Adjust minimal intensity level so no negative cells are seen and use the same intensity scale for the F30-2xdBroccoli cells. Typical bacteria imaging data is presented on Fig. 4A.
Figure 4.
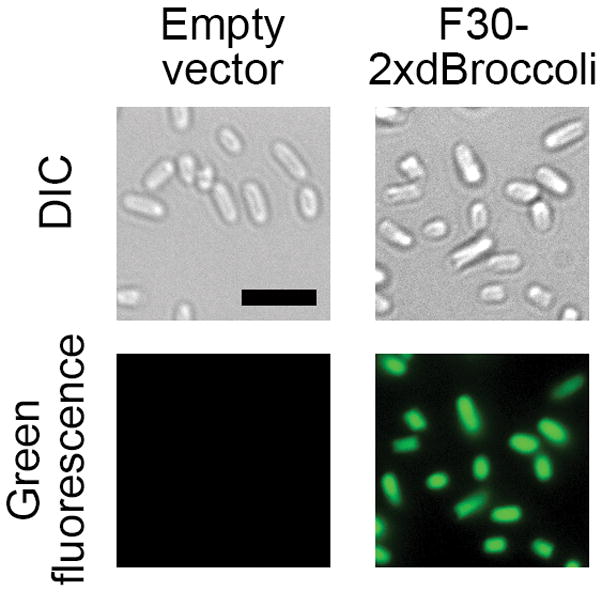
F30-2xdBroccoli imaging in E. coli. The pET28c or pET28c-F30-2xdBroccoli plasmids were transformed into bacteria, then RNA was expressed for 4 h and the cells were processed as described in the Basic Protocol 3 and imaged. Broccoli fluorescence was detected in the FITC channel (excitation filter 470±20 nm and emission filter 525±25 nm). Exposure time 200 ms. Scale bar, 5 μm.
ALTERNATE PROTOCOL 3
This alternate protocol describes Broccoli imaging in mammalian cells. Aside from the RNA expression and cell preparation steps, the major difference is that mammalian cells are generally dimmer and thus it is harder to find bright fluorescent cells. Care should be taken to minimize photobleaching and, if the photobleaching happened, cells should we allowed to rest in the dark before imaging again. Also, mammalian cells are sensitive to the temperature and media pH, so experiments should be performed at 37°C and with CO2 level of 5% (or in a buffered media).
Preparing HEK293T cells for imaging
-
1
Transfect HEK293T cells and express RNA following steps 1–12 in the Alternate Protocol 1.
-
2
48 h after transfection, in the morning prepare five 3.5 cm glass-bottom dishes by coating them with 50 μl of laminin (1:100 in PBS) for 2 hours at 37°C.
-
3
At the end of 2 h coating time start preparing cells. Aspirate media from the transfected cells.
-
4
Gently wash them with 1 ml of 1xPBS, aspirate PBS.
-
5
Detach the cells off the plate by incubating with 200 μl of TrypLE for 5 min at RT.
-
6
Add 1 ml of clear cell culture media, resuspend by pipetting cells up and down until no cell clumps are seen.
-
7
Aspirate the coating solution from the glass-bottom dishes and wash twice with 2 ml dH2O.
-
8
Add 1.6 ml of clear cell culture media to the glass-bottom dishes.
-
9
Add 400 μl of the cells from step 6 to the glass-bottom dishes, gently mix by rocking the dishes.
The rest of the cells (~800 μl) can stay in the same well and be collected the next day to isolate total RNA or for other applications. -
10
Incubate for another ~24 h at 37°C.
Imaging F30-2xdBroccoli
-
11
Turn on the environmental chamber to pre-warm the microscope to 37 °C for imaging. Open the CO2 valve to fill the chamber to 5%. Allow 30 min for the chamber to equilibrate.
If using a heated stage instead, make sure that the media on cells is buffered with 25 mM HEPES, pH 7.4 rather than a bicarbonate-containing buffer. -
12
Thirty minutes prior to experiment, remove 1 ml of media from the glass-bottom dishes with transfected cells.
-
13
Add DFHBI-1T and Hoechst till the final concentrations of 40 μM and 5 μg/ml, respectively. Incubate for 30 min in a cell culture incubator at 37°C.
-
14
Bring the cells to the microscope. Use 60X oil objective to find green fluorescent cells.
Green fluorescent cells can be found by scanning the fields. Since dBroccoli, like Broccoli and Spinach, bleaches very fast, expect the fluorescent signal to quickly disappear once you find it. Center the field on a promising cell and let it stay in the dark for couple minutes so the bleached fluorophore can be exchanged for the fresh one in solution. -
15
Take image of DFHBI-1T using the FITC filter with exposure of 0.5–1 s. If Hoechst 33342 was used, image nuclei with the DAPI filter (~100 ms exposure). If mCherry or other red fluorescent protein was co-expressed, image it in Texas Red filter set (~400 ms exposure time).
-
16
Take images of DFHBI-1T-treated untransfected and 5S- and U6+27-expressing cells (negative controls) under the same conditions and with the same settings as for the Broccoli-containing cells.
