

Abstract
Objective:
To identify the genetic defect for adult-onset primary lateral sclerosis (PLS) in a family with 5 patients.
Methods:
Whole-exome sequencing was performed to identify the shared genetic variants in 3 affected members in a PLS family with 5 affected individuals. Sanger sequencing was used for validation of the variants and for cosegregation analysis. Mitochondrial activity for both patients and unaffected siblings was measured using a SeaHorse metabolic analyzer.
Results:
Whole-exome sequencing and subsequent cosegregation analysis demonstrated that compound heterozygous missense variants L695P and I743T in SPG7 were the only mutations cosegregating with the disease in an autosomal recessive fashion in this family. The parents and siblings are genetically heterozygous and clinically unaffected. Functional studies suggested that the PLS-associated SPG7 mutants affect mitochondrial function when glucose is reduced.
Conclusions:
Compound heterozygote mutations in SPG7 are associated with adult-onset PLS, extending the spectrum of SPG7-linked neurologic diseases. Patients with the PLS phenotype should have genetic testing for paraplegin, especially when the condition is familial.
Primary lateral sclerosis (PLS) is a relatively rare upper motor neuron (UMN) disease characterized by slowly progressive spasticity and weakness in voluntary muscles of 3 regions of the body, including those controlling legs, arms, and muscles of speech, swallowing, and chewing. Although the disease can occur in children, caused by mutations in the gene ALSIN,1 it usually occurs between ages 40 and 60 years. The causes and pathogenic mechanism of adult-onset PLS are largely unknown. We describe a family with 5 patients with PLS who had disease onset in adulthood, ranging from 20 to 37 years old. The initial symptoms were either gait disturbance or stiffness in lower extremities and were followed by progressive arm and hand dysfunction and pseudobulbar involvement (table e-1 at Neurology.org/ng). We identified compound heterozygous missense mutations in SPG7, expanding the clinical spectrum of SPG7-linked neurologic diseases.
METHODS
Clinical presentation.
A 2-generation Caucasian family with 5 PLS cases was identified (figure 1A). The proband (II-1) was a 58-year-old man who developed progressive gait dysfunction at age 35. When seen at age 45, he showed spastic gait, baclofen-responsive spasticity in the lower extremities, mild spastic dysarthria, minimal upper extremity dysfunction, and normal extraocular movements. No features of peripheral neuropathy were observed. His reflexes were diffusely increased, but there was no tremor. Imaging studies of his brain and complete spine were normal. Over the next 13 years, he developed progressive spasticity and inability to move his legs, with increasing dysfunction in his hands and a brisk jaw jerk. By age 53, he had developed progressive loss of weight and muscle bulk, with a decrease in deep tendon reflexes. His speech became weaker and hypophonic. He also developed mild nystagmus on lateral gaze. A younger brother and 2 younger sisters developed a similar syndrome (figure 1A). A third sister developed more progressive spasticity. The family has been followed over the years, and no family members have developed any sensory involvement or bladder problems. Both parents and the other 2 siblings are free of neurologic symptoms.
Figure 1. Mutations in SPG7 in a family with primary lateral sclerosis.

(A) Pedigree with primary lateral sclerosis (PLS). DNA samples from 3 affected individuals (II-1, II-4, and II-7) were used for whole-exome sequencing. Candidate variants were validated, and cosegregation analysis was performed using the DNA samples from all 9 members of this family. The compound missense mutations, L695P and I743T, cosegregated with PLS in this family. (B) Sanger sequencing of SPG7. DNA and messenger RNA samples from lymphoblastoid cells from patients with PLS were Sanger-sequenced; heterozygous mutations are indicated by arrows. (+) = positive; (−) = absence; CF = cerebellar feature; LMN = lower motor neuron signs; NN = neurologically normal; WCB = wheelchair bound.
Standard protocol approvals, registrations, and patient consents.
The family history, clinical data, consent forms, and blood samples from 5 affected and 4 unaffected family members were collected, and permanent lymphoblast cell lines were established. This study was approved by our institutional review board.
Genetic mutation identification.
The Illumina HQ-Seq 2500 NGS platform was used to sequence the exomes of 3 patients from the family. Sanger sequencing and Western blot were performed.
Mitochondrial activity.
