

Abstract
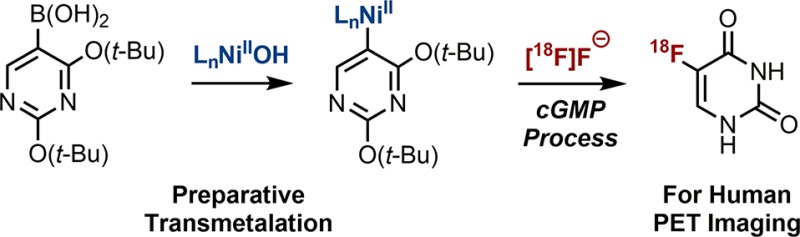
Translation of new 18F-fluorination reactions to produce radiotracers for human positron emission tomography (PET) imaging is rare because the chemistry must have useful scope and the process for 18F-labeled tracer production must be robust and simple to execute. The application of transition metal mediators has enabled impactful 18F-fluorination methods, but to date none of these reactions have been applied to produce a human-injectable PET tracer. In this article we present chemistry and process innovations that culminate in the first production from [18F]fluoride of human doses of [18F]5-fluorouracil, a PET tracer for cancer imaging in humans. The first preparation of nickel σ-aryl complexes by transmetalation from arylboronic acids or esters was developed and enabled the synthesis of the [18F]5-fluorouracil precursor. Routine production of >10 mCi doses of [18F]5-fluorouracil was accomplished with a new instrument for azeotrope-free [18F]fluoride concentration in a process that leverages the tolerance of water in nickel-mediated 18F-fluorination.
Introduction
Transition-metal-mediated fluorination reactions have increased in scope and application, particularly for the synthesis of 18F-labeled molecules for positron emission tomography (PET).1−3 However, no such reaction has yet been translated to enable human PET imaging. Here we report the cGMP4 production of human doses of [18F]5-fluorouracil ([18F]5-FU), a PET tracer for clinical research in oncology,5 by nickel-mediated oxidative fluorination with [18F]fluoride (Scheme 1).2d The synthesis of the [18F]5-FU nickel precursor was enabled by the first transmetalation reaction from arylboronic acid derivatives that yields isolated nickel σ-aryl complexes. The transmetalation reaction was developed because other methods for nickel σ-aryl preparation were inadequate. All previously reported syntheses of [18F]5-FU for human use have employed [18F]F2 gas,6 which is challenging to produce and handle and is less preferable as a starting material compared to [18F]fluoride.7
Scheme 1. Strategy for the Synthesis of [18F]5-Fluorouracil.

Nickel σ-aryl complexes are employed as precatalysts,8 substrates for mechanistic investigation,9 and precursors for late-stage 18F-fluorination.10 Nickel σ-aryl complexes react with [18F]fluoride and oxidant in less than 1 min at 23 °C in the presence of water to afford [18F]aryl fluorides.2d In contrast, competing methods such as the 18F-fluorination of aryliodine(III) precursors2k,3a,3b,3d−3f and copper-mediated fluorination of arylboronate esters2j utilize extended reaction times at elevated temperatures with azeotropically dried [18F]fluoride. A drawback of nickel σ-aryl complexes is that limited methods exist for their preparation, which we sought to address with this investigation. Nickel σ-aryl complexes are commonly prepared by oxidative addition of Ni(0) to aryl halides11 or by transmetalation from aryllithium, Grignard, and arylzinc reagents.12,13 However, the range of accessible nickel σ-aryl complexes is limited due to the strong reducing activity of Ni(0) reagents and the basicity and nucleophilicity of organometallic Li, Mg, and Zn precursors. There are no general methods for nickel σ-aryl preparation from more functional-group-tolerant p-block organometallics such as arylboron, aryltin, or arylsilicon reagents.14−16 The preparation of other first-row transition metal σ-aryl complexes from such reagents is rare.17 Development of a versatile nickel(II) σ-aryl synthesis by transmetalation was further motivated by the failure of previously reported oxidative addition methodology2d to afford the desired nickel(II) σ-aryl precursor to [18F]5-FU (see the Supporting Information).
Arylboronic acids/esters were desired as the aryl source for transmetalation, because they are easily prepared and may contain functional groups not tolerated in organometallic reagents of Li, Mg, and Zn.18 As a starting point for the development of the synthesis of Ni(II) σ-aryl complexes by transmetalation from arylboronic acids, the reduction of inorganic LnNiIIX2 precatalysts (for example, (PCy3)2NiCl2) by boronic acid in Suzuki–Miyaura coupling was considered (Scheme 2).19 Reduction to low-valent Ni is likely initiated by transmetalation from arylboronic acid to LnNiIIX2 to form LnNiII(Aryl)X, which is relevant because the desired 18F-fluorination precursors are LnNiII(Aryl)X complexes. However, to our knowledge, direct observation of a nickel(II) σ-aryl complex formed by transmetalation from an arylboronic acid or ester has not been reported. Instability of LnNiII(Aryl)X compared to LnNiIIX2, in the presence of boronic acid and base, may prevent the accumulation of LnNiII(Aryl)X. To develop a preparative synthesis of LnNiII(Aryl)X from arylboronic acids, a fundamental challenge of relative rates must be addressed. Transmetalation to form LnNiII(Aryl)X must be more facile than its destruction by homocoupling or protiodemetalation.
Scheme 2. Plausible Pathway for Reduction of Nickel(II) by Arylboronic Acid.

To maximize the transmetalation rate, an LnNiIIX2 precursor that contains a basic X-type ligand, such as hydroxide, that could induce transmetalation from arylboron reagents was desired. In support of this design, a study has implicated that transmetalation to a nickel(II) hydroxide is faster than transmetalation to the analogous nickel(II) halide in the presence of base.9c Incorporation of the required pyridylsulfonamide ligand as the second X-type ligand in the LnNiIIX2 precursor would increase its solubility in organic solvent, prevent a second transmetalation and hence homocoupling from occurring due to the weak basicity of the sulfonamide, and eliminate the need for additional ligand exchange steps after transmetalation in order to access the desired precursors for 18F-fluorination.