-
17
Process cells in ImageJ or other similar program. Adjust the minimal intensity of the negative control cells so almost no background fluorescence is seen and then use the same intensity scale for the F30-2xdBroccoli-expression cells. This will show the actual Broccoli signal. Cell images generated using this protocol are presented at Fig. 4B.
SUPPORT PROTOCOL 1: In vitro transcription of RNA-Broccoli fusions and their fluorescence measurements
This protocol describes how to synthetize RNA-Broccoli fusions in vitro. These RNAs then can be used to assess chimeric RNA’s fluorescence and folding, as described in Strategic Planning. The protocol follows three general steps: PCR products generation, in vitro transcription and fluorescence measurement.
Materials
AmpliScribe T7-Flash Transcription Kit (Epicenter, cat. no. ASF3257), or similar
Micro Bio-Spin P30 gel columns (Bio-Rad, cat. no. 7326223)
5x buffer for fluorescence measurement (see recipe)
DFHBI-1T (Lucerna, cat. no. 410)
NanoDrop (Thermo Scientific) or similar instrument
1.5 ml microcentrifuge tubes (USA Scientific, cat. no. 1615–5500)
FluoroMax-4 fluorometer (Horiba Scientific), or similar instrument
Cuvette for fluorescence measurements, Quartz SUPRASIL Ultra-micro with PTFE Stopper (PerkinElmer, cat. no. B0631124), or similar
In vitro transcription
-
1
Generate PCR products, encoding your RNA-Broccoli fusions. Make sure that 5′ end of the PCR contains the T7 promoter sequence (5′ GTATAATACGACTCACTATAGGG 3′, transcription starts after TATA).
Have PCR products that encode your RNA-Broccoli (or F30-2xdBroccoli) fusion and Broccoli only. The latter will be used as a positive control. -
2
Purify PCR products using commercially available spin columns. Aim to have final concentration of 50–100 ng/μl.
-
3
Prepare the in vitro transcription reaction mixture following the protocol from the kit. 20 μl total volume is sufficient to generate enough RNA for in vitro experiments. Use 500–1000 ng of PCR template for each reaction.
-
4
Incubate for 2 h at 37 °C, for the highest yield incubate overnight.
-
5
Purify using Micro Bio-Spin P30 gel columns following the manufacturer’s protocol.
-
6
Measure RNA concentration using NanoDrop or similar instrument that enables nucleic acid quantification in small volumes.
For more accurate concentration determination consider using fluorescence-based kits, such as Quant-iT RiboGreen RNA Assay Kit (ThermoFisher Scientific, cat. no. R11490).
Fluorescence measurement
-
7
Mix 1 μM RNA and 10 μM DFHBI-1T (or DFHBI) in 1x buffer for fluorescence measurement, incubate at room temperature for 30 minutes. Prepare also the dye only solution to serve as a background signal control. For the cuvette suggested, prepare 130 μl of solution.
-
8
Transfer solutions to the cuvette and measure fluorescent signal on a fluorometer. Subtract the background signal and then compare the signal from RNA-Broccoli to the signal from Broccoli only.
If the RNA-Broccoli fusion folds well, then expect to have its signal being not less than 50% of that for Broccoli alone. Fluorescent signal lower than 50% of that of Broccoli indicates that the construct misfolds and its redesigning may be needed.
REAGENTS AND SOLUTIONS
LB media
10 g tryptone (Sigma Aldrich, cat. no. T9410)
5 g yeast extract (Fisher, cat. no. BP9727)
10 g NaCl (US Biological, cat. no. S5000)
Deionized water up to 1 L
Sterilize by autoclaving. Supplement with 50 mg/ml of kanamycin sulfate when culturing cells (Sigma Aldrich, cat. no. K4000)
Store up to several month at 4°C
LB/agar dishes
10 g tryptone (Sigma Aldrich, cat. no. T9410)
5 g yeast extract (Fisher, cat. no. BP9727)
10 g NaCl (US Biological, cat. no. S5000)
20 g agar (Thermo Scientific, cat. no. ICN19461590)
Deionized water up to 1 l
Sterilize by autoclaving and pouring into 10 cm petri dishes, 25 ml per dish.
