

Abstract
Objective:
The aim of this study was to analyze the association between the variations of coenzyme Q2 4-hydroxybenzoate polyprenyltransferase gene (COQ2) and Japanese patients with multiple system atrophy (MSA).
Methods:
We investigated the genetic variations in exons 1, 2, 6, and 7 of the COQ2 gene in 133 Japanese patients with MSA and 200 controls and analyzed the association between the variations and MSA.
Results:
Six DNA variations (G21S, L25V, V66L, P157S, V393A, and X422K) were found in the 133 patients with MSA, and G21S and X422K were new variations that had never been reported. V66L was a common variation that was found in all 133 patients with MSA. G21S, P157S, V393A, and X422K did not show gene frequency differences between patients with MSA and controls. On the other hand, L25V was newly proven to be the only risk factor of sporadic MSA with predominant olivopontocerebellar ataxia.
Conclusions:
The present study suggests L25V variant of COQ2 gene as a genetic risk factor in Japanese patients with MSA with cerebellar ataxia.
Multiple system atrophy (MSA) is an adult-onset, fatal, and essentially sporadic neurodegenerative disease with poor prognosis.1 MSA is clinically characterized by progressive cerebellar, parkinsonian, and autonomic symptoms as initial and primary features in various combinations, which basically correspond to olivopontocerebellar ataxia (OPCA), striatonigral degeneration (SND), and Shy-Drager syndrome (SDS), respectively.1–3 MSA is also classified into 2 main subtypes, MSA with predominant cerebellar ataxia (MSA-C) corresponding to OPCA and MSA with predominant parkinsonism (MSA-P) corresponding to SND; these are more common than SDS.2–4 The common pathologic feature of MSA is the excessive aggregation of α-synuclein, primarily in oligodendroglia.1,4,5 However, the exact pathogenic mechanisms of MSA still remain unknown.
Although MSA is basically a nongenetic disease, previous reports indicated genetic variations for susceptibility to MSA. A common variant V393A in coenzyme Q2 4-hydroxybenzoate polyprenyltransferase (COQ2) gene was further detected in 363 Japanese patients with sporadic MSA as a risk factor of MSA, especially MSA-C,5 suggesting that genetic factors may play an important role in MSA. In the present study, therefore, we performed DNA variant analysis of COQ2 gene in 133 Japanese patients with sporadic MSA and compared the results with 200 controls.
METHODS
Standard protocol approvals, registrations, and patient consents.
All participants were enrolled in the present study retrospectively on the basis of research protocols that were approved by the institutional review board at Okayama University (approval no. 22). Written informed consent was obtained from all participants.
Patients with MSA and controls.
A total of 133 clinically confirmed patients with sporadic probable or possible MSA and 200 non-neurodegenerative disease controls were included in the present study. The diagnosis of sporadic MSA was made on the basis of the current consensus criteria.1,2 Of the 133 patients with MSA, 87 were diagnosed as having OPCA (MSA-C), 28 as having SND (MSA-P), and 18 as having SDS.2–4 Non-neurodegenerative disease controls (n = 200) included patients with peripheral neuropathy (n = 51), CNS infection (n = 25), collagen neurologic disease (n = 23), stroke (n = 19), myopathy (n = 18), cervical or lumbar spondylosis (n = 12), neurosis (n = 8), epilepsy (n = 7), headache (n = 5), and other neurologic diseases without neurodegenerative disease (n = 32). All individuals were unrelated within the sample groups.
Genetic analysis.
Genomic DNA was extracted from peripheral lymphocytes obtained from the patients with MSA and controls. DNA sequence analysis was performed for exons 1, 2, 6, and 7 of all 133 patients with MSA after amplification by PCR using the primers for COQ2.5 We used sequence scanner software to align and detect variations against the human reference genome (figure 1).
Figure 1. DNA variations of COQ2 gene in patients with multiple system atrophy.