Results and Discussion
Complex 1 was synthesized as a reagent for preparative transmetalation (Scheme 3). Consistent with our design for a transmetalation reagent, complex 1 has a hydroxide ligand to activate the arylboron reaction partner for transmetalation, dissolves in organic solvents such as pyridine and DMSO, and contains the pyridylsulfonamide ligand required for oxidative arene fluorination. Complex 1 was prepared on a multigram scale starting from nickel(II) acetate tetrahydrate, potassium tert-butoxide, and the bidentate pyridylsulfonamide ligand in a 1:2:1 molar ratio in pyridine solvent. After evaporation of pyridine and dissolving in THF, 1 precipitated by treatment with just 3% water (by volume), likely by hydrolysis of a more soluble nickel(II) tert-butoxide intermediate. The use of pyridine as solvent for the synthesis of 1 is essential. In all other solvents evaluated, an insoluble orange solid identified as the square-planar bispyridylsulfonamide nickel(II) complex 2 precipitates after mixing inorganic Ni(II) precursors with a pyridylsulfonamide ligand. The rapid formation of 2 by association with two ligands stands in contrast with reactivity observed for Pd(II), where a complex with a single pyridylsulfonamide ligand can be isolated in 99% yield by concentration of a CH2Cl2 solution of Pd(OAc)2, pyridylsulfonamide, and 3 equiv of pyridine.20 Complex 1 is a green solid that appears to be stable when stored in a sealed vial under air at room temperature for several months. In pyridine-d5 solution, 1 exhibits a set of characteristic proton NMR signals between −10 and 50 ppm. The structure of 1 was determined by X-ray crystallography and consists of a tetrameric nickel(II) hydroxide core, with one hydroxide and one pyridylsulfonamide ligand per nickel atom. Two of the nickel atoms are bound to acetate derived from the nickel(II) acetate tetrahydrate precursor, with almost equivalent Ni–O bond lengths (2.05 and 2.07 Å).
Scheme 3. Synthesis of Nickel Hydroxide Cubane 1, a Reagent for Preparative Transmetalation.

ORTEP structures of 1 and 2 (50% probability ellipsoids) are displayed. Hydrogen and solvent atoms are omitted for clarity, as are the counterion and pyridylsulfonamide ligand atoms in 1 (except nitrogens bound to nickel). Ar = 2-nitrophenyl.
Transmetalation occurs when 1 is treated with arylboronic acid (or ester) in pyridine at 70 °C for 1 h, to generate the desired complexes (Scheme 4). The preparation of complexes 3a–3i represents the first example of preparative nickel(II) σ-aryl synthesis starting from boronic acids or esters. In addition to the previously inaccessible [18F]5-fluorouracil precursor 3a, diverse nickel(II) σ-aryl and heteroaryl complexes 3b–3i were synthesized. While the previous synthesis of pyridylsulfonamido nickel(II) σ-aryl complexes by oxidative addition with nickel(0) biscyclooctadiene required a glovebox,2d synthesis by transmetalation with 1 does not and is conveniently set up at the bench. Purification of the nickel(II) σ-aryl complexes is accomplished without exclusion of water or oxygen. The transmetalation reaction likely proceeds by dissociation of 1 in pyridine solution, so that the oxygen atom of the nickel hydroxide will be coordinatively unsaturated and, therefore, be able to bond to boron for transmetalation.
Scheme 4. Synthesis of Nickel(II) σ-Aryl Complexes by Transmetalation from Boronic Acids and Esters.
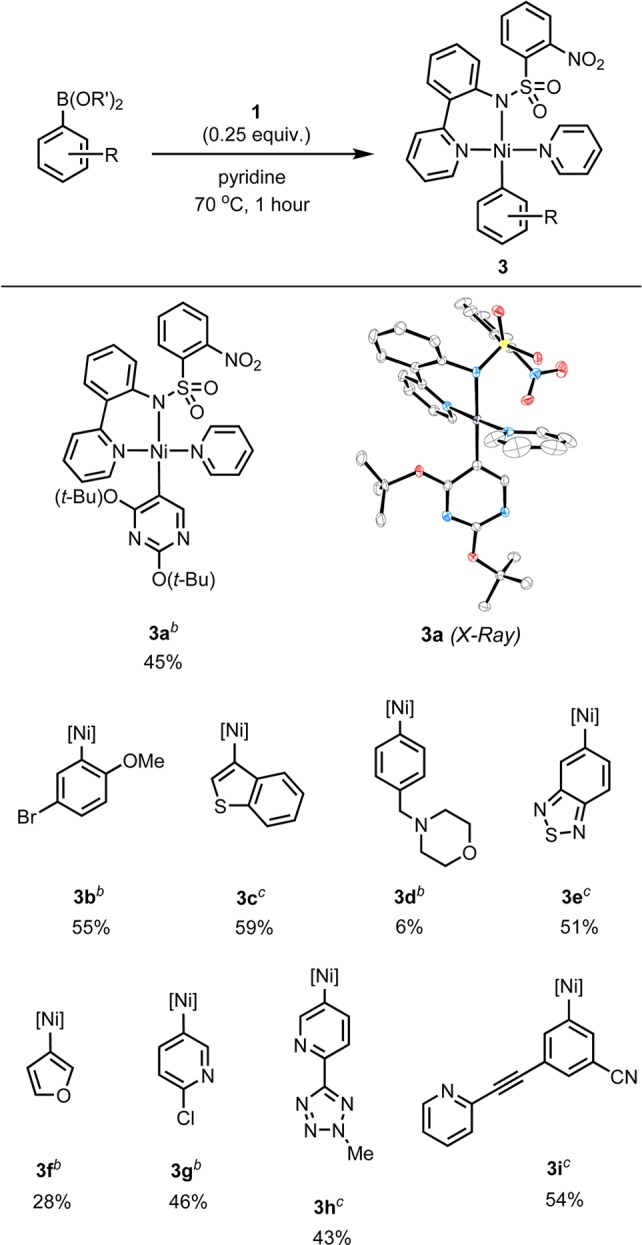
Yields of isolated products are given.