Store up to several month at 4° C
1 M IPTG solution
239 mg IPTG (Sigma Aldrich, cat. no. I6758)
Water up to 1 ml
Filter sterilize
Store up to several month at −20° C
70% ethanol
35 ml 100% ethanol (KOPTEC, cat. no. V1016)
15 ml DNase, RNase-free water (Gibco, cat. no. 10977)
Mammalian cell culture media
450 ml Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, cat. no. 11995073)
50 ml fetal bovine serum (FBS) (Life Technologies, cat. no. 10437028)
5 ml Penicillin-Streptomycin (10,000 U/mL) (Life Technologies, cat. no. 15140122)
Store up to several months at 4° C
Clear mammalian cell culture media
450 ml Dulbecco’s modified Eagle’s medium (DMEM) without phenol red (Life Technologies, cat. no. 31053036)
50 ml fetal bovine serum (Life Technologies, cat. no. 10437028)
10 ml 200 mM glutamine (Life Technologies, cat. no. 25030081)
5 ml Penicillin-Streptomycin (10,000 U/mL) (Life Technologies, cat. no. 15140122)
Store up to several months at 4° C
1xPBS
100 ml of 10x PBS (Gibco, cat. No 70011-044)
900 ml of water
Filter sterilize
Store up to several months at 4° C
PBS with 4% FBS
5 ml of 10x PBS (Gibco, cat. No 70011-044)
2 ml of fetal bovine serum (Life Technologies, cat. no. 10437028)
43 ml of sterile water
Store up to several months at 4° C
1T stock
5 mg DFHBI-1T (Lucerna, cat. no. 410)
500 μl dimethyl sulfoxide (Sigma Aldrich, cat. no. 472301)
Protect from light, store indefinitely at 4° C or –20° C
TE buffer
1 mM Tris-HCl pH 7.5
1 mM EDTA pH 8.0
1x TBE buffer
10.8 g Tris (US biological, cat. no. T8600)
5.5 g Boric acid (J.T.Baker, cat. no. 4035-01)
Deionized water up to 900 ml
4 ml 0.5 M Na2EDTA (pH 8.0)
Water up to 1 l
Store indefinitely at room temperature.
DFHBI-1T staining buffer
40 mM HEPES pH 7.4
100 mM KCl
1 mM MgCl2
10 μM DFHBI-1T
Store up to several months at 4° C
5x buffer for fluorescence measurement
200 mM HEPES pH 7.4
500 mM KCl
5 mM MgCl2
COMMENTARY
Background Information
In recent years many different approaches for RNA imaging in cells have been developed, each with their own advantages and limitations.
One of the extensively used approaches to image RNA in cells is based on exogenously added labeled antisense probes (Molenaar et al., 2004; Molenaar et al., 2001) and molecular beacons (Bao et al., 2004; Bratu et al., 2003; Nitin et al., 2004; Tyagi and Alsmadi, 2004). The major advantage of these protocols is their ability to target endogenous, non-modified RNAs. The major drawback of this approach, though, is the perturbing and often complex nature of these probes’ delivery into cells (Santangelo et al., 2012). This problem necessitates development of more convenient, genetically-encoded probes for RNA imaging.
Currently, there are three major types of genetically encoded systems for live-cell RNA imaging: the MS2-GFP imaging system, the Pumilio-split-FP imaging system, and fusing an RNA of interest to RNA mimic of GFP.
MS2-GFP was the first example of a genetically encoded system for RNA imaging (Bertrand et al., 1998). This system consists of the MS2 RNA hairpin, which is appended to an RNA of interest, and the MS2 coat protein, which binds to the MS2 hairpin as a dimer with high affinity. Labelling MS2 protein with GFP allows tethering the latter to RNA and reporting RNA localization. Utilizing up to 24 copies of MS2 hairpin attracts 48 GFP molecules allowing imaging of single transcripts (Fusco et al., 2003; Shav-Tal et al., 2004). This technique was recently used to image individual β-actin mRNAs in the neuronal cells of a living mouse (Park et al., 2014).
Although a very powerful approach, the MS2-GFP approach has its drawbacks. GFP-MS2 fusions not bound to RNA remain fluorescent and generate high fluorescent background in cells. To mitigate this problem, two approaches were proposed. Initially, MS2-GFP was fused with nuclear localization signal so free MS2-GFP molecules would be concentrated in nucleus, leaving the cytoplasm relatively background-free. However, this excludes nuclear RNAs from the imaging experiment due to high background fluorescence in that compartment. Additionally, and more importantly, the presence of 48 nuclear localization signals on a single transcript can severely interfere with the trafficking behavior of the tagged RNA (Tyagi, 2009). Another way to deal with the high background problem is to utilize a split protein fluorescent approach. In a relatively recent design, an RNA of interest was labeled with two types of small hairpins: MS2 and PP7, each positioned next to each other and binding to a specific protein, MS2 coat protein and PP7 coat protein, respectively. Each of these proteins is linked not to a fluorescent protein, but rather to a half of a fluorescent protein. These non-fluorescent halves can interact with each other and reconstitute a functional fluorescent protein only when MS2 and PP7 coat proteins bind to their respective RNA hairpins and are brought into proximity (Wu et al., 2014). While solving the background problem, these proteins still retain nuclear localization elements and may thus influence the trafficking of the RNA to which they are attached.
Another genetically encoded approach for RNA imaging is based on employing Pumilio homology domains (PUM-HDs) (Ozawa et al., 2007). PUM-HDs are unique proteins which bind to a specific sequence, rather than to a structural element of RNA, and they can potentially be engineered to interact with any custom 8 nt RNA site. PUM-HDs were utilized as a part of a split fluorescent protein imaging approach. Two halves of a fluorescent protein are fused to two different PUM-HDs that target two adjacent 8 nt sites within one transcript. Again, this brings two halves of a fluorescent protein together thus reconstituting fluorescent signal. This approach benefits from the low background, since the initial fluorescent protein halves are dark, and from the ability to target endogenous RNAs. The major disadvantage is that PUM-HD domains need to be engineered every time a new transcript is targeted.