(A–C) Direct DNA sequencings detect G21S (A), L25V (B), and X422K (C) variations. Arrows point out the position of each variation. (D) G21S DNA variation confirmed by PCR restriction fragment length polymorphism (RFLP) with the arrow as normal band and the arrowhead as variant band. (E) L25V DNA variation confirmed by allele-specific (AS)-PCR with the variant case positive both for common forward (CF) plus normal reverse (NR) primers and CF plus variant reverse (VR) primers. (F) X422K DNA variation confirmed by PCR-RFLP with the variant allele (arrowhead).
PCR–restriction fragment length polymorphism analysis.
To verify the specificity of the sequence analysis and DNA variations, PCR–restriction fragment length polymorphism (PCR-RFLP) analysis method was implemented to check the DNA samples of patients with MSA carrying variations and all 200 controls. The selectivity of restriction enzyme, which can cut DNA at the variant site, was achieved using Takara Cut-site Navigator online. BfaⅠ, AluⅠ, and AseⅠ were applied for G21S, V393A, and X422K, respectively. Twenty microliters of the PCR mixture was digested in a final volume of 25 μL with 1- to 3-μL units of enzyme and 2.5-μL units of buffer for more than 4 hours. Digested samples were then resolved by 3% or 5% agarose gel electrophoresis in Tris-borate-EDTA (TBE) buffer at 50 or 100 V for approximately 1–5 hours.
Allele-specific PCR analysis.
Because the corresponding restriction enzyme site was not found to confirm L25V and P157S variations, allele-specific PCR (AS-PCR) was performed for the patients' DNA samples carrying these variations and for all controls. A common forward primer was designed about 200 base pairs upstream from the variation sites (5′-GGGGCTGCAAGTCACCAC-3′ for L25V and 5′-GAGCCTCTCTATTCCTTTTAGG-3′ for P157S). As for the reverse primer sets, 2 reverse primers were designed for detecting both the normal and variant sequences at single PCR, matching only 1 of the biallelic single-nucleotide substitution (5′-CAGCCTGGCAGACTAGGGTT-3′ as normal reverse primer and 5′-CAGCCTGGCAGACTAGGCGT-3′ as variant reverse primer for L25V and 5′-GCTGAACCAGGTTGTTTACC-3′ as normal reverse primer and 5′-GCTGAACCAGGTTGTTGTTC-3′ as variant reverse primer for P157S). The underlined amino acids of the above primers are complementary to the normal site or variant site. Double underlined amino acids of the above primers enhance the specificity of the PCR by destabilizing the extension of the doubly mismatched primers. The amplification products are separated on a 3% agarose gel and run in TBE buffer at 100 V for 30 minutes (figure 1).
Sequence analysis and statistical analysis.
All variations in all 133 patients with MSA were investigated in the Single Nucleotide Polymorphism Database, Human Genetic Variation Database, and Human Gene Mutation Database.
For G21S, L25V, V66L, P157S, V393A, and X422K, statistical analysis was performed using χ2 text and Fisher exact test to calculate the significance of the difference in the number of variation carriers and noncarriers between patients with MSA and controls, with contingency tables and standard methods used to calculate odds ratios and the corresponding 95% confidence intervals. Statistical significance was set at p < 0.05. All statistical analysis was computed using SPSS software.
RESULTS
DNA variations of patients with MSA.