Synthesized from arylboronic acid.
Synthesized from the arylboronic ester of 2,2-dimethyl-1,3-propanediol.
Fluorination of the nickel(II) σ-aryl complexes 3a–3e occurs rapidly with [18F]fluoride and iodine(III) oxidant2d at 23 °C (Scheme 5). Incorporation of 18F at the 3-position of a thiophene ring system was observed in the synthesis of [18F]4c. Electron-rich O/S-heteroarenes are difficult to fluorinate on the same ring as the heteroatom with conventional nucleophilic radiofluorination chemistry.21 Furthermore, oxidative [18F]fluorination occurs in the presence of a tertiary amine to afford [18F]4d, which is notable because of the reactivity of amines toward oxidation with iodine(III) oxidants.22 Formation of [18F]4d can be rationalized on the basis of oxidative fluorination being faster than amine oxidation and that product [18F]4d is spared from oxidation because the starting material 3d is present in slight excess compared to oxidant. Complexes 3g, 3h, and 3i, all of which contain unbound Lewis basic pyridyl substituents, did not undergo radiofluorination to the desired products. In contrast, bisalkoxypyrimidine complex 3a underwent fluorination to form [18F]4a in 15% radiochemical conversion (RCC). This result served as a starting point for our synthesis of [18F]5-FU from [18F]fluoride.
Scheme 5. Oxidative Fluorination of Nickel(II) σ-Aryl Complexes.

Conditions: 0.5 mL of 18F– solution (0.50 mL of MeCN, 5.0 mg of 18-crown-6, 1.0 μL of 0.56 M K3PO4(aq.), 1.5 μL of 18F–(aq)), 1.0 mg of Ni complex, 1.0 mg of PhI(4-OMe-pyridine)2(OTf)2.
Our ultimate goal was to prepare human doses of [18F]5-FU from [18F]fluoride to enable PET imaging in oncology. Therefore, an isolated yield of at least 10 mCi was desired.5 Purity to match USP guidelines and production in a cGMP environment were also required. Oxidative fluorination of 3a with [18F]fluoride affords [18F]4a, which is protected with hydrophobic tert-butyl groups that facilitate purification by conventional reverse-phase chromatographic methods. All previous preparations of human doses of [18F]5-FU started from [18F]F2 gas and uracil, and a challenging separation of [18F]5-FU from uracil and other polar byproducts was required.6 Purified [18F]4a undergoes rapid and clean conversion to [18F]5-FU upon mixing with HCl(aq) in ethanol at room temperature. Neutralization of HCl with NaHCO3 forms a buffered saline solution for in vivo application.
Preliminary efforts to produce [18F]5-FU on a large scale by [18F]fluorination of 3a with an established automation platform2i resulted in the isolation of 2.4 mCi of [18F]5-FU (0.2% yield, see the Supporting Information). The yield was prohibitively low for human dose production and was diminished by inefficient separation of [18F]fluoride from the 2.4 mL [18O]water in which it was produced in a cyclotron (26% yield of dry [18F]fluoride). To increase the yield, a more efficient [18F]fluoride concentration process was developed that afforded 81% yield of [18F]fluoride (Scheme 6, top). This process is performed with an instrument (see the Supporting Information for full details) that leverages a miniaturized ion-exchange cartridge and microfluidic lines in order to elute [18F]fluoride with a total of 4 μL of water. The [18F]fluoride was eluted into 1 mL of dry MeCN to afford a 0.4% aqueous MeCN solution that has sufficiently low water content for oxidative [18F]fluorination, without the need for evaporation steps. Because azeotropic drying and fluoride resolubilization were not necessary, time was saved and radiochemical yield improved.
Scheme 6. cGMP Synthesis of [18F]5-Fluorouracil (>10 mCi) for Use in Human PET Imaging.

Based on decay-corrected measurement of trapped activity on a microcartridge, relative to initial [18F]fluoride in [18O]water.
Three elutions were applied, with 0.5 μmol of K3PO4 and 3.0 μmol of 18-crown-6 ([18c6]) in 6.2 μL of MeCN/H2O (4/1, v/v) per elution.
Based on decay-corrected measurement of eluted activity relative to trapped activity.
Yield (not decay corrected) based on [18F]fluoride (range: 7–14 mCi) in 2.4 mL of water at the start of synthesis. Starting from 1.7 Ci of [18F]fluoride, the yield (not decay corrected) was 79%.
SPE: solid phase extraction; see Supporting Information for full purification details.
Isolated yield (not decay corrected) is based on activity of [18F]fluoride (range: 1.4–1.8 Ci) in [18O]water at the start of synthesis.
The streamlined [18F]fluoride concentration was incorporated into a cGMP process for the synthesis of [18F]5-FU (Scheme 6, bottom). Formulated [18F]5-FU was isolated with a yield range of 13.5–18.8 mCi (over three runs), starting from 1.4–1.8 Ci of [18F]fluoride, with a synthesis time of about 1.5 h. A specific activity of 34.3 ± 18.0 Ci/μmol was observed for isolated [18F]5-FU.23 The percent yield of 0.92% ± 0.18% is low due to the RCC of [18F]fluoride to [18F]4a (2.9% ± 0.5%, n = 3).24 Nearly all of the remaining 18F eluted near the beginning of reverse-phase HPLC runs and at the baseline of silica TLC analysis runs, consistent with the presence of unreacted [18F]fluoride that remains after the rapid fluorination reaction ceases. The origin of low RCC may involve side reactions of the water- and base-sensitive biscationic iodine(III) oxidant or of high-valent nickel intermediates. Application of PPTS to buffer the [18F]fluoride solution2i did not substantially improve the RCC (2.96%). Development of robust alternative oxidants or high-valent nickel σ-aryl fluorination precursors may lead to a more efficient fluorination reaction. Nevertheless, at this stage the yield of [18F]5-FU is sufficient for human PET imaging because only 5–10 mCi is needed for a human dose.5 [18F]5-FU is obtained as a sterile, colorless saline solution, with >99% radiochemical purity, <10 μg of impurities not arising from USP formulation ingredients, and <0.1 ppm of Ni. The doses of [18F]5-FU pass quality control protocols for radiopharmaceuticals for human use and are validated for human in vivo application. No other preparation from [18F]fluoride of [18F]5-FU for human use has been reported.
