

Abstract
Objective:
Muscle carnitine palmitoyltransferase (CPT) II deficiency, the most common defect of lipid metabolism in muscle, is characterized by attacks of myoglobinuria without persistent muscle weakness.
Methods:
His6-N-hCPT2 (wild-type) and His6-N-hCPT2/S113L (variant) were produced recombinantly in prokaryotic host and characterized according to their functional and regulatory properties.
Results:
The wild-type and the variant S113L showed the same enzymatic activity and thermostability at 30°C. The mutated enzyme, however, revealed an abnormal thermal destabilization at 40°C and 45°C. This was consistent with an increased flexibility (B-factor) of the variant at 40°C compared with that of the wild-type shown by molecular dynamics analysis. Preincubation of the enzymes with l-carnitine and acyl-l-carnitines containing more than 10 carbons in the acyl side-chain stabilized the mutated enzyme against thermal inactivation. In contrast, palmitoyl-CoA destabilized both enzymes.
Conclusions:
The problems in CPT II deficiency originating from the S113L mutation are not caused by the loss of catalytically active enzyme. They might be at least partially related to an impaired thermal stability of the protein. The lower thermodynamic stability of the variant might explain why fever and prolonged exertion provoke attacks of myoglobinuria in CPT II deficiency. The stabilization by acyl-l-carnitines might provide the basis for possible preventive therapy of CPT II deficiency.
Carnitine palmitoyltransferase (CPT) II deficiency is regarded as the most common defect of lipid metabolism in skeletal muscle.1 There are 3 phenotypes of CPT II deficiency: (1) a lethal neonatal form, (2) a severe infantile hepatocardiomuscular form, and (3) a mild myopathic form.2 The symptoms of the latter usually consist of recurrent attacks of myalgia and muscle weakness leading to myoglobinuria, which might result in kidney failure. The attacks are provoked by prolonged exercise, fasting, fever, or exposure to cold.3
The mutation S113L has been found on at least 1 allele in about 90% of patients with muscle CPT II deficiency (allele frequency about 75%).3
Biochemical consequences of the mutation S113L are still controversial and comprise the following hypotheses:
Reduced enzyme activity with normal or reduced protein content.4,5
Abnormally regulated enzyme rather than a reduced catalytic activity.6–8
Decreased protein stability leading to a marked reduction in the steady-state level of the enzyme.9,10
Decreased thermal stability.4
All previous studies dealing with CPT II are based on experiments using vertebrate enzyme or human cell lysate. The present work on the recombinant enzymes His6-N-hCPT2 and His6-N-hCPT2/S113L evaluates their thermal stability.
METHODS
Recombinant production and purification of human CPT II (wild-type and variant S113L).
The genes coding for wild-type enzyme (His6-N-hCPT2) and the most prominent variant of CPT II deficiency, S113L (His6-N-hCPT2/S113L), were created by site-directed mutagenesis from the gene coding for the triple variant (His6-N-hCPT2/S113L/V368I/M647V). The genes were then cloned into (pET28a(+)). The plasmids were transformed into chemo-competent Escherichia coli BL21Gold DE3 cells, and the recombinant protein was realized by heterologous gene expression in the prokaryotic host. Cells carrying the plasmids were grown in LURIA-BERTANI medium (LB-medium) supplemented with kanamycin (30 μg/mL) in a preculture overnight at 37°C. After centrifugation (30 minutes at 6,000 rpm at 10°C), cells were resuspended in fresh LB-medium for inoculation of 5-L sterile LB-medium supplemented with kanamycin (30 μg/mL) in a Biostat C-DCU 3 fermenter (B. Braun Biotech International GmbH, Melsungen, Germany). Cells were grown at oxygen content of 25–30% and feeding control (yeast extract, glycerol) for 10 hours. Gene expression was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG) (1 mM), and recombinant protein was produced for about 12–14 hours. Cells were then harvested by centrifugation (30 minutes at 6,000 rpm and 10°C) and shock frozen in liquid nitrogen. One hundred grams of frozen cells were thawed in suspension buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 150 mM NaCl, 3 mM tris (2-chloroethyl) phosphate [TCEP], 10 mM MgCl2, 3 mM benzamidine, 1 μg/mL leupeptine, 0.7 μg/mL pepstatin A, pH 8.0) and lysed by adding lysozyme (50 μg/mL) using a French Press at 1,200 bar. Nucleic acids were digested by incubation of DNase I. After centrifugation of the lysed cells (45 minutes at 45,000 rpm and 4°C, using a 45Ti rotor in a Beckman L8-60M ultracentrifuge [Beckman Instruments Inc., Brea, CA]), the supernatant was directly applied to a Ni-nitrilotriacetic acid (NTA) affinity chromatography column (3 × 5 mL His-Trap columns). The bound target protein carrying a His-tag was eluted using an imidazole gradient. Recombinant CPT II was further purified by gel filtration on Superdex S 200 (26/60) and finally by ion-exchange chromatography on Q-Sepharose HP (10/16) according to supplier's recommendations. The purification was verified by SDS-PAGE and protein identity was confirmed by mass spectrometry.