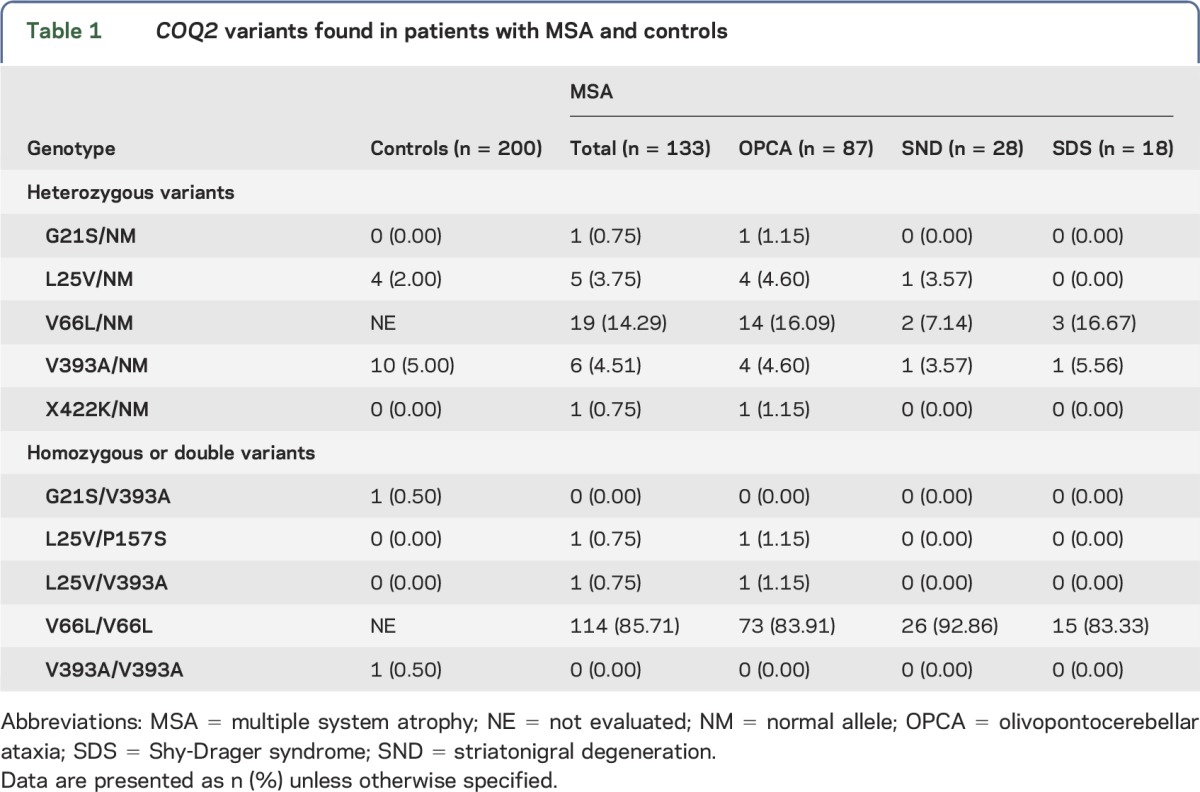
Among our 133 patients with probable or possible MSA, direct DNA sequencing of the COQ2 gene found the following 6 variations: G21S (figure 1A), L25V (figure 1B), V66L, P157S, V393A, and X422K (figure 1C). G21S and X422K were new variations that have never been reported and were confirmed by PCR-RFLP for G21S (figure 1D) and X422K (figure 1F). AS-PCR also confirmed the genetic variation of L25V in patients with MSA (figure 1E). P157S and V393A were also confirmed by AS-PCR and PCR-RFLP, respectively.
Five DNA variations (G21S, L25V, P157S, V393A, and X422K) were also simultaneously examined by PCR-RFLP or AS-PCR in 200 control participants. V66L was not examined because all 133 patients with MSA carried V66L variation in the direct DNA sequence (the allele frequency of heterozygous V66L is 0.07 and of homozygous V66L is 0.93), which suggested that V66L variation was common in the Japanese participants.5 Among the 5 DNA variations, G21S, L25V, and V393A were also found in controls by AS-PCR or PCR-RFLP. One control carried homozygous V393A and another carried a combinational variation (G21S/V393A). The numbers and frequencies of patients having each variation are displayed in table 1.
Table 1.