Conclusions
The first preparative synthesis of nickel(II) σ-aryl complexes by transmetalation from arylboronic acids and esters was developed and enabled the synthesis of previously inaccessible complex 3a. Oxidative fluorination of 3a allowed for the first synthesis of [18F]5-fluorouracil from [18F]fluoride that affords doses suitable for in vivo use in humans. The nickel-mediated synthesis of [18F]5-fluorouracil represents the first clinical translation of transition-metal-mediated fluorination to enable PET imaging in humans. We aim to advance oncology clinical research by routinely supplying human doses of [18F]5-fluorouracil.
Experimental Section
Synthesis of Nickel Hydroxide Cubane 1
To a 1 L round-bottomed flask were added nickel(II) acetate tetrahydrate (2.80 g, 11.3 mmol, 1.00 equiv) and a Teflon-coated stirbar. The flask was fitted with a septum, and the headspace was filled with nitrogen. Anhydrous pyridine (114 mL) was added, and a blue solution was observed after mixing. To this solution was added 2-nitro-N-(2-(pyridin-2-yl)phenyl)benzenesulfonamide2d (4.00 g, 11.3 mmol, 1.00 equiv, as a solution in 166 mL of anhydrous pyridine) by cannula over 3 min, and a green-blue solution was observed. To this solution was added potassium tert-butoxide (2.53 g, 22.5 mmol, 2.00 equiv, as a solution in 80 mL of anhydrous pyridine) by cannula over 5 min. A yellow-green solution with a colorless precipitate was observed, which was stirred at 23 °C for 45 min, before being concentrated in vacuo (by rotary evaporation at 60 °C until all liquid pyridine was removed and then under high vacuum at 23 °C) to give a mixture of green and orange solids. These solid residues were ground with a spatula under anhydrous THF (130 mL) in order to dissolve the green solid. The mixture was filtered through Celite on a glass frit, which was then rinsed with anhydrous THF (2 × 20 mL). The THF filtrates were combined to give a dark green solution, which was treated dropwise with H2O (5.0 mL) over 30 min, with magnetic stirring, which caused a light green solid to precipitate. The solid was collected by filtration on a glass frit, rinsed with THF (2 × 20 mL), and dried in vacuo (0.2 Torr, 50 °C, 40 min; then 0.2 Torr, 150 °C, 2 h) to afford 2.66 g of the title compound (as a solvate with 4 water molecules) as a green solid (50% yield). NMR spectroscopy: 1H NMR (600 MHz, pyridine-d5, 23 °C, δ): 46.1, 45.2, 44.8, 44.5, 43.0, 41.8, 40.9, 40.7, 39.6, 39.4, 39.2, 37.4, 36.1, 33.6, 33.1, 32.2, 30.4, 29.4, 24.4, 22.0–19.0, 20.3, 19.9, 19.6, 18.3, 17.9, 17.7, 17.5, 16.7, 16.0. 14.8, 14.7, 14.4, 14.1, 13.8, 13.5, 13.1, 12.6, 11.9, 11.4, 10.9, 10.7, 10.6, 10.2, 9.9, 8.3, 8.1, 7.5, 6.9, 6.6, 6.3, 6.2, 6.1, 6.0, 5.8, 5.4, 1.9, 1.0, 0.5, 0.2, −0.2, −0.4, −0.7, −1.0, −1.9, −2.0, −2.6, −3.6, −4.0, −4.6, −5.0, −5.5, −6.5. Due to gradual decomposition in solution, 13C NMR analysis was not performed. Anal. Calcd for C70H55N12S4O22KNi4(H2O)4: C, 44.47; H, 3.36; N, 8.89. Found: C, 44.63; H, 3.07; N, 8.77. IR (neat, ν, cm–1): 1594 (w), 1575 (w), 1535 (s), 1489 (m), 1476 (w), 1428 (m), 1367 (m), 1275 (m), 1234 (m), 1147 (s), 1129 (s), 1117 (s), 1061 (m), 972 (s), 852 (w), 824 (m), 753 (s), 730 (s), 651 (m), 630 (w), 593 (s), 562 (s), 528 (m), 434 (m).
General Procedure for Preparation of Complexes 3a–3i by Transmetalation
Complex 1 (142 mg, 75.0 μmol, 0.250 equiv) and arylboron reagent (1.0 equiv) were dissolved in dry pyridine (12 mL) under N2. The mixture was heated with stirring at 70 °C for 1 h. After cooling to 23 °C, hexanes (120 mL) was added, and the precipitated product was collected by filtration on Celite, dissolved in dichloromethane/pyridine (95:5, v/v), and then passed through the Celite. The filtrate was concentrated in vacuo to afford a residue, which was purified by chromatography on SiO2/K2CO3 (9:1 w/w), eluting with dichloromethane/pyridine (95:5, v/v). The collected fractions were concentrated in vacuo to remove dichloromethane, and hexanes was added. The supernatant was decanted after centrifugation, and the remaining residue was triturated with hexanes. The title compound was obtained after centrifugation and decantation of the supernatant and drying of the remaining solid in vacuo. For some complexes, the solvents used in the workup procedure were varied (see the Supporting Information for full details).