Enzyme assay of CPT II.
CPT II activity was determined spectroscopically according to Rufer et al.11 with some modifications. The assay is based on the reduction of 5,5′-dithio-bis-(2-nitrobenzoic acid) by CoA-SH and subsequent formation of 5-mercapto-2-nitrobenzoic acid, which absorbs at 410 nm with a molar extinction coefficient of 13,600/M/cm.12 The formation of CoA-SH was monitored by addition of CPT II to a solution containing l-carnitine and palmitoyl-CoA. The 1-mL reaction volume contained 50 mM potassium phosphate, 120 mM KCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM 5,5′-dithio-bis-(2-nitrobenzoic acid), 30 μM palmitoyl-CoA, and 12 mM l-carnitine (pH 7.4). After temperature equilibration, the reaction was initiated by the addition of recombinant enzyme to a final concentration of 15 nM and monitored for 120 seconds. Activity measurements were conducted at 25°C.
Thermal and effector-mediated inactivation of CPT II.
The temperature transitions (melting curves) of recombinant CPT II wild-type and the variant S113L were measured by circular dichroism at 210 nm in 25 mM phosphate buffer (pH 8.0) containing 50 mM NaCl and 2 mM TCEP.
Data of effector-mediated and temperature-induced inactivation of CPT II were investigated according to the reaction scheme depicted in figure 1 considering a negligible reactivation of the inactivated enzyme only.
Figure 1. Effector-mediated thermal inactivation of carnitine palmitoyltransferase II (considering only 1 binding site of the effector).
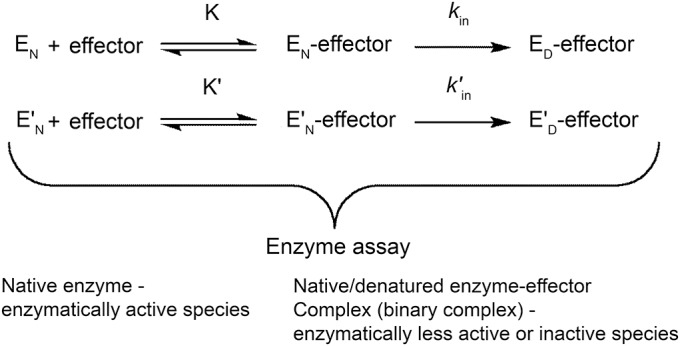
E represents the wild-type, E′ the variant S113L, N the native state, D the denatured state, K the equilibrium constant of the initial enzyme-effector binary complex, and kin the microscopic first-order rate constant of the inactivation. The apostrophe indicates the parameters of the variant.
The decrease of the enzymatic activity was analyzed according to a single-exponential first-order reaction using equation 1, yielding the apparent rate constant of inactivation:
 |
where A is the residual activity of the enzyme measured at time t of inactivation, A0 is the initial activity of the enzyme, kin is the first-order rate constant of inactivation, kre is the first-order rate constant of reactivation, and kapp is the apparent first-order rate constant of inactivation. The apparent rate constants of inactivation were determined by fitting the time course of the change of the enzymatic activities according to equation 1 using the program Kaleidagraph. For better data investigation and comparison, the activity changes were evaluated as a percentage of total initial activity (relative activities). Fitting parameters are presented with mean square errors.
Simultaneous incubation with palmitoyl-l-carnitine.
To evaluate the stabilizing effect of palmitoyl-l-carnitine on CPT II, the activities of His6-N-hCPT2 and His6-N-hCPT2/S113L were measured in the presence of 25 μM palmitoyl-l-carnitine in the reaction mixture.
Preincubation with acyl-CoA and acyl-l-carnitine derivatives.
To evaluate a putative substrate protection effect on the kinetic stability of the enzymes before activity measurements, His6-N-hCPT2 and His6-N-hCPT2/S113L were preincubated with the indicated amounts of l-carnitine or palmitoyl-CoA at 30°C, 40°C, and 45°C in different series of experiments. Acyl-l-carnitines (C2, C4, C5, C6, C8, C10, C12, C14, and C16) at a concentration of 25 μM were preincubated with the wild-type or the mutated enzyme at 40°C and 45°C, respectively. Activity measurements were performed after different incubation times.
Computational methods.
Homology modeling.
A homology model of human CPT II was built using the crystal structure of rat CPT II (PDB code 2DEB)11 as a template. Sequence alignment of both proteins (aa 32–658 showing 95.2% sequence identity) was performed using the molecular modeling package Molecular Operating Environment (Chemical Computing Group, Montreal, Quebec, Canada).13 The program MODELLER9v1114 was used to generate the homology model and the model of the variant S113L.
Molecular dynamics.