To determine whether the P695 and T743 compound mutations in SPG7 are associated with altered mitochondrial activity, we performed metabolic measurements on lymphoblasts from both patients and unaffected heterozygous individuals using a SeaHorse XF24 instrument. The SeaHorse metabolic analyzer measures oxygen consumption rates under 4 conditions: basal, complex V inhibited (oligomycin), uncoupler stimulated (carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone [FCCP]), and in the presence of complex I and III inhibitors (Rotenone, Antimycin A). We followed the manufacturer's protocols, with steps and conditions optimized for lymphoblasts.2 In a typical experiment, 2 patient samples and 2 samples from unaffected heterozygous individuals were analyzed in a 5-well per sample setting.
RESULTS
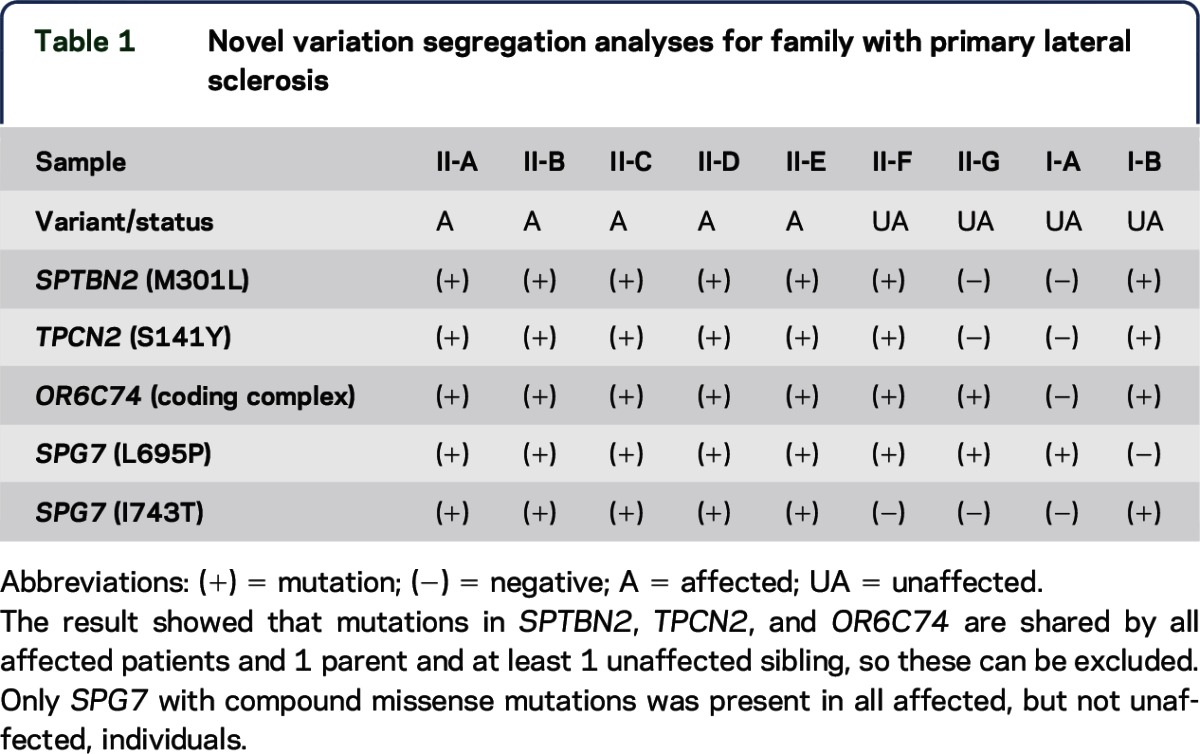
The exome sequencing data from 3 affected individuals showed that 21 variants in 20 genes were shared by these 3 patients. Subsequent Sanger sequencing and cosegregation analysis demonstrated that 5 variants in 4 genes were present in all 5 patients. Variants in 3 genes, including SPTN2, TPCN2, and ORC74, were also present in the unaffected individuals (table 1). Uniquely, 2 missense variants in SPG7, c.2084T>C/p.L695P and c.2228T>C/p.I743T, were present in all 5 patients. The L695P was present in the unaffected father and 2 unaffected siblings, and the I743T was present in the unaffected mother (figure 1A). We performed Sanger sequencing of a cohort of 480 Caucasian controls and did not find these variants. The L695P variant was not present in the Exome Aggregation Consortium (ExAC), which includes 60,706 unrelated individuals sequenced as part of various disease-specific and population genetic studies. The I743T was observed in 6 individuals in a heterozygous condition but in none in a homozygous condition in the ExAC. These data indicate that the compound heterozygous mutations L695P and I743T in SPG7 are linked to PLS in this family.
Table 1.