General Procedure for Preparation of [18F]Aryl Fluorides [18F]4a–[18F]4e
In a nitrogen-filled glovebox, to an oven-dried 1-dram (4 mL) glass vial were added nickel(II) aryl complex 3 and PhI(4-OMe-pyridine)2(OTf)22d in a 1:1 mass ratio, and the two solids were mixed gently with a metal spatula to give a homogeneous admixture. To an oven-dried 1-dram glass vial was added 2.0 mg of this admixture, and the vial was sealed with a screw cap with a Teflon-lined septum insert under nitrogen and removed from the glovebox. An [18F]fluoride solution with 18-crown-6 and potassium phosphate tribasic was prepared as follows. To an oven-dried 1-dram (4 mL) glass vial was added dry 18-crown-6 (20.0–44.0 mg) under nitrogen, and this vial was sealed with a Teflon-lined cap. The vial was opened under air, dry MeCN (1.0 mL per 10.0 mg of 18-crown-6) was added quickly, and the vial was sealed and mixed until all 18-crown-6 had dissolved. The vial was opened, aqueous potassium phosphate (0.561 M K3PO4 in water, 2.0 μL per 10.0 mg of 18-crown-6) was added quickly, and the vial was sealed, shaken, and then vortexed for 10 s. The vial was opened, aqueous [18F]fluoride from the cyclotron (3.0 μL per 10.0 mg of 18-crown-6) was added quickly, and the vial was sealed, shaken, and then vortexed for 10 s. A portion of the resulting solution (0.50 mL) was added as rapidly as possible, with a 1 mL plastic syringe with an 18 G disposable metal needle, to the vial containing nickel(II) aryl complex and oxidant through the septum. After 1 min at 23 °C, the radiochemical conversion was then measured by radioTLC, and HPLC analysis was performed to confirm the formation of the title compound.
cGMP Synthesis of [18F]5-Fluorouracil from [18F]Fluoride
All chemicals, equipment, facilities, staff, procedures, and documentation were controlled in accordance with cGMP guidelines. All operations were conducted behind lead shielding and were remote-controlled. [18F]Fluoride (1.4–1.8 Ci) in [18O]water (2.4 mL) was passed through a miniature ion-exchange cartridge, which was then dried with MeCN (1.0 mL). The trapped [18F]fluoride was released with three elutions of 0.5 μmol of K3PO4 and 3.0 μmol of 18-crown-6 in 6.2 μL of MeCN/H2O (4:1, v/v) per elution. The eluted [18F]fluoride was diluted in a vial containing 1.0 mL of dry MeCN and 10.0 mg of 18-crown-6, and the resulting solution was rapidly added to a solid mixture of complex 3a (10.0 mg) and PhI(4-OMe-pyridine)2(OTf)2 (10.0 mg). After 1 min at 23 °C, the reaction solution was diluted with 4.0 mL of water and purified by semipreparative HPLC. The collected fractions were passed through two C18 silica cartridges, which were washed with water and then eluted with ethanol into a vial with concentrated HCl(aq). After 2 min at 23 °C, the reaction solution was neutralized with NaHCO3(aq) and filtered through solid-phase extraction cartridges. The resulting solution was transferred to a clean room, where it was passed through a sterilizing filter to afford [18F]5-fluorouracil for human injection (13.5–18.8 mCi) as a 23 mL saline solution with <10% ethanol.
Acknowledgments
Funding provided by NIH-NIGMS (GM088237), NIH-NIBIB (EB013042), the Blavatnik Biomedical Accelerator, DOE BER (DE-SC0001249), the Harvard/MGH Nuclear Medicine Training Program from the Department of Energy (DE-SC0008430, in support of H.R.), National Center for Research Resources (1S10RR017208-01A1), and the DOE SCGF (graduate fellowship to A.J.H.) is gratefully acknowledged. Acknowledgment is given to Dr. Shao-Liang Zheng, Dr. Michael Campbell, and Heejun Lee (Harvard University) for assistance with X-ray crystallographic analysis, Judit Sore, Kari Phan, and Garima Gautham (Massachusetts General Hospital) for assistance with cGMP validation, Dr. Nickeisha Stephenson (Vasdev Lab, Harvard Medical School and Massachusetts General Hospital) for the aryl bromide precursor to 3i, and Martin G. Strebl for attempted synthesis of a nickel complex by oxidative addition.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.6b00059.
The authors declare no competing financial interest.