The sander module in AMBER1215 was applied for the energy minimization steps, whereas Particle-Mesh-Ewald molecular dynamics (MD) was used for the MD simulation. Particle-Mesh-Ewald method was used to treat electrostatic interactions, and a cut-off radius of 10 Å was set for nonbonded interactions. Preparation of the input files was conducted as follows. The General Amber Force Field with AM1-BCC charges was applied to assign atom types to the ligands (palmitoyl-l-carnitine), whereas Amber03 force field was used for the protein. The protein or protein-acyl-l-carnitine complexes were solvated in an octahedral box of TIP3P water with 13 Å between the complex surface and the box boundary, and counter ions were added to neutralize the system. The solvated system was then subjected to 3 steps of energy minimization: a 1,600-step minimization (800 cycles of steepest descent followed by 800 cycles of conjugate gradient) while keeping the solute fixed by a force of 10 kcal/mol/Å; 1,600 cycles of minimization using 800-step steepest descent and subsequent 800-step conjugate gradient while keeping the solute fixed by a force of 2 kcal/mol/Å; and a 2,000-step minimization (1,000-step steepest descent and 1,000-step conjugate gradient). The system was gradually heated to the studied temperature (277 K, 299 K, and 313 K) over 100 picoseconds, and then a 22-nanosecond MD simulation was performed in the NPT ensemble (pressure 1.0 bar and temperature 300 K). During MD, all covalent bonds involving H-atoms were constrained using the SHAKE procedure, and the time step was set to 2 femtoseconds. The root-mean-square deviation (RMSD) of the backbone atoms of the protein was calculated using the input structure as reference. The B-factor for the backbone of the protein residues was calculated using the program ptraj from AMBER12 over the 22-nanosecond MD simulation.
Docking studies.
The human CPT II homology model was used for molecular docking studies. First, we tested whether the docking program GOLD16 was able to redock the cocrystallized ligands in the CPT II and carnitine acetyltransferase crystal structures taken from the Protein Database (2RCU, 4EP9, 1TQ7). Good agreement between experimental and docking structures with RMSD values below 1.5 Å was obtained.
RESULTS
Activities of recombinant CPT II.
The expression of recombinantly produced human CPT II wild-type and its variant S113L was confirmed by mass spectrometry and their structural integrity was proven by enzymatic assays. At 30°C, both the wild-type and the variant S113L displayed similar specific activities of at least 39 and 33 U/mg, respectively.
Thermal inactivation of CPT II.
At 30°C, both enzymes exhibited the same thermal stability; however, the incubation of the enzymes at 40°C and at 45°C (figure 2) resulted in a considerably faster inactivation of the mutated enzyme compared with the wild-type at both temperatures.
Figure 2. Thermal inactivation of His6-N-hCPT2 and His6-N-hCPT2/S113L at different temperatures.
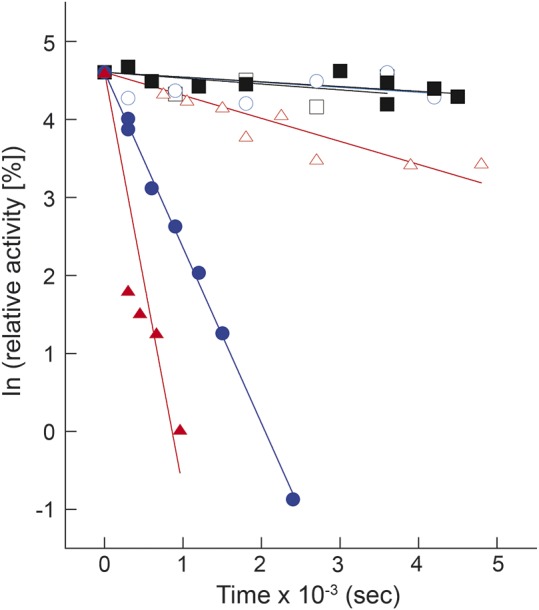
Black squares, blue circles, and red triangles represent values at 30°C, 40°C, and 45°C, respectively. Open symbols show the wild-type recombinant carnitine palmitoyltransferase II and the closed symbols show His6-N-hCPT2/S113L. Data are presented as time-dependent changes of natural-log–transformed relative activities. The experiments were performed as explained in the Methods section.
The thermal denaturation of the proteins turned out to be irreversible. Hence, with the transition midpoint dependent on the protein concentration, no thermodynamic parameters of thermal equilibrium unfolding could be determined. During thermal unfolding, no aggregation and precipitation of the proteins occurred in the unfolded state at elevated temperatures, which was indicated by the photomultiplier voltage of the dichrograph and by light scattering (data not shown).