Novel variation segregation analyses for family with primary lateral sclerosis
To test whether the PLS-linked SPG7 mutations affect its messenger RNA (mRNA) and protein stability, we performed mRNA analysis using the lymphoblastoid cells and found that both alleles are expressed at a similar level (figure 1B). Both P695 and I743 are conserved in several vertebrate species (figure 2A) and are located within the SPG7 C-terminal domain that is required for interaction with binding partner AFG3L2. Western blots using cell lysates from lymphoblastoid cells suggested an increased amount of SPG7 protein in the compound heterozygous patients compared with the unaffected single heterozygous siblings (figure 2B).
Figure 2. Increased mitochondrial sensitivity to glucose reduction in lymphoblastoid cells from patients with primary lateral sclerosis.

(A) The mutated amino acids L695 and I743 are highly conserved in vertebrate species. Mutant amino acid residues are indicated by arrows. (B) Western blot was performed using the lysates of the lymphoblastoid cells from the unaffected heterozygous (lanes 1 and 2) and the affected compound heterozygous (lanes 3 and 4) individuals in this family. GAPDH was used as a loading control. The relative ratios of band density for SPG7 over GAPDH are shown in the right panel. (C, D) Oxygen consumption rate (OCR) assays. The OCR was measured under 4 conditions: basal, complex V inhibited, uncoupler stimulated, and in the presence of complex I and III inhibitors. The lymphoblastoid cells from 3 different patients and 2 unaffected heterozygous individuals were used in normal (C) or stress (D) conditions using the standard SeaHorse protocols. In all assays, cell adhesion was facilitated by using Cell-Tak-treated assay plates. The cells were allowed to attach for 1 hour before beginning the assay. *p < 0.05.
The SeaHorse experiment's data showed that the patient cells exhibit a higher level (p = 0.037) of oxygen consumption in the presence of the uncoupler FCCP than do the unaffected cells (figure 2C). In a second experiment, the patient and unaffected cells were treated with a glycolytic antagonist 2-deoxyglucose. This stress condition partially inhibits glycolysis and glucose uptake.2 As shown in figure 2D, the basal level and the FCCP-induced oxygen consumption are greatly reduced (p = 0.003) in the patient-derived cells but do not change significantly in the unaffected individuals, which suggests that mitochondria with the compound heterozygous mutants were more sensitive to glucose reduction.
DISCUSSION
Mutations in SPG7, which encodes a mitochondrial protein paraplegin, typically cause a recessive form of spastic paraplegia.3 Recently, it has been shown that homozygous or compound heterozygous mutations in SPG7 also lead to chronic progressive external ophthalmoplegia (CPEO)4 and are a major cause of previously unexplained adult-onset ataxia.5 Several mutations were found in patients with sporadic adult-onset UMN syndrome, none of which produced symptoms in the bulbar region.6 We report compound heterozygous missense mutations that lead to PLS in an autosomal recessive fashion in a large family with 5 affected individuals, therefore expanding the spectrum of SPG7-linked diseases.
PLS typically occurs sporadically, and its etiology and pathogenic mechanisms are not well understood. We previously found that loss-of-function mutations in ALSIN cause recessive amyotrophic lateral sclerosis (ALS) type 2 and juvenile primary lateral sclerosis (JPLS).1 Alsin has 2 protein isoforms due to alternative splicing. We observed that the mutations affecting both isoforms led to ALS, whereas the mutations affecting only the long form, but leaving the short form intact, resulted in JPLS. This observation led us to propose that the short isoform of alsin may retain some functions that protect lower motor neurons, therefore resulting in the phenotype differences.1 In mice, there are 2 paraplegia isoforms. The long isoform is targeted to the mitochondria, and the short isoform is targeted to the endoplasmic reticulum. Knockout of the long isoform causes mitochondrial abnormalities. In the knockout model, the short isoform is retained and is not affected.7
Of note, SPG7 has 6 isoforms due to alternative splicing; the expression profile and functional importance of the 6 isoforms are not clear. It is interesting that in all the patients reported with CPEO or adult-onset ataxia, 1 SPG7 allele was a missense or a truncation mutation affecting the 2 longest isoforms and not affecting the remaining 4 short isoforms, whereas the mutations on the other SPG7 allele varied greatly.4,5 These observations raise the possibility that full-length paraplegin with these missense mutations and the 4 short paraplegin isoforms may retain partial functions. The most prevalent missense mutation in SPG7 identified in CPEO or adult-onset ataxia is A510V, which accounts for more than half of the mutant alleles in these patients. Although the