The third approach, as described above, is based on a small RNA aptamer, called Spinach, and a fluorophore, DFHBI, which is non-fluorescent in free form but becomes highly fluorescent when bound to Spinach (Paige et al., 2011). This system benefits from very low background fluorescence, allowing imaging of RNA localization throughout the cell, and from a very small size of Spinach (and especially Broccoli), and DFHBI. The small size of the Broccoli-DFHBI complex implies that there may be minimal, if any, influence on the dynamics of a tagged RNA. While DFHBI has to be added to the cells externally, this fluorophore is non-toxic and readily cell-permeable and does not appear to have negative effects on cellular physiology. The major disadvantage of this system so far is its inability to detect single transcripts or low abundance RNAs. This problem can potentially be solved by creating cassettes of Broccoli so each transcript in study has at least 24–48 Broccoli units appended to it. Another problem is relatively low photostability of all RNA-DFHBI fluorescent complexes. While severely affecting the fluorescent signal, this problem can in part be mitigated by the fact that, unlike for fluorescent proteins or small organic fluorophores, photobleaching of RNA-DFHBI is reversible. The fluorescent signal of a bleached complex can be recovered after a short incubation in the dark. Indeed, a pulsed illumination approach has been described that utilizes fluorescence recovery to obtain increased imaging sensitivity (Han et al., 2013).
Overall, a researcher should consider all the options before choosing an imaging approach. The MS2-GFP system at this point may be more suitable for imaging of a single transcript. The PUM-HD approach, although requiring more optimization, should allow imaging of endogenous, non-engineered cellular RNAs. However, if you expect you RNA to be highly expressed, the Spinach/Broccoli system can serve as a relatively simple and efficient alternative.
Critical Parameters and troubleshooting
The most important considerations are covered in the Strategic Planning section of this protocol. In this section, we will re-iterate some of them and add a few more points which are critical for the success of reproducing of this protocol.
RNases
A general problem which can impact in vitro experiments is that RNA is prone to degradation by ubiquitous RNases. Researchers who do not have experience handling RNA should familiarize themselves with numerous “working with RNA” guides that are abundant on the internet, such as (Nielsen, 2011). Poor RNA handling techniques may result in contamination with RNases or metal ions which in turn will result in rapid RNA degradation. This can ruin all in vitro experiments, such as assessing the RNA-Broccoli construct folding efficiency or preparation of total RNA samples, as described in the Basic Protocol 1. In general, it is strongly advised to use gloves and ensure an RNase-free environment by using RNase-free plasticware, water and reagents.
Verify Broccoli fusion fluorescence in vitro
We encourage researchers to test the fluorescence of their RNA-Broccoli fusions in vitro before proceeding with the in-cell experiments for two reasons. First, it provides information about the Broccoli (or F30-2xdBroccoli) folding efficiency within their specific transcript. Second, it provides a sense of how the tagged RNA will perform in cells. Poor Broccoli folding when appended to your transcript is usually a “no go” decision maker and should prompt optimization of the linker between the transcript and Broccoli, or inserting Broccoli into a different site of your RNA. In general, varying the Broccoli insertion point (5′, 3′ or within the sequence) and varying a Broccoli-RNA connector sequence should be one of the first things to try when in vitro folding or, later, in-cell expression is not successful. As mentioned above, appending your RNA to the 5′ end of F30-2xdBroccoli sequence is recommended as a starting point.
Expression levels
In mammalian cells Broccoli has been used to image RNAs expressed off the strong promoters only, such as U6 and 5S. If you plan to express your transcript off another promoter, it is very important that you have high enough expression level so you can detect your RNA in cells. In-gel staining can be very helpful at this stage as it offers a simple, rapid, and literally “on the fly” method of assessing RNA level in total cellular RNA. Thus, comparison of your transcript’s level with that of U6+27-F30-2xdBroccoli should also be a major decision point. A substantially lower expression level of your transcript, compared to U6+27-F30-2xdBroccoli, hints that detection of Broccoli fluorescence in cells using flow cytometry or fluorescence imaging can be problematic. It should be noted, though, that bright Broccoli-containing band on a gel does not necessarily ensures that bright cellular fluorescence will be observed. At this point, it seems that there may be other variables affecting the intensity of Broccoli signal in vivo. For example, it is conceivable, that an RNA which Broccoli is appended to can attract protein partners which can mask or unfold Broccoli and thus prevent DFHBI binding.