MD simulations were performed to study the stability of the wild-type and the variant S113L at 3 different temperatures: 4°C (277 K), 20°C (293 K), and 40°C (313 K). For this purpose, the generated homology model of human CPT II was used. To assess the thermal stability of the enzyme, the RMSD of the protein backbone (figure e-1 at Neurology.org/ng) and the B-factor (indicating the flexibility of the backbone) over the length of the MD simulation were measured. MD simulations showed that the calculated B-factor for the residues neighboring the mutation site (S110-L121) reveal in a considerably higher fluctuation for the mutant's residues at 313 K (40°C) compared with 277 K (4°C) and to a lesser extent with 293 K (20°C). This could be indicative of the lower thermal stability of the mutated enzyme at 40°C. Conversely, the calculated B-factor for the wild-type did not show any noticeable differences at the 3 temperatures studied (figure 3A). The hydroxyl group of S113 can form several H-bond interactions. These include a water-mediated H-bond with R498 as found in the homology model or an H-bond interaction with the backbone carbonyl group of H496 (figure 3B). The reason behind the instability of the variant S113L might be the inability of L113 to form these H-bonds, which would induce conformational changes, especially at the residues neighboring the mutation site.
Figure 3. Flexibility of carnitine palmitoyltransferase (CPT) II studied by molecular dynamics simulation.

(A) B-factors of residues S110-L121 of CPT II calculated over 22 nanoseconds of MD simulation of wild-type (left) and the variant S113L (right) at 277 K, 293 K, and 313 K. (B) S113 (magenta) forms H-bond interactions with neighboring residues as seen in the generated homology model of human CPT II. The α-helix–bearing residue S113 is shown as magenta ribbon, and the rest of the enzyme is shown as pink ribbon. H-bond interactions are shown as blue dotted lines. Water molecules are depicted as red balls. (C) B-factors of residues S110-L121 of CPT II were calculated over 22 nanoseconds of MD simulation for the wild-type (left) and the variant S113L (right) in complex with palmitoyl-l-carnitine at 277 K, 293 K, and 313 K.