COQ2 variants found in patients with MSA and controls
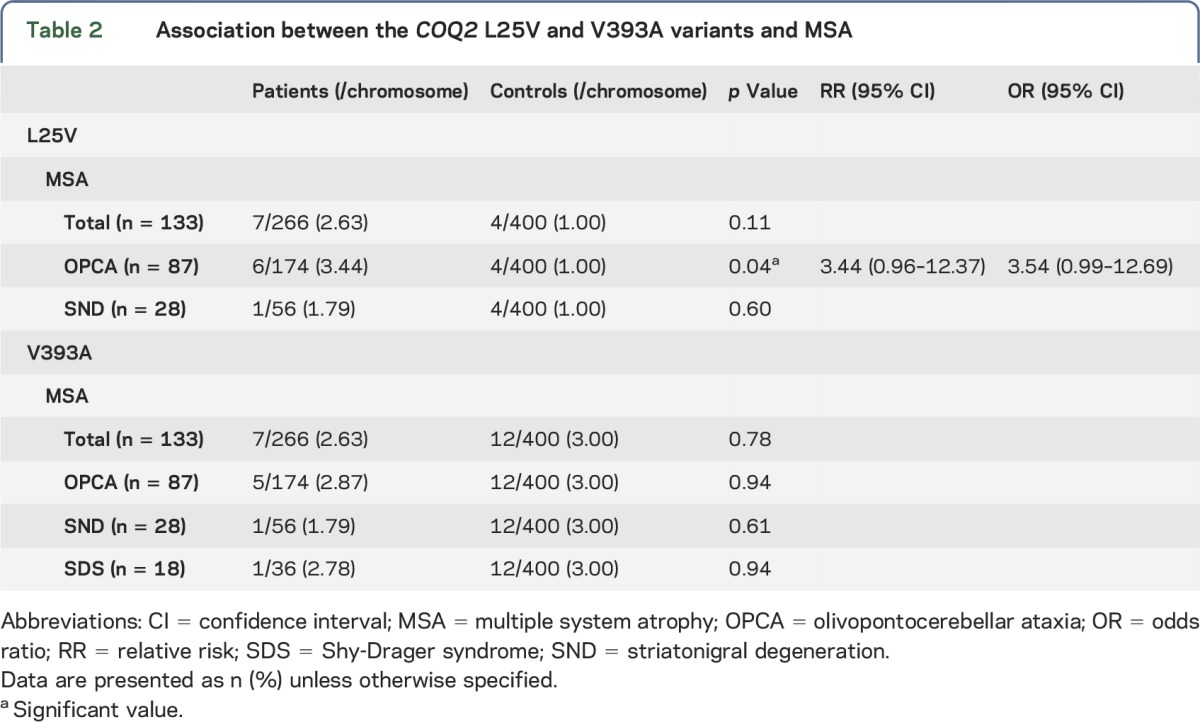
Association analysis of COQ2 variations.
The frequencies of L25V and V393A variation in patients with MSA were larger than other variations of COQ2 gene except for V66L variation. Although there was no difference in the allelic L25V frequencies between 133 patients with MSA and 200 controls (2.63% and 1.00%, respectively), the allelic L25V variation in patients with OPCA showed a 3.44-fold greater relative risk of developing the disease (3.44%, p = 0.04 vs control) compared with controls (1.00%) (table 2). On the other hand, SND showed no such difference in the allelic L25V frequencies (1.79% in patients with SND vs 1.00% in controls). As for allelic V393A frequencies, there was no difference in total MSA, OPCA, SND, or SDS in patients compared with controls (2.63%, 2.78%, 2.87%, 1.79%, and 3.00%, respectively). G21S, P157S, and X422K variations were detected in 1 chromosome in each of 3 patients with OPCA, but frequencies of chromosomes were not different compared with controls (data not shown).
Table 2.
Association between the COQ2 L25V and V393A variants and MSA
Amino acid sequences of COQ2 gene in different species.
The amino acid sequences of COQ2 gene in humans were compared with those of different species (figure 2). Although G21S and L25V were conserved in human, gorilla, and monkey, they were not located in COQ2 gene of other species. Although variations of V66L and V393A were conserved as valine among primates, other mammals of lower order showed different amino acids. P157S was conserved from human to zebrafish as proline. X422K was conserved as X in human, gorilla, cattle, and sheep but not in monkey and the other mammals.