Low expression of an RNA-Broccoli construct expressed from a promoter other U6 or 5S may indicate that the promoter strength is not sufficient. However, if the level of expression is low even when the U6+27 or 5S promoter is used, then the Broccoli-RNA construct may be targeted for rapid cleavage and/or degradation. RNA cleavage can sometimes be detected by observing additional shorter bands on the gel, compared to the full-length transcript. Rapid degradation can be verified by measuring the RNA turnover rate upon actinomycin D treatment. This protocol is described in detail in Filonov et al., 2015. Again, the F30-2xdBroccoli transcript expressed from the U6+27 promoter can be used as a control. If cleavage and/or rapid transcript degradation are confirmed, the transcript should be closely inspected for potential RNA processing sites or destabilization elements. One example of an undesired sequence is a stretch of five or more Us which serves as signal for Pol III transcription termination (Filonov et al., 2015).
A more general reason for the low expression level of an RNA-Broccoli construct can be poor transfection efficiency. This can be tested by co-transfecting red fluorescent protein and assessing red cellular fluorescence using flow cytometry or fluorescent microscopy. A good transfection should result in at least 20–30% of cells being fluorescent. If, however, very few red fluorescent cells are observed, two possible reasons should be considered. First, transfection efficiency is greatly influenced by cell health and confluence. Make sure the cells are free of mycoplasma or other types of contamination and are 50–90% confluent on the day of transfection. Poor cellular health or low confluence will result in cell death while cell confluence close to 100% will result in very low transfection efficiency. If using cells other than the HEK293T cell line, it is also recommended to optimize the transfection protocol. Refer to the transfection reagent manufacturer’s protocol for more details. Second, the plasmids used for transfection should be of the highest purity. Verify the plasmid purity by measuring the A260/A280 ratio. If this ratio is not around 1.8, re-purify the plasmids.
Flow cytometry
Normally, flow cytometric analysis of mammalian cells does not pose any technical difficulties, as long as the transcript is expressed at a high level and Broccoli folds well. The only recommendation will be to include a known positive control, such as U6+27-F30-2xdBroccoli- or 5S-F30-2xdBroccoli-expressing cells. Bacterial cell flow cytometry may be a little challenging since bacteria are substantially smaller than mammalian cells and the threshold setting for flow cytometers is often set rather high so bacterial cells will be excluded from analysis. If you do not have any experience analyzing bacteria on a flow cytometer, we recommend consulting with a Flow Cytometry Core member in your institution to ensure the success of your experiment.
Photostability
The final recommendation is related to the low photostability of Broccoli, mentioned in the Strategic Planning section. While this does not prevent visualization of bright bacterial cells, this property makes it challenging to spot Broccoli-expressing mammalian cells if the imaging field has been exposed to the excitation light for an extended time. To find bright cells, we normally set a medium excitation light lamp intensity (Neutral Density setting 4 or 8) and move the stage to scan different fields. Green fluorescent cells will enter the field of view and then quickly bleach to almost background fluorescence levels. To image these cells, note their location, center the optical field on them, and then allow the fluorescence to recover for 1–2 minutes in the dark. During this time, images of the red fluorescent protein, if present, can be obtained. It is not recommended, however, to illuminate the field with violet light, such as used for Hoechst imaging, since high power violet light can potentially bleach Broccoli. After 1–2 minutes take an image in the green channel. You should expect to see a green fluorescent signal that is substantially brighter than the background.
The possible problems and their solutions are summarized in Table 1 and 2.
Table 1.
Troubleshooting guide for F30-2xdBroccoli expression and imaging in E. coli
| Problem | Possible reason | Solution |
|---|---|---|
| No RNA pellet observed after centrifugation (Basic Protocol 1, step 24) | RNA degraded following contamination with
RNase Too little cells were taken for RNA isolation |
Ensure RNase-free environment. Use only
RNase-free solutions and plasticware. Use more cells |
| Total RNA concentration is too low | RNA degraded following contamination with RNase | See above |
| No fluorescent bands observed after DFHBI staining | RNA degraded following contamination with
RNase F30-2xdBroccoli does not fold when fused to your RNA No F30-2xdBroccoli expression Too weak F30- 2xdBroccoli expression Too short staining time with DFHBI Low concentration of DFHBI in the staining buffer or buffer was used for too many times |
See above Test if F30-2xdBroccoli folds and fluoresces when fused to the RNA in study in vitro. Optimize F30- 2xdBroccoli insertion point if needed. Make sure that the strain expresses T7 RNA polymerase Re-make IPTG stock. IPTG, if stored improperly, quickly loses its activity The promoter is too weak. Use T7 promoter. Extend DFHBI staining time Re-make the staining buffer |
| No distinct individual bands observed after SYBR Gold staining, smear seen in lanes | RNA degraded following contamination with RNase | See above |
| No fluorescent population observed when using a flow cytometer | Poor RNA-F30- 2xdBroccoli folding in
cells Too high threshold setting Incorrect DFHBI concentration Cell incubation time with DFHBI is too short |
Optimize F30-2xdBroccoli insertion
point Low the threshold Verify that cells were pretreated with 40 μM DFHBI or DFHBI-1T Incubate cell with DFHBI or DFHBI-1T for at least 5 min |
| Difficulty focusing on cells | Small size of E. coli cells
/low cell density Poor magnification of microscope |
Focus on cells expressing F30-2xdBroccoli
using fluorescence in the FITC channel Make sure the microscope has an objective capable of imaging E. coli (i.e. 60–100X with >1.4 Numeric Aperture) |
| No adhered cells | Residual LB medium in samples can inhibit cell
adhesion No poly-lysine on the dishes |
Wash cells with PBS twice before adding cells
to dishes Verify that the dishes are pre-coated with poly-lysine |
| No green fluorescence signal in cells under the microscope | Improper concentration of
DFHBI Photobleaching |
Check stock solutions and remake imaging
media Let the fluorescence to recover in the dark and then re-acquire an image |
Table 2.