Effects of substrates on thermal inactivation of CPT II.
The kinetic stability of the enzymes was measured after their preincubation with various substrates at different temperatures.
Destabilization by palmitoyl-CoA.
Preincubation of His6-N-hCPT2 and His6-N-hCPT2/S113L with the native substrate palmitoyl-CoA before the addition of l-carnitine generally increased the rate constants of thermal inactivation at 40°C and 45°C and revealed no substrate protection. At both temperatures, the thermal inactivation was faster in the variant than in wild-type (figure 4, A and B; table).
Figure 4. Effect of substrates on kinetic stability of recombinant carnitine palmitoyltransferase (CPT) II enzymes.
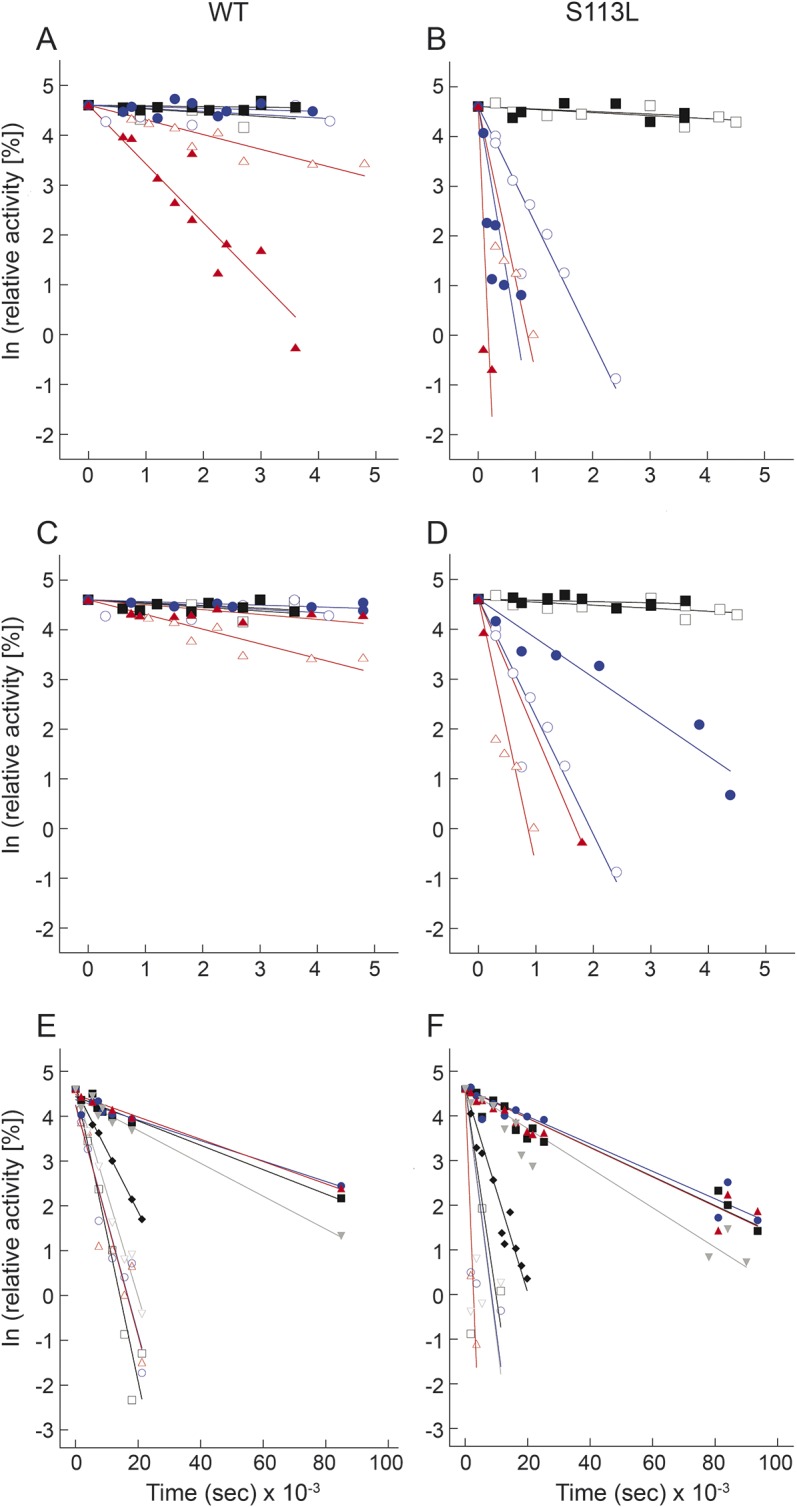
A and B show the heat inactivation of His6-N-hCPT2 and His6-N-hCPT2/S113L, respectively, at different temperatures supported by palmitoyl-CoA. C and D show the heat inactivation of His6-N-hCPT2 and His6-N-hCPT2/S113L, respectively, at different temperatures suppressed by l-carnitine. Black squares, blue circles, and red triangles represent values at 30°C, 40°C, and 45°C, respectively. Open and closed symbols show the results of experiments performed in the absence or in the presence of palmitoyl-CoA or l-carnitine, respectively. E and F present the stabilization of His6-N-hCPT2 and His6-N-hCPT2/S113L, respectively, against heat inactivation by preincubation with acyl-l-carnitines at 45°C. Black open squares, blue open circles, red open triangles, gray open inverted triangles, black closed diamonds, black closed squares, blue closed circles, red closed triangles, and gray closed inverted triangles represent acetyl-l-carnitine (C2), isobutyryl-l-carnitine (C4), valeryl-l-carnitine (C5), hexanoyl-l-carnitine (C6), octanoyl-l-carnitine (C8), decanoyl-l-carnitine (C10), lauroyl-l-carnitine (C12), myristoyl-l-carnitine (C14), and palmitoyl-l-carnitine (C16), respectively. Data are presented as time-dependent changes of natural-log–transformed relative activities. The experiments were performed as explained in the Methods section. S113L = the variant S113L; WT = wild-type.
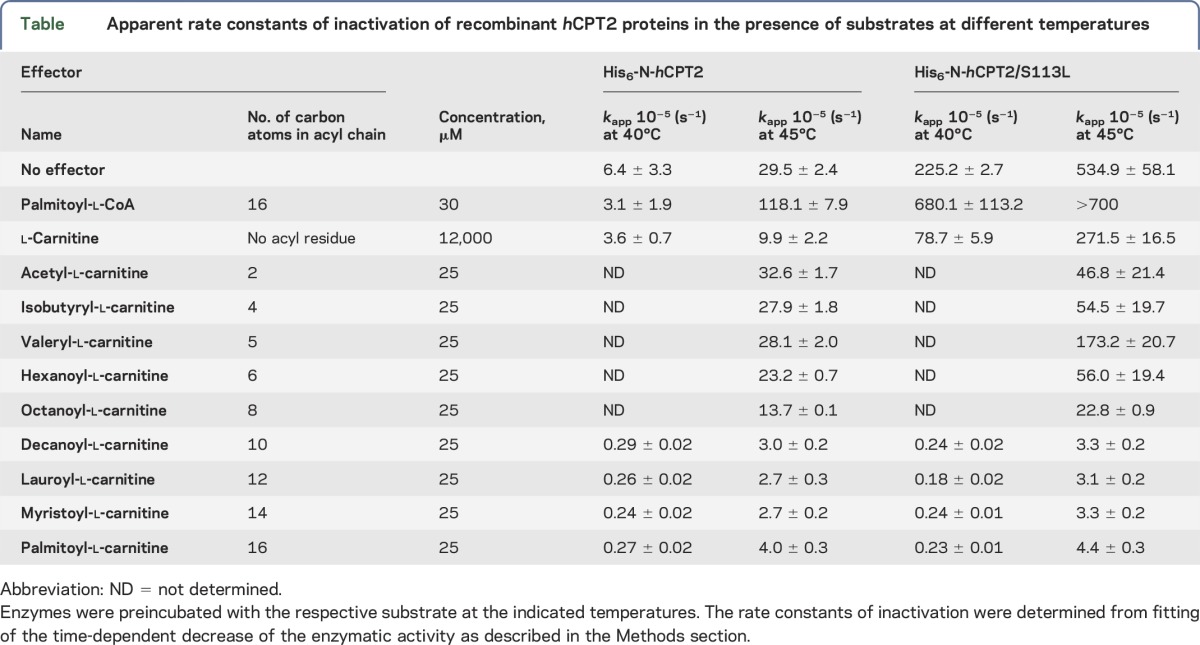
Table.