Figure 2. Comparison of amino acid sequence for COQ2 variations in different species.

DISCUSSION
Our 133 patients with sporadic MSA showed 6 DNA variations in COQ2 gene: G21S (figure 1A), L25V (figure 1B), V66L, P157S, V393A, and X422K (figure 1C). G21S (figure 1A) and X422K (figure 1C) variations have not been reported previously but did not show differences in the allelic frequencies between 133 patients with MSA and 200 controls (table 1). On the other hand, the allele frequency of L25V (figure 1B) in patients with OPCA (3.44%) was significantly higher than that in the controls (1.00%), with a relative risk of 3.44 (table 2). All 133 patients with MSA carried V66L variation, and both P157S and V393A showed no difference between patients with MSA and controls (table 2). Amino acid sequences of COQ2 gene in different species showed that the above 6 variations were relatively conserved in primates but not in the lower order mammals (figure 2).
We performed genetic analyses for exons 1, 2, 6, and 7 because previous reports showed that the DNA variations of COQ2 were located in exons 1, 2, 6, and 7.3,6–8 Another Japanese group also reported 3 variations (V66L, P157S, and V393A) in Japanese patients with MSA.5 V66L variation was also found in British7 and American patients with MSA,9 P157S in European patients with MSA,8 and V393A in European,8 Korean,10 and Chinese patients with MSA.11 Among the previous reports, only the Japanese report suggested a risk of MSA-C (OPCA) for V393A,5,12 but this was not proven in the patients with MSA considered in the present study (table 2). In the current study, L25V was found to be a risk factor for OPCA corresponding to MSA-C, which has not been reported in the previous reports from Japan,5 the United States,9 Europe,8 Korea,10 or China.11 Most of the patients with MSA and controls included in the present study were living in the same Chugoku area of Japan, and both the groups may have similar genetic backgrounds. Although the patients with MSA included in the present study were diagnosed as having probable or possible MSA, the number of participants in our study was small and inhomogeneous between OPCA (n = 87), SND (n = 28), and SDS (n = 18). There were no studies containing patients with MSA and controls in the same pedigree and the segregation study was not done. Therefore, further studies with additional numbers and containing first-degree relatives will be needed to confirm this result.
COQ2 is a mitochondrial enzyme that is essential for the biosynthesis of antioxidative coenzyme Q10 in the mitochondrial respiratory chain.5,10,11,13 A primary deficiency of coenzyme Q10 caused by COQ2 gene mutations was associated with infantile-onset multisystem disorder, renal disease,5,6,14,15 and cardiovascular failure.16,17 The decrease of coenzyme Q10 induced by COQ2 variations can cause the decline of ATP level in mitochondria.6,14,18 The mitochondrial dysfunction characterized as an increased fission, an impaired fusion, and a fragmentation of mitochondria is associated with the pathogenesis of several neurodegenerative diseases,19 such as Parkinson disease, Alzheimer disease, Huntington disease, amyotrophic lateral sclerosis,20 and Charcot-Marie-Tooth disease. These reports may support the association between COQ2 variants and MSA. However, to confirm the influence for COQ10 expression due to L25V COQ2 variation, further studies should be done.
In the present study, L25V variation in the COQ2 gene was newly found as a risk factor of MSA-C (OPCA), but V393A variation was not. Although previous reports showed no association of variations of COQ2 with MSA (except for the Japanese report), our study could suggest a race-specific pathologic role of COQ2 gene variations in MSA.
ACKNOWLEDGMENT
The authors thank all the patients for their participation in the study.
