

Abstract
Objective:
To identify the underlying molecular basis of a familial developmental disorder characterized by chorea, marked speech delay, and learning difficulties in 4 sisters from a consanguineous family.
Methods:
Whole-exome analysis of DNA of the 2 older patients followed by Sanger sequencing of the mutated exon in all family members.
Results:
A homozygous deleterious mutation, p.C291X, was identified in the GPR88 gene in both exome analyses. The mutation segregated with the disease in the family and was absent from a large cohort of controls.
Conclusions:
Homozygous deleterious mutation in GPR88 in humans is associated with marked speech delay, learning disabilities, and chorea, which manifest at 8–9 years of age. The finding is consistent with the reported abundant expression of GPR88 in the striatum and the hyperkinetic activity and learning impairment observed in GPR88 knockout mice. Although further functional characterization is needed, the finding underscores the importance of GPR88 in movement control and learning.
Chorea, defined as involuntary movements of limbs, trunk, neck, or face that rapidly flit from region to region in an irregular, flowing, nonstereotyped pattern, may be underlined by genetic, pharmacologic, metabolic, or anatomic causes. A practical approach to genetic childhood chorea is consideration of the mode of inheritance.1 X-linked chorea is seen in Lesch-Nyhan and Rett syndromes, and maternally inherited chorea is part of Leigh and MELAS syndromes. Autosomal dominant transmitted forms of chorea are usually part of a more complicated disorder, such as GLUT1 deficiency or PRRT2 mutation. A relatively “pure” chorea transmitted in an autosomal dominant mode is “benign hereditary chorea,” attributed to mutations in the TITF1 gene.2 Autosomal recessive transmitted forms of chorea are also part of a complex neurologic presentation, as in inborn errors of organic and amino acid metabolism, lysosomal storage diseases, mitochondrial respiratory chain defects, and brain iron accumulation (reviewed in reference 1).
We now report the results of the molecular investigation of patients from a consanguineous family who presented in childhood with choreiform movements, speech delay, and learning disabilities.
METHODS
Patients.
Patient II-1 was the first daughter of first-cousin Palestinian parents (figure, A). Global developmental delay was noted in early infancy. She started walking at 24 months and uttered her first words at 3 years. Abnormal involuntary movements were first noted at the age of 9. These were initially mild and intermittent and confined to the face, predominantly around the mouth, as twitching of the lips. Within several months, the movements became more persistent and spread to the fingers and thighs (video at Neurology.org/ng). No further progression was observed beyond 11 years of age. Neurologic examination at 15 years revealed purposeless insuppressible choreiform movements involving the face and hands. The facial movements were mainly perioral and consisted of involuntary movements of the upper lip upward, grimacing, and movement of the lower jaw from side to side. The hand movements were continuous choreiform movements mainly of the fingers. These movements partly subsided with action but persisted despite distraction. Purposeless movements of the thighs and the head were also observed but at a lesser frequency. Motor impersistence of the tongue was noted. The rest of the neurologic examination was unremarkable. At 15 years, patient II-1 could read fluently but had marked dyscalculia. Her formal IQ was estimated at 40.
Figure. Family pedigree and genotype.
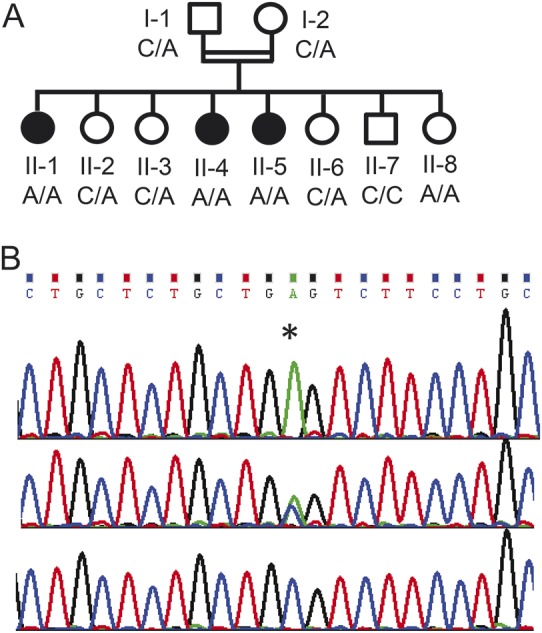
(A) The family pedigree showing symptomatic patients (filled symbols) and genotype of the c.C873A, p.C291X mutation in the GPR88 gene. (B) The chromatogram of the mutation (asterisk), a patient (upper panel), a carrier (middle panel), and a healthy control (lower panel).
Patient II-4 had essentially a similar presentation and course. Psychomotor delay was evident in infancy; she walked independently at 2 years and had markedly retarded speech acquisition, with short sentences uttered only at 8 years of age. Her school performance was poor because of severe learning difficulties, and at 10 years, she could read only simple words and had marked dyscalculia. Involuntary movements were first noted at 8 years and consisted of intermittent rising of the eyebrows and forward head movements. At 10 years, she developed choreiform movements with constant flexion/extension of the fingers and hands, grimacing, side-to-side movement of the pelvis and trunk, upward shrugging of the shoulders, and flexion of the thighs (see video). Her speech was slow but the rest of the neurologic examination was unremarkable.
Patient II-5 had a similar course; she started walking at 18 months and her speech acquisition was delayed, with first words uttered at 3 years of age. Repetitive rising of the eyebrows and