Apparent rate constants of inactivation of recombinant hCPT2 proteins in the presence of substrates at different temperatures
Effects of l-carnitine and acyl-l-carnitines.
Simultaneous addition of palmitoyl-l-carnitine.
The presence of additional palmitoyl-l-carnitine at 25 μM in the assay mixture had no effect on the enzyme activity. The simultaneous addition of higher concentrations of palmitoyl-l-carnitine as 1 product of the forward CPT II assay reaction progressively inhibited both enzymes. This inhibition, however, was much stronger for the variant S113L than for the wild-type. The residual activity of the variant S113L in the presence of 200 μM palmitoyl-l-carnitine was only 60% that of the wild-type under the same conditions.
Preincubation with l-carnitine and acyl-l-carnitines.
Both the wild-type and the mutated recombinant enzyme displayed a much higher kinetic stability on preincubation with l-carnitine at 45°C (figure 4, C and D; table).
The short-chain acyl-l-carnitines C2, C4, C5, and C6 showed a slight stabilizing effect on His6-N-hCPT2 and His6-N-hCPT2/S113L at 45°C (figure 4, E and F; table). The middle-chain acyl-l-carnitines C10, C12, and C14 and the long-chain C16 stabilized the mutated enzyme to the level of the wild-type. Thus, the inactivation rate constants of His6-N-hCPT2/S113L were smaller by a factor of about 150 at 45°C. Octanoyl-l-carnitine (C8) displayed an intermediate stabilizing effect on both enzymes against thermal inactivation (figure 4, E and F; table).
The acyl-l-carnitines with the most pronounced stabilizing effect on the enzymes were also tested at 40°C. Here, the acyl-l-carnitines C10, C12, C14, and C16 decreased the inactivation rate constant of His6-N-hCPT2/S113L and His6-N-hCPT2 by a factor of about 1,000 and 25, respectively. At 40°C, the mutated enzyme showed a very similar kinetic stability as the wild-type in the presence of these acyl-l-carnitines (table).
MD studies on CPT II in complex with palmitoyl-l-carnitine showed similar behavior of the wild-type and the variant S113L with respect to temperature. The B-factor of the residues of the mutant surrounding the mutation site did not show any increase at higher temperature (313 K, 40°C) (figure 3C). Generally, the flexibility of the mutation site (S110-L121) of the acyl-l-carnitine–binding site (figures e-2 and e-3) and of the whole protein (figure e-1) was lower for the complex of the variant S113L with palmitoyl-l-carnitine than for the protein without substrate at 313 K.
DISCUSSION
The enzymatic activities of the recombinant wild-type CPT II and its variant S113L were similar. This shows that the hypothesis of reduced enzyme activity (deficiency) of the S113L variant can no longer be maintained. This is consistent with the clinical observation that patients harboring the S113L mutation show only attacks of muscle weakness and myoglobinuria but no persistent or progressive muscle weakness (in contrast to l-carnitine–deficient patients). This is also consistent with recent findings of normal protein concentrations of the CPT II variant in skeletal muscle,7 indicating that there is no decreased steady-state level of the protein, as previously suggested.9 The phenotype of the S113L mutation restricted to muscle without any other organ involvement indicates that a mutant enzyme can be variously represented and function in vivo in various tissues.
Between the attacks, the patients have no symptoms of myalgia and muscle weakness. This is consistent with the findings of the normal activity and stability of the variant at body temperature. Attacks of myoglobinuria in CPT II deficiency are usually provoked by fever, infections, fasting, prolonged physical stress, and exposure to cold. All of these trigger factors except fasting might in some way be related to a changed thermal stability of the mutated enzyme. It could be speculated that even exposure to cold might provoke symptoms, probably because of increased intramuscular temperature due to compensatory mitochondrial heat production. This is based on the observation that exposure to cold stimulates thermogenesis by increased mitochondrial metabolic rate and increased total energy expenditure not only in brown adipose tissue but also in rodent and human skeletal muscle and in bird plasma.17–19
The data reveal that the S113L mutation impairs the kinetic stability of human CPT II at increased temperatures. This is consistent with the lower heat resistance of the mutated enzyme in cultured fibroblasts.4 The results are also consistent with the MD simulations of the S113L variant, which show an increase in the flexibility of some residues at 40°C, especially the residues in the vicinity of the mutation site. This in turn might cause a change in the conformational state of the mutated enzyme, resulting in a decreased thermal stability.