Troubleshooting guide for F30-2xdBroccoli imaging in mammalian cells
| Problem | Possible reason | Solution |
|---|---|---|
| No RNA pellet observed after centrifugation (Alternate protocol 1, step 19) | RNA degraded following contamination with
RNase Too little cells were taken for RNA isolation |
Ensure RNase-free environment. Use only
RNase-free solutions and plasticware Make sure that cells are confluent in the well and did not dye after transfection. Use larger well to grow and transfect cells. |
| Total RNA concentration is too low | RNA degraded following contamination with RNase | See above |
| No fluorescent bands observed after DFHBI staining | RNA degraded following contamination with
RNase F30-2xdBroccoli does not fold when fused to the RNA in study Transfection failure Too low F30-2xdBroccoli expression level Too short staining time with DFHBI Low concentration of DFHBI in the staining buffer or buffer was used for too many times |
See above Test if dBroccoli folds and fluoresces when fused to the RNA in study in vitro. Optimize dBroccoli insertion point if needed. Co-transfect a red fluorescent protein to confirm it. Ensure that the cells are healthy and in a low passage number. Thaw fresh cell batch if needed. Verify the plasmids purity. Re-elute the plasmids if needed. Promoter is too weak. Use stronger promoter, such as U6 or 5S High rate of RNA-F30-2xdBroccoli turnover in cells or undesirable cleavage of the transcript. Inspect your transcript’s sequence for cryptic transcription termination sites or potential destabilizing elements. Insert F30-2xdBroccoli into a different site within your RNA. Extend DFHBI staining time Re-make the staining buffer |
| No distinct individual bands observed after SYBR Gold staining, smear seen in lanes | RNA degraded following contamination with RNase | See above |
| No fluorescent population observed when using a flow cytometer | Poor RNA-F30- 2xdBroccoli folding in
cells Incorrect DFHBI concentration Cell incubation time with DFHBI is too short |
Optimize F30-2xdBroccoli insertion
point. Verify that the cells are incubated with 40 μM of DFHBI or DFHBI-1T Extend cell incubation time with DFHBI (DFHBI-1T) |
| Cell did not attach to the glass-bottom dish | Poor cell health Insufficient glass surface coating |
Ensure that cells are healthy and free of
contamination. Change the media when it turns yellow. Do not transfect
cells when their confluence is low. Ensure that the glass surface is properly coated with poly-lysine and laminin. Increase laminin coating time if needed. |
| Cells die during the imaging | Media pH change Phototoxicity of the excitation light |
Use either an environmental chamber with a
controlled level of CO2 or buffer the imaging media with HEPES to
maintain pH of 7.4 Do not expose cells to high intensity light for an extended period of time. Aim to expose cells to light only for brief time periods when quickly scanning the fields and then during actual images taking. |
| No green fluorescence signal in cells under the microscope | Low expression level of RNA-F30-2xdBroccoli in
cells Photobleaching Improper concentration of DFHBI |
Even when a fluorescent band is seen on a gel
after DFHBI staining and a positive population is observed during the
flow cytometry analysis, the dBroccoli brightness level may still not be
enough for successful fluorescence detection using fluorescence
microscopy. Increase expression level by utilization of the strongest
promoters, such as U6 or 5S, and ensure no degradation or cleavage of
the transcript. Photobleaching is more noticeable when imaging dBroccoli in mammalian cells due to the low expression level of the transcript. Leaving the cells in the dark to allow new fluorophore to bind to Broccoli is critical to restore the signal prior to acquiring an image. Check stock solutions and remake imaging media |
Anticipated Results
The protocols described here should result in robust detection of F30-2xdBroccoli or F30-2xdBroccoli-fused transcripts in bacterial and mammalian cells.
In our experience, the in-gel staining protocol is so sensitive that it almost never fails to report the presence of a Broccoli-tagged RNA in cellular extracts unless a technical failure has occurred. Bacterial cells produce so much of exogenous RNA off the T7 promoter that this RNA can be easily detected even by using the nonselective RNA dye SYBR Gold. For mammalian cells DFHBI-1T staining is critical. However, this protocol allows detection of very small RNA amounts, e.g., Broccoli transcribed off the CMV promoter, which is substantially weaker than U6 (Filonov et al., 2015).
Flow cytometry is also a very robust way to detect Broccoli fluorescence. One of the reasons is that signal acquisition during analysis occurs for such a short period of time and under such a strong light intensity that Broccoli bleaching likely does not occur at all and so the cell generates a very bright signal. Thus, as long as the Broccoli transcript folds well and is expressed at sufficiently high levels, flow cytometry should allow observation of a bright population of the Broccoli-positive cells.