GLOSSARY
- AS
allele-specific
- COQ2
coenzyme Q2 4-hydroxybenzoate polyprenyltransferase
- MSA
multiple system atrophy
- MSA-C
multiple system atrophy with predominant cerebellar ataxia
- MSA-P
multiple system atrophy with predominant parkinsonism
- OPCA
olivopontocerebellar ataxia
- RFLP
restriction fragment length polymorphism
- SDS
Shy-Drager syndrome
- SND
striatonigral degeneration
- TBE
Tris-borate-EDTA
AUTHOR CONTRIBUTIONS
Prof. Abe designed the study and revised the manuscript. Dr. Sun and Dr. Ohta performed the experiments, conducted the statistical analysis, and drafted the manuscript. Dr. Yamashita, Dr. Sato, Dr. Takemoto, and Dr. Hishikawa collected the DNA samples and analyzed the data.
STUDY FUNDING
Prof. Abe received funding for this study from Grand-in-Aid for Scientific Research (B) 2529320216, (C) 24591263, and Challenging Research 24659651, and by Grants-Aid from the Research Committees (Mizusawa H, Nakano I, Nishizawa M, Sasaki H, and Aoki M) from the Ministry of Health, Labour and Welfare of Japan.
DISCLOSURE
Koji Abe has received research support from the Ministry of Health, Labour and Welfare in Japan. The other authors report no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med 2015;372:1375–1376. [DOI] [PubMed] [Google Scholar]
- 2.Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999;163:94–98. [DOI] [PubMed] [Google Scholar]
- 3.Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study. Arch Neurol 1960;2:511–527. [DOI] [PubMed] [Google Scholar]
- 4.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitsui J, Matsukawa T, Ishiura H, et al. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 2013;369:233–244. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Martin JM, Salviati L, Trevisson E, et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet 2007;16:1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schottlaender LV, Houlden H; Multiple-System Atrophy (MSA) Brain Bank Collaboration. Mutant COQ2 in multiple-system atrophy. N Engl J Med 2014;371:81. [DOI] [PubMed] [Google Scholar]
- 8.Sharma M, Wenning G, Kruger R; European Multiple-System Atrophy Study Group (EMSA-SG). Mutant COQ2 in multiple-system atrophy. N Engl J Med 2014;371:80–81. [DOI] [PubMed] [Google Scholar]
- 9.Ogaki K, Fujioka S, Heckman MG, et al. Analysis of COQ2 gene in multiple system atrophy. Mol Neurodegener 2014;9:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeon BS, Farrer MJ, Bortnick SF; Korean Canadian Alliance on Parkinson's Disease and Related Disorders. Mutant COQ2 in multiple-system atrophy. N Engl J Med 2014;371:80. [DOI] [PubMed] [Google Scholar]
- 11.Chen YP, Zhao B, Cao B, et al. Mutation scanning of the COQ2 gene in ethnic Chinese patients with multiple-system atrophy. Neurobiol Aging 2015;36:1222 e1211–e1227. [DOI] [PubMed] [Google Scholar]
- 12.Mitsui J, Tsuji S. Mutant COQ2 in multiple-system atrophy. N Engl J Med 2014;371:82–83. [DOI] [PubMed] [Google Scholar]
- 13.Spindler M, Beal MF, Henchcliffe C. Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatr Dis Treat 2009;5:597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duberley KE, Abramov AY, Chalasani A, Heales SJ, Rahman S, Hargreaves IP. Human neuronal coenzyme Q10 deficiency results in global loss of mitochondrial respiratory chain activity, increased mitochondrial oxidative stress and reversal of ATP synthase activity: implications for pathogenesis and treatment. J Inherit Metab Dis 2013;36:63–73. [DOI] [PubMed] [Google Scholar]
- 15.Quinzii C, Naini A, Salviati L, et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 2006;78:345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007;7(suppl):S154–S167. [DOI] [PubMed] [Google Scholar]
- 17.Sarter B. Coenzyme Q10 and cardiovascular disease: a review. J Cardiovasc Nurs 2002;16:9–20. [DOI] [PubMed] [Google Scholar]
- 18.Quinzii CM, Garone C, Emmanuele V, et al. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J 2013;27:612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 2013;369:2236–2251. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Yamashita T, Tian F, et al. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr Neurovasc Res 2013;10:222–230. [DOI] [PubMed] [Google Scholar]