Preincubation of the enzymes with the native substrate palmitoyl-CoA at 40°C before the addition of l-carnitine further increased the thermal inactivation rate of CPT II, which was more pronounced for the variant than for the wild-type. The mechanism of this destabilizing effect and its pathophysiologic impact remain enigmatic.
Acyl-l-carnitines with more than 10 carbons in the acyl side-chain stabilized the variant S113L against the temperature-induced inactivation. The stabilizing effect occurred only when the enzyme was preincubated with acyl-l-carnitine before starting the assay. A possible explanation for this stabilization is the formation of a stabilized binary complex after preincubation of the apoenzyme of CPT II with acyl(palmitoyl)-l-carnitine.
It has been shown that administration of a bolus of middle-chain fatty acids just before intensive exercise might prevent attacks of myoglobinuria in CPT II deficiency.20 Increased middle-chain acyl-CoA levels might lead to an increase of middle-chain acyl-l-carnitines. This will help not only to provide the required energy but also to stabilize this essential enzyme against inactivation.
However, there is not only a stabilizing effect by long-chain acyl-l-carnitine but also a progressively inhibiting effect on CPT II activity. In contrast to the stabilization by preincubation of the enzyme with acyl(palmitoyl)-l-carnitine, this inhibitory effect was observed only when the enzyme was simultaneously exposed to the substrates palmitoyl-CoA, palmitoyl-l-carnitine, and l-carnitine. The formation of a stabilized binary complex of the apoenzyme of CPT II with acyl(palmitoyl)-l-carnitine might be prevented by this simultaneous exposure. Under this condition, acyl(palmitoyl)-l-carnitine will impair the enzymatic turnover by product inhibition.
Both effects (inhibition and stabilization of the enzyme) by palmitoyl-l-carnitine were more pronounced for the S113L variant than for the wild-type, which further supports the view of an abnormal regulation of the mutated enzyme.21
Fasting is another trigger factor for attacks in CPT II–deficient patients. It is characterized by a decrease of free l-carnitine and an increase in acyl-l-carnitine level in plasma.22,23 In the present study, the simultaneous addition of palmitoyl-l-carnitine to the assay might mimic this effect. The preincubation of middle- and long-chain acyl-l-carnitines before starting the assay, however, led to stabilization of the enzyme against thermal inactivation. Thus, acyl-l-carnitines can exert both effects (stabilization and inhibition) in vitro. It can be hypothesized that in vivo the time profile of changes in substrate concentration might determine which of these effects will prevail. In serum of CPT II–deficient patients, the concentration of long-chain acyl-l-carnitines is persistently elevated, even between the attacks. This can be used as a marker for diagnosis of this enzyme disorder.24 However, this might also exert the stabilizing effect in most physiologically relevant situations. Thus, the attacks might occur only when this mechanism is counteracted in extreme situations, such as by severe fever or under extreme dietary conditions with rapid increase of palmitoyl-l-carnitine concentrations (inhibition effect). Both aspects might explain the rare occurrence of attacks caused by the S113L variant of CPT II deficiency. The biochemical consequences of other CPT II mutations have to be characterized in separate future studies.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Dina Robaa for carrying out molecular dynamics from the Institute of Pharmacy, Martin Luther University Halle-Wittenberg. We also thank Prof. Dr. Sven-Erik Behrens from the Institute of Biochemistry and Biotechnology, Martin Luther University Halle-Wittenberg; Prof. Dr. Marcus Deschauer and Dr. Pushpa Raj Joshi from the Department of Neurology, Martin Luther University Halle-Wittenberg; and Prof. Dr. Reinhard Neubert from the Institute of Pharmacy, Martin Luther University Halle-Wittenberg for providing excellent discussion and support.