Fluorescence imaging is also very straightforward for bacterial cells. You should be able to observe a large number of bright Broccoli-positive cells even after photobleaching. Mammalian cell imaging, however, has its own challenges. This is due to the substantially lower expression level of Broccoli-containing transcripts in mammalian cells, which makes the signal dimmer. Thus, photobleaching in mammalian cells is much more noticeable than in bacterial cells. However, using the approach described above, bright Broccoli-expressing cells can be located, such as those presented in Fig. 5. These cells are bright enough to be easily spotted while scanning fields. Conceivably, dimmer and more abundant cells can be imaged “blindly”, i.e., by choosing a random field of view and then taking an image in the FITC channel. This image is then processed to subtract the background fluorescence, which reveals the specific Broccoli signal.
Figure 5.

5S-F30-2xdBroccoli and U6+27-F30-2xdBroccoli imaging in HEK293T cells. Cells were transfected and processed as described in the Alternate Protocol 3, treated with 40 μM DFHBI-1T and 5 μg/ml Hoechst 33342 and imaged using the microscope. Broccoli fluorescence was detected in the FITC channel (excitation filter 470±20 nm and emission filter 525±25 nm), mCherry-filled cells were observed in the Texas Red channel (excitation filter 560±20 nm and emission filter 630±37.5 nm) and Hoechst 33342-stained nuclei were imaged in the DAPI channel (excitation filter 350±25 nm and emission filter 460±25 nm). Exposure times are 0.5–1 s for Broccoli and 100–400 ms for Hoechst 33342 and mCherry. Scale bar, 20 μm.
Time Considerations
Any of the protocols described above can be completed within one week or a little longer. One can consider starting experiments on Monday by doing a bacterial cell transformation or seeding mammalian cells for transfection the next day. On Tuesday, either inoculate bacterial cells in the evening or transfect mammalian cells during the day. On Wednesday, express Broccoli in bacteria and then collect bacterial cells and proceed with RNA isolation or bacteria flow cytometry or imaging. No mammalian cell work is needed on Wednesday. On Thursday, transfer mammalian cells to a glass-bottom dish, if planning imaging. If not, consider changing the media if it turns yellow. On Friday, harvest the cells to isolate total RNA or to analyze with flow cytometry, or proceed with imaging. Total cellular RNA can be isolated on that day while PAGE and gel staining can be done the next Monday.
Acknowledgments
We thank J. McCormick and S.Z. Merlin (Department of Pathology and Laboratory Medicine cell sorter core) for their help with flow cytometry. This work was supported by NIH grants to S.R.J. (R01 NS064516 and R01 EB010249). Additionally, the authors disclose that S.R.J. and G.S.F. are authors of a patent application (provisional patent USPTO# 61/874,819) related to technology described in this paper.
List of abbreviations
- RNA
ribonucleic acid
- GFP
green fluorescent protein
- DFHBI
3,5-difluoro-4-hydroxybenzylidene imidazolinone
- nt
nucleotide
- DFHBI-1T
3,5-difluoro-4-hydroxybenzylidene 1-trifluoroethyl-imidazolinone
Contributor Information
Grigory S. Filonov, Email: gf2004@med.cornell.edu.
Samie R. Jaffrey, Email: srj2003@med.cornell.edu.
LITERATURE CITED
- Bao G, Tsourkas A, Santangelo PJ. Engineering nanostructured probes for sensitive intracellular gene detection. Mech Chem Biosyst. 2004;1:23–36. [PubMed] [Google Scholar]
- Belly A, Bodon G, Blot B, Bouron A, Sadoul R, Goldberg Y. CHMP2B mutants linked to frontotemporal dementia impair maturation of dendritic spines. J Cell Sci. 2010;123:2943–2954. doi: 10.1242/jcs.068817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. Localization of ASH1 mRNA particles in living yeast. Molecular Cell. 1998;2:437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- Bratu DP, Cha BJ, Mhlanga MM, Kramer FR, Tyagi S. Visualizing the distribution and transport of mRNAs in living cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13308–13313. doi: 10.1073/pnas.2233244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filonov GS, Kam CW, Song W, Jaffrey SR. In-gel imaging of RNA processing using broccoli reveals optimal aptamer expression strategies. Chemistry & biology. 2015;22:649–660. doi: 10.1016/j.chembiol.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filonov GS, Moon JD, Svensen N, Jaffrey SR. Broccoli: Rapid Selection of an RNA Mimic of Green Fluorescent Protein by Fluorescence-Based Selection and Directed Evolution. Journal of the American Chemical Society. 2014 doi: 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH, Bertrand E. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Current biology : CB. 2003;13:161–167. doi: 10.1016/s0960-9822(02)01436-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubbs RD. Intracellular magnesium and magnesium buffering. Biometals : an international journal on the role of metal ions in biology, biochemistry, and medicine. 2002;15:251–259. doi: 10.1023/a:1016026831789. [DOI] [PubMed] [Google Scholar]