Supplementary Material
References
- Campbell M. G.; Hoover A. J.; Ritter T. Top. Organomet. Chem. 2014, 52, 1–54. 10.1007/3418_2014_88. [DOI] [Google Scholar]
- Transition-metal-mediated 18F-fluorination:; a Teare H.; Robins E. G.; Kirjavainen A.; Forsback S.; Sandford G.; Solin O.; Luthra S. K.; Gouverneur V. Angew. Chem., Int. Ed. 2010, 49, 6821–6824. 10.1002/anie.201002310. [DOI] [PubMed] [Google Scholar]; b Hollingworth C.; Hazari A.; Hopkinson M. N.; Tredwell M.; Benedetto E.; Huiban M.; Gee A. D.; Brown J. M.; Gouverneur V. Angew. Chem., Int. Ed. 2011, 50, 2613–2617. 10.1002/anie.201007307. [DOI] [PubMed] [Google Scholar]; c Lee E.; Kamlet A. S.; Powers D. C.; Neumann C. N.; Boursalian G. B.; Furuya T.; Choi D. C.; Hooker J. M.; Ritter T. Science 2011, 334, 639–642. 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee E.; Hooker J. M.; Ritter T. J. Am. Chem. Soc. 2012, 134, 17456–17458. 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Stenhagen I. S. R.; Kirjavainen A. K.; Forsback S. J.; Jørgensen C. G.; Robins E. G.; Luthra S. K.; Solin O.; Gouverneur V. Chem. Commun. 2013, 49, 1386–1388. 10.1039/c2cc38646a. [DOI] [PubMed] [Google Scholar]; f Kamlet A. S.; Neumann C. N.; Lee E.; Carlin S. M.; Moseley C. K.; Stephenson N.; Hooker J. M.; Ritter T. PLoS One 2013, 8, e59187. 10.1371/journal.pone.0059187. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Graham T. J. A.; Lambert R. F.; Ploessl K.; Kung H. F.; Doyle A. G. J. Am. Chem. Soc. 2014, 136, 5291–5294. 10.1021/ja5025645. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Huang X.; Liu W.; Ren H.; Neelamegam R.; Hooker J. M.; Groves J. T. J. Am. Chem. Soc. 2014, 136, 6842–6845. 10.1021/ja5039819. [DOI] [PubMed] [Google Scholar]; i Ren H.; Wey H.-Y.; Strebl M.; Neelamegam R.; Ritter T.; Hooker J. M. ACS Chem. Neurosci. 2014, 5, 611–615. 10.1021/cn500078e. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Tredwell M.; Preshlock S. M.; Taylor N. J.; Gruber S.; Huiban M.; Passchier J.; Mercier J.; Génicot C.; Gouverneur V. Angew. Chem., Int. Ed. 2014, 53, 7751–7755. 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]; k Ichiishi N.; Brooks A. F.; Topczewski J. J.; Rodnick M. E.; Sanford M. S.; Scott P. J. H. Org. Lett. 2014, 16, 3224–3227. 10.1021/ol501243g. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Zlatopolskiy B. D.; Zischler J.; Urusova E. A.; Endepols H.; Kordys E.; Frauendorf H.; Mottaghy F. M.; Neumaier B. ChemistryOpen 2015, 4, 457. 10.1002/open.201500056. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Huang X.; Liu W.; Hooker J. M.; Groves J. T. Angew. Chem., Int. Ed. 2015, 54, 5241–5245. 10.1002/anie.201500399. [DOI] [PubMed] [Google Scholar]
- For recent advances in arene fluorination (not transition-metal-mediated) with [18F]fluoride, see:; a Pike V. W.; Aigbirhio F. I. J. Chem. Soc., Chem. Commun. 1995, 2215–2216. 10.1039/c39950002215. [DOI] [Google Scholar]; b Ross T. L.; Ermert J.; Hocke C.; Coenen H. H. J. Am. Chem. Soc. 2007, 129, 8018–8025. 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]; c Gao Z.; Lim Y. H.; Tredwell M.; Li L.; Verhoog S.; Hopkinson M.; Kaluza W.; Collier T. L.; Passchier J.; Huiban M.; Gouverneur V. Angew. Chem., Int. Ed. 2012, 51, 6733–6737. 10.1002/anie.201201502. [DOI] [PubMed] [Google Scholar]; d Cardinale J.; Ermert J.; Humpert S.; Coenen H. H. RSC Adv. 2014, 4, 17293–17299. 10.1039/c4ra00674g. [DOI] [Google Scholar]; e Rotstein B. H.; Stephenson N. A.; Vasdev N.; Liang S. H. Nat. Commun. 2014, 5, 4365. 10.1038/ncomms5365. [DOI] [PubMed] [Google Scholar]; f Stephenson N. A.; Holland J. P.; Kassenbrock A.; Yokell D. L.; Livni E.; Liang S. H.; Vasdev N. J. Nucl. Med. 2015, 56, 489–492. 10.2967/jnumed.114.151332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA Center for Drug Evaluation and Research, Guidance, PET Drugs–Current Good Manufacturing Practice (CGMP), August 2011. URL:http://www.fda.gov/downloads/Dmgs/GuidanceComplianceRegulatoryInformation/Guidances/UCM266640.pdf (retrieved July 13, 2015).