GLOSSARY
- CPT
carnitine palmitoyltransferase
- LB-medium
LURIA-BERTANI medium
- MD
molecular dynamics
- RMSD
root-mean-square deviation
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
L. Motlagh designed and performed the experiments and wrote the manuscript. R. Golbik designed and performed the experiments and wrote the manuscript. W. Sippl designed and performed the experiments and wrote the manuscript. S. Zierz designed the experiments and wrote the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
Dr. L. Motlagh and Dr. R. Golbik report no disclosures. Prof. W. Sippl has served on the scientific advisory board of the Journal of Computer Aided Molecular Design and has received research support from EU FP7 (“A-PARADDISE” EC-GA no. 602080) DFG Germany. Prof. S. Zierz has received speaker honoraria from Genzyme, Germany. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem 1997;244:1–14. [DOI] [PubMed] [Google Scholar]
- 2.Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med 2004;25:495–520. [DOI] [PubMed] [Google Scholar]
- 3.Joshi PR, Deschauer M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype-phenotype analysis of 50 patients. J Neurol Sci 2014;338:107–111. [DOI] [PubMed] [Google Scholar]
- 4.Olpin SE, Afifi A, Clark S, et al. Mutation and biochemical analysis in carnitine palmitoyltransferase type II (CPT II) deficiency. J Inherit Metab Dis 2003;26:543–557. [DOI] [PubMed] [Google Scholar]
- 5.Fanin M, Anichini A, Cassandrini D, et al. Allelic and phenotypic heterogeneity in 49 Italian patients with the muscle form of CPT-II deficiency. Clin Genet 2012;82:232–239. [DOI] [PubMed] [Google Scholar]
- 6.Zierz S. Limited trypsin proteolysis renders carnitine palmitoyltransferase insensitive to inhibition by malonyl-CoA in patients with muscle carnitine palmitoyltransferase deficiency. Clin Investig 1994;72:957–960. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann D, Zierz S. Normal protein content but abnormally inhibited enzyme activity in muscle carnitine palmitoyltransferase II deficiency. J Neurol Sci 2014;339:183–188. [DOI] [PubMed] [Google Scholar]
- 8.Motlagh L, Golbik R, Sippl W, Zierz S. Malony-CoA inhibits the S113L variant of carnitine-palmitoyltransferase II. Biochim Biophys Acta 2016;1861:34–40. [DOI] [PubMed] [Google Scholar]
- 9.Taroni F, Verderio E, Dworzak F, Willems PJ, Cavadini P, DiDonato S. Identification of a common mutation in the carnitine palmitoyltransferase II gene in familial recurrent myoglobinuria patients. Nat Genet 1993;4:314–320. [DOI] [PubMed] [Google Scholar]
- 10.Verderio E, Cavadini P, Montermini L, et al. Carnitine palmitoyltransferase II deficiency: structure of the gene and characterization of two novel disease-causing mutations. Hum Mol Genet 1995;4:19–29. [DOI] [PubMed] [Google Scholar]
- 11.Rufer AC, Thoma R, Benz J, et al. The crystal structure of carnitine palmitoyltransferase 2 and implications for diabetes treatment. Structure 2006;14:713–723. [DOI] [PubMed] [Google Scholar]
- 12.Ellman GL. A colorimetric method for determining low concentrations of mercaptans. Arch Biochem Biophys 1958;74:443–450. [DOI] [PubMed] [Google Scholar]
- 13.Molecular Operating Environment (MOE), 2013.08. Montreal: Chemical Computing Group Inc.; 2015. [Google Scholar]
- 14.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234:779–815. [DOI] [PubMed] [Google Scholar]
- 15.Case D, Darden T, Cheatham TE, III, et al. AMBER 12. San Francisco: University of California; 2012. [Google Scholar]
- 16.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–748. [DOI] [PubMed] [Google Scholar]
- 17.Stier A, Massemin S, Criscuolo F. Chronic mitochondrial uncoupling treatment prevents acute cold-induced oxidative stress in birds. J Comp Physiol B 2014;184:1021–1029. [DOI] [PubMed] [Google Scholar]
- 18.Lim S, Honek J, Xue Y, et al. Cold-induced activation of brown adipose tissue and adipose angiogenesis in mice. Nat Protoc 2012;7:606–615. [DOI] [PubMed] [Google Scholar]
- 19.van den Berg SA, van Marken Lichtenbelt W, van Dijk KW, Schrauwen P. Skeletal muscle mitochondrial uncoupling, adaptive thermogenesis and energy expenditure. Curr Opin Clin Nutr Metab Care 2011;14:243–249. [DOI] [PubMed] [Google Scholar]
- 20.Tucci S, Primassin S, Ter Veld F, Spiekerkoetter U. Medium-chain triglycerides impair lipid metabolism and induce hepatic steatosis in very long-chain acyl-CoA dehydrogenase (VLCAD)-deficient mice. Mol Genet Metab 2010;101:40–47. [DOI] [PubMed] [Google Scholar]
- 21.Zierz S, Engel AG. Regulatory properties of a mutant carnitine palmitoyltransferase in human skeletal muscle. Eur J Biochem 1985;149:207–214. [DOI] [PubMed] [Google Scholar]
- 22.Frohlich J, Seccombe DW, Hahn P, Dodek P, Hynie I. Effect of fasting on free and esterified carnitine levels in human serum and urine: correlation with serum levels of free fatty acids and beta-hydroxybutyrate. Metabolism 1978;27:555–561. [DOI] [PubMed] [Google Scholar]
- 23.Hoppel CL, Genuth SM. Carnitine metabolism in normal-weight and obese human subjects during fasting. Am J Physiol 1980;238:E409–E415. [DOI] [PubMed] [Google Scholar]
- 24.Gempel K, Kiechl S, Hofmann S, et al. Screening for carnitine palmitoyltransferase II deficiency by tandem mass spectrometry. J Inherit Metab Dis 2002;25:17–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.