- Han KY, Leslie BJ, Fei J, Zhang J, Ha T. Understanding the photophysics of the spinach-DFHBI RNA aptamer-fluorogen complex to improve live-cell RNA imaging. Journal of the American Chemical Society. 2013;135:19033–19038. doi: 10.1021/ja411060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger CA, Chen C, Whiteley AT, Portnoy DA, Hammond MC. RNA-Based Fluorescent Biosensors for Live Cell Imaging of Second Messenger Cyclic di-AMP. Journal of the American Chemical Society. 2015;137:6432–6435. doi: 10.1021/jacs.5b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC. RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J Am Chem Soc. 2013;135:4906–4909. doi: 10.1021/ja311960g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell RE, Nevins JR, Sullenger BA. Optimizing aptamer activity for gene therapy applications using expression cassette SELEX. Molecular therapy : the journal of the American Society of Gene Therapy. 2002;6:30–34. doi: 10.1006/mthe.2002.0624. [DOI] [PubMed] [Google Scholar]
- Molenaar C, Abdulle A, Gena A, Tanke HJ, Dirks RW. Poly(A)+ RNAs roam the cell nucleus and pass through speckle domains in transcriptionally active and inactive cells. J Cell Biol. 2004;165:191–202. doi: 10.1083/jcb.200310139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar C, Marras SA, Slats JC, Truffert JC, Lemaitre M, Raap AK, Dirks RW, Tanke HJ. Linear 2′ O-Methyl RNA probes for the visualization of RNA in living cells. Nucleic acids research. 2001;29:E89–89. doi: 10.1093/nar/29.17.e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H. Working with RNA. Methods Mol Biol. 2011;703:15–28. doi: 10.1007/978-1-59745-248-9_2. [DOI] [PubMed] [Google Scholar]
- Nitin N, Santangelo PJ, Kim G, Nie S, Bao G. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic acids research. 2004;32:e58. doi: 10.1093/nar/gnh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Natori Y, Sato M, Umezawa Y. Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nature Methods. 2007;4:413–419. doi: 10.1038/nmeth1030. [DOI] [PubMed] [Google Scholar]
- Paige JS, Nguyen-Duc T, Song W, Jaffrey SR. Fluorescence imaging of cellular metabolites with RNA. Science. 2012;335:1194. doi: 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HY, Lim H, Yoon YJ, Follenzi A, Nwokafor C, Lopez-Jones M, Meng X, Singer RH. Visualization of dynamics of single endogenous mRNA labeled in live mouse. Science. 2014;343:422–424. doi: 10.1126/science.1239200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul CP, Good PD, Li SX, Kleihauer A, Rossi JJ, Engelke DR. Localized expression of small RNA inhibitors in human cells. Molecular therapy : the journal of the American Society of Gene Therapy. 2003;7:237–247. doi: 10.1016/s1525-0016(02)00038-2. [DOI] [PubMed] [Google Scholar]
- Ponchon L, Dardel F. Recombinant RNA technology: the tRNA scaffold. Nat Methods. 2007;4:571–576. doi: 10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- Romani AM. Magnesium homeostasis in Mammalian cells. Metal ions in life sciences. 2013;12:69–118. doi: 10.1007/978-94-007-5561-1_4. [DOI] [PubMed] [Google Scholar]
- Santangelo PJ, Alonas E, Jung J, Lifland AW, Zurla C. Probes for intracellular RNA imaging in live cells. Methods Enzymol. 2012;505:383–399. doi: 10.1016/B978-0-12-388448-0.00028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, Spector DL, Singer RH. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–1800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu D, Khisamutdinov EF, Zhang L, Guo P. Programmable folding of fusion RNA in vivo and in vitro driven by pRNA 3WJ motif of phi29 DNA packaging motor. Nucleic acids research. 2014;42:e10. doi: 10.1093/nar/gkt885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Strack RL, Jaffrey SR. Imaging bacterial protein expression using genetically encoded RNA sensors. Nat Methods. 2013;10:873–875. doi: 10.1038/nmeth.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Strack RL, Svensen N, Jaffrey SR. Plug-and-play fluorophores extend the spectral properties of Spinach. J Am Chem Soc. 2014;136:1198–1201. doi: 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack RL, Disney MD, Jaffrey SR. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat RNA. Nature Methods. 2013;10:1219–1224. doi: 10.1038/nmeth.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nat Methods. 2009;6:331–338. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Alsmadi O. Imaging native beta-actin mRNA in motile fibroblasts. Biophys J. 2004;87:4153–4162. doi: 10.1529/biophysj.104.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chen J, Singer RH. Background free imaging of single mRNAs in live cells using split fluorescent proteins. Sci Rep. 2014;4:3615. doi: 10.1038/srep03615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Litke JL, Jaffrey SR. Imaging metabolite dynamics in living cells using a Spinach-based riboswitch. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E2756–2765. doi: 10.1073/pnas.1504354112. [DOI] [PMC free article] [PubMed] [Google Scholar]