- a Lieberman L. M.; Wessels B. W.; Wiley A. L. Jr.; Gatley S. J.; Nickles R. J. Jr.; Young D.; Wolberg W. H.; Bogden A. E. Int. J. Radiat. Oncol., Biol., Phys. 1980, 6, 505–509. 10.1016/0360-3016(80)90067-X. [DOI] [PubMed] [Google Scholar]; b Dimitrakopoulou A.; Strauss L. G.; Clorius J. H.; Ostertag H.; Schlag P.; Heim M.; Oberdorfer F.; Helus F.; Haberkorn U.; van Kaick G. J. Nucl. Med. 1993, 34, 1075–1081. [PubMed] [Google Scholar]; c Kissel J.; Brix G.; Bellemann M. E.; Strauss L. G.; Dimitrakopoulou-Strauss A.; Port R.; Haberkorn U.; Lorenz W. J. Cancer Res. 1997, 57, 3415–3423. [PubMed] [Google Scholar]; d Moehler M.; Dimitrakopoulou-Strauss A.; Gutzler F.; Raeth U.; Strauss L. G.; Stremmel W. Cancer 1998, 83, 245–253. 10.1002/(SICI)1097-0142(19980715)83:2<245::AID-CNCR7>3.3.CO;2-4. [DOI] [PubMed] [Google Scholar]; e Harte R. J. A.; Matthews J. C.; O’Reilly S. M.; Price P. M. J. Nucl. Med. 1998, 39, 1370–1376. [PubMed] [Google Scholar]; f Dimitrakopoulou-Strauss A.; Strauss L. G.; Schlag P.; Hohenberger P.; Irngartinger G.; Oberdorfer F.; Doll J.; van Kaick G. J. Nucl. Med. 1998, 39, 465–473. [PubMed] [Google Scholar]; g Kissel J.; Port R. E.; Zaers J.; Bellemann M. E.; Strauss L. G.; Haberkorn U.; Brix G. Med. Phys. 1999, 26, 609–615. 10.1118/1.598560. [DOI] [PubMed] [Google Scholar]; h Saleem A.; Yap J.; Osman S.; Brady F.; Lucas S. V.; Jones T.; Price P. M.; Aboagye E. O. Lancet 2000, 355, 2125–2131. 10.1016/S0140-6736(00)02380-1. [DOI] [PubMed] [Google Scholar]; i Hino-Shishikura A.; Suzuki A.; Minamimoto R.; Shizukuishi K.; Oka T.; Tateishi U.; Sugae S.; Ichikawa Y.; Horiuchi C.; Inoue T. Appl. Radiat. Isot. 2013, 75, 11–17. 10.1016/j.apradiso.2013.01.014. [DOI] [PubMed] [Google Scholar]
- a Fowler J. S.; Finn R. D.; Lambrecht R. M.; Wolf A. P. VII J. Nucl. Med. 1973, 14, 63–64. [PubMed] [Google Scholar]; b Diksic M.; Farrokhzad S.; Yamamoto Y. L.; Feindel W. Int. J. Nucl. Med. Biol. 1984, 11, 141–142. 10.1016/0047-0740(84)90049-4. [DOI] [PubMed] [Google Scholar]; c Visser G. M.; Gorree G. M.; Braakhuis B. M.; Herscheid J. M. Eur. J. Nucl. Med. 1989, 15, 225–229. 10.1007/BF00257538. [DOI] [PubMed] [Google Scholar]
- Bergman J.; Solin O. Nucl. Med. Biol. 1997, 24, 677–683. 10.1016/S0969-8051(97)00078-4. [DOI] [PubMed] [Google Scholar]
- a Chen C.; Yang L.-M. J. Org. Chem. 2007, 72, 6324–6327. 10.1021/jo0709448. [DOI] [PubMed] [Google Scholar]; b Chen C.; Yang L.-M. Tetrahedron Lett. 2007, 48, 2427–2430. 10.1016/j.tetlet.2007.01.175. [DOI] [Google Scholar]; c Rodriguez B. A.; Delferro M.; Marks T. J. J. Am. Chem. Soc. 2009, 131, 5902–5919. 10.1021/ja900257k. [DOI] [PubMed] [Google Scholar]; d Fan X.-H.; Yang L.-M. Eur. J. Org. Chem. 2011, 2011, 1467–1471. 10.1002/ejoc.201001519. [DOI] [Google Scholar]; e Ge S.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 12837–41. 10.1002/anie.201207428. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Chakraborty S.; Zhang J.; Patel Y. J.; Krause J. A.; Guan H. Inorg. Chem. 2013, 52, 37–47. 10.1021/ic300587b. [DOI] [PubMed] [Google Scholar]; g Gutsulyak D. V.; Gott A. L.; Piers W. E.; Parvez M. Organometallics 2013, 32, 3363–3370. 10.1021/om400288u. [DOI] [Google Scholar]; h Standley E. A.; Jamison T. F. J. Am. Chem. Soc. 2013, 135, 1585–1592. 10.1021/ja3116718. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Jezorek R. L.; Zhang N.; Leowanawat P.; Bunner M. H.; Gutsche N.; Pesti A. K. R.; Olsen J. T.; Percec V. Org. Lett. 2014, 16, 6326–6329. 10.1021/ol503061c. [DOI] [PubMed] [Google Scholar]; j Park N. H.; Teverovskiy G.; Buchwald S. L. Org. Lett. 2014, 16, 220–223. 10.1021/ol403209k. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Standley E. A.; Smith S. J.; Müller P.; Jamison T. F. Organometallics 2014, 33, 2012–2018. 10.1021/om500156q. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Shields J. D.; Gray E. E.; Doyle A. G. Org. Lett. 2015, 17, 2166–2169. 10.1021/acs.orglett.5b00766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tsou T. T.; Kochi J. K. J. Am. Chem. Soc. 1979, 101, 7547–7560. 10.1021/ja00519a015. [DOI] [Google Scholar]; b Keen A. L.; Doster M.; Johnson S. A. J. Am. Chem. Soc. 2007, 129, 810–819. 10.1021/ja067112w. [DOI] [PubMed] [Google Scholar]; c Christian A. H.; Müller P.; Monfette S. Organometallics 2014, 33, 2134–2137. 10.1021/om5001327. [DOI] [Google Scholar]; d Schley N. D.; Fu G. C. J. Am. Chem. Soc. 2014, 136, 16588–16593. 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zheng B.; Tang F.; Luo J.; Schultz J. W.; Rath N. P.; Mirica L. M. J. Am. Chem. Soc. 2014, 136, 6499–6504. 10.1021/ja5024749. [DOI] [PubMed] [Google Scholar]; f Camasso N. M.; Sanford M. S. Science 2015, 347, 1218–1220. 10.1126/science.aaa4526. [DOI] [PubMed] [Google Scholar]
- See refs (2d), (2i), and (2l).