initial symptoms vary, patients with these mutations may eventually develop spasticity after a long duration of disease, resulting in an overlap of symptoms with complicated or syndromic spastic paraplegia.4,5 The mutations identified in the PLS family in our report include L695P and I743T. The L695P mutation does not affect the 4 short isoforms, whereas the I743T affects only the longest isoform, leaving the 5 short isoforms intact. Therefore, it is plausible that the differential expression profile and relative functional importance of the paraplegin isoforms, together with age and other genetic and environmental factors, may determine the clinical variance of the SPG7-linked symptoms, especially in the early stages of the diseases. Further studies are warranted.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Navdeep Chandel for the use of the SeaHorse metabolic analyzer and Janice Caliendo for editing and language help on the manuscript.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- CPEO
chronic progressive external ophthalmoplegia
- ExAC
Exome Aggregation Consortium
- FCCP
carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone
- JPLS
juvenile primary lateral sclerosis
- mRNA
messenger RNA
- PLS
primary lateral sclerosis
- UMN
upper motor neuron
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Yi Yang: study concept and design, involved in all of the experiments and analyses related to this manuscript, and wrote the draft of this manuscript. Lei Zhang: study concept and design. David Lynch: the clinical and diagnostic workup of the patients and collection of the blood samples from family. Thomas Lukas: carried out mitochondrial oxygen consumption experiments. Kreshnik Ahmeti: exome sequencing data analyses. Eanna Ryan: carried out SPG7 protein experiments. Nailah Siddique: organized clinical features and DNA sampling. Kimberly Schadt: organized clinical features and DNA sampling. Jordan Newman: performed Sanger sequencing on normal controls. Patrick Sleiman: performed Sanger sequencing on normal controls. Han-Xiang Deng: analyzed sequencing data and helped write the draft of this manuscript. Teepu Siddique: study concept and design, analysis of data, and manuscript finalization. The experiments were carried out in Teepu Siddique's laboratory and in other laboratories at Northwestern University Feinberg School of Medicine. Yi Yang, Thomas Lukas, Han-Xiang Deng, and Teepu Siddique wrote the manuscript.
STUDY FUNDING
Les Turner ALS Foundation, Les Turner ALS Foundation/Herbert C. Wenske Foundation Professorship, Vena E. Schaff ALS Research Fund, Foglia Family Fund for ALS Research, SP Foundation, and NIH Grants List: RO1 ES022310.
DISCLOSURE
Yi Yang and Lei Zhang report no disclosures. David R. Lynch has served on the editorial board of Journal of Neurogenetics; holds a patent for the creation of a test for anti-NMDA receptor encephalitis; has consulted for Bracket Inc; and has received research support from Edison Pharmaceutical, Shire Pharmaceutical, Reata Pharmaceutical, Horizon Pharma, NIH (R21), and FARA. Thomas Lukas holds a patent for Anti-inflammatory and protein kinase inhibitor compositions and related methods for downregulation of detrimental cellular responses and inhibition of cell death (US8088774-B2). Kreshnik Ahmeti, Patrick M.A. Sleiman, Eanna Ryan, Kimberly A. Schadt, Jordan H. Newman, and Han-Xiang Deng report no disclosures. Nailah Siddique has received research support from NINDS (NS050641) and Les Turner ALS Foundation. Teepu Siddique has served on the editorial board of Neurogenetics and Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration and has received funding from NIEHS (RO1 NS078504), NINDS (RO1 NS078504), Les Turner ALS Foundation, the Foglia Family Fund for ALS Research, Les Turner ALS Foundation/Herbert C. Wenske Foundation Professorship, Vena E. Schaff ALS Research Fund, and SP Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 2001;29:160–165. [DOI] [PubMed] [Google Scholar]
- 2.Jitschin R, Hofmann AD, Bruns H, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014;123:2663–2672. [DOI] [PubMed] [Google Scholar]
- 3.Casari G, De Fusco M, Ciarmatori S, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998;93:973–983. [DOI] [PubMed] [Google Scholar]
- 4.Pfeffer G, Gorman GS, Griffin H, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014;137:1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeffer G, Pyle A, Griffin H, et al. SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 2015;84:1174–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brugman F, Scheffer H, Wokke JH, et al. Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology 2008;71:1500–1505. [DOI] [PubMed] [Google Scholar]
- 7.Mancuso G, Barth E, Crivello P, Rugarli EI. Alternative splicing of Spg7, a gene involved in hereditary spastic paraplegia, encodes a variant of paraplegin targeted to the endoplasmic reticulum. PLoS One 2012;7:e36337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.