- a Fahey D. R. J. Am. Chem. Soc. 1970, 92, 402–404. 10.1021/ja00705a612. [DOI] [Google Scholar]; b Fahey D. R.; Mahan J. E. J. Am. Chem. Soc. 1977, 99, 2501–2508. 10.1021/ja00450a017. [DOI] [Google Scholar]; c Tsou T. T.; Kochi J. K. J. Am. Chem. Soc. 1979, 101, 6319–6332. 10.1021/ja00515a028. [DOI] [Google Scholar]
- Early report of transmetalation from aryllithium and Grignard reagents:Chatt J.; Shaw B. L. J. Chem. Soc. 1960, 345, 1718–1729. 10.1039/jr9600001718. [DOI] [Google Scholar]
- Reports of transmetalation from arylzinc reagents:; a Kurosawa H.; Ohnishi H.; Emoto M.; Chatani N.; Kawasaki Y.; Murai S.; Ikeda I. Organometallics 1990, 9, 3038–3042. 10.1021/om00162a014. [DOI] [Google Scholar]; b Volpe E. C.; Manke D. R.; Bartholomew E. R.; Wolczanski P. T.; Lobkovsky E. B. Organometallics 2010, 29, 6642–6652. (see also ref (9d)) 10.1021/om100420z. [DOI] [Google Scholar]
- Transmetalation of phenyl from BPh4– to Ni(II):; a Treichel P. M.; Shubkin R. L. Inorg. Chim. Acta 1968, 2, 482–484. 10.1016/S0020-1693(00)87091-7. [DOI] [Google Scholar]; b Sacconi L.; Dapporto P.; Stoppioni P. Inorg. Chem. 1976, 15, 325–329. 10.1021/ic50156a017. [DOI] [Google Scholar]; c Chadwell S. J.; Coles S. J.; Edwards P. G.; Hursthouse M. B.; Imran A. Polyhedron 1995, 14, 1057–1065. 10.1016/0277-5387(94)00343-D. [DOI] [Google Scholar]
- Transmetalation of pentafluorophenyl from B(C6F5)3 to Ni(II):; a Kalamarides H. A.; Iyer S.; Lipian J.; Rhodes L. F.; Day C. Organometallics 2000, 19, 3983–3990. 10.1021/om000606w. [DOI] [Google Scholar]; b Liang L.-C.; Chien P.-S.; Huang Y.-L. J. Am. Chem. Soc. 2006, 128, 15562–15563. 10.1021/ja065505p. [DOI] [PubMed] [Google Scholar]; c Yamamoto T.; Shikada C.; Kaita S.; Olivier T.; Maruyama Y.; Wakatsuki Y. J. Mol. Catal. A: Chem. 2009, 300, 1–7. 10.1016/j.molcata.2008.11.037. [DOI] [Google Scholar]
- Transmetalation of phenyl from LiSnPh3 to Ni(II):Müller P. Z. Chem. 1973, 13, 141–142. 10.1002/zfch.19730130416. [DOI] [Google Scholar]
- Preparation of copper(I) σ-aryl complexes from (NHC)CuI(Ot-Bu) precursors and arylboronic esters:; a Ohishi T.; Nishiura M.; Hou Z. Angew. Chem., Int. Ed. 2008, 47, 5792–5795. 10.1002/anie.200801857. [DOI] [PubMed] [Google Scholar]; b Whittaker A. M.; Rucker R. P.; Lalic G. Org. Lett. 2010, 12, 3216–3218. 10.1021/ol101171v. [DOI] [PubMed] [Google Scholar]; c Takatsu K.; Shintani R.; Hayashi T. Angew. Chem., Int. Ed. 2011, 50, 5548–5552. 10.1002/anie.201008196. [DOI] [PubMed] [Google Scholar]; d Saijo H.; Ohashi M.; Ogoshi S. J. Am. Chem. Soc. 2014, 136, 15158–15161. 10.1021/ja5093776. [DOI] [PubMed] [Google Scholar]
- Hall D. G.Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine; Wiley, 2006. [Google Scholar]
- a Percec V.; Bae J.-Y.; Hill D. H. J. Org. Chem. 1995, 60, 1060–1065. 10.1021/jo00109a044. [DOI] [Google Scholar]; b Indolese A. F. Tetrahedron Lett. 1997, 38, 3513–3516. 10.1016/S0040-4039(97)00707-7. [DOI] [Google Scholar]; c Leadbeater N. E.; Resouly S. M. Tetrahedron 1999, 55, 11889–11894. 10.1016/S0040-4020(99)00676-6. [DOI] [Google Scholar]; d Inada K.; Miyaura N. Tetrahedron 2000, 56, 8657–8660. 10.1016/S0040-4020(00)00814-0. [DOI] [Google Scholar]; e Zim D.; Lando V. R.; Dupont J.; Monteiro A. L. Org. Lett. 2001, 3, 3049–3051. 10.1021/ol016526l. [DOI] [PubMed] [Google Scholar]; f Zim D.; Monteiro A. L. Tetrahedron Lett. 2002, 43, 4009–4011. 10.1016/S0040-4039(02)00716-5. [DOI] [Google Scholar]; g Kobayashi Y.; William A. D.; Mizojiri R. J. Organomet. Chem. 2002, 653, 91–97. 10.1016/S0022-328X(02)01174-9. [DOI] [Google Scholar]; h Percec V.; Golding G. M.; Smidrkal J.; Weichold O. J. Org. Chem. 2004, 69, 3447–3452. 10.1021/jo049940i. [DOI] [PubMed] [Google Scholar]; i González-Bobes F.; Fu G. C. J. Am. Chem. Soc. 2006, 128, 5360–5361. 10.1021/ja0613761. [DOI] [PubMed] [Google Scholar]; j Paulose T. A. P.; Wu S.-C.; Olson J. A.; Chau T.; Theaker N.; Hassler M.; Quail J. W.; Foley S. R. Dalton Trans. 2012, 41, 251–260. 10.1039/C1DT10858A. [DOI] [PubMed] [Google Scholar]
- Furuya T.; Ritter T. J. Am. Chem. Soc. 2008, 130, 10060–10061. 10.1021/ja803187x. [DOI] [PubMed] [Google Scholar]
- Martin-Santamaria S.; Carroll M. A.; Carroll C. M.; Carter C. D.; Pike V. W.; Rzepa H. S.; Widdowson D. A. Chem. Commun. 2000, 649–650. 10.1039/b000868k. [DOI] [Google Scholar]
- a Magnus P.; Lacour J.; Weber W. J. Am. Chem. Soc. 1993, 115, 9347–9348. 10.1021/ja00073a084. [DOI] [Google Scholar]; b Desjardins S.; Jacquemot G.; Canesi S. Synlett 2012, 23, 1497–1500. 10.1055/s-0031-1290676. [DOI] [Google Scholar]
- Specific activity was determined using 5-FU UV absorption values that were near the limit of detection, hence the large variance in observed values.
- RCC was determined in reactions with about 10 mCi of [18F]fluoride; reaction setup was otherwise identical to the 1.4–1.8 Ci scale reaction, where radiation safety precautions prohibited direct measurement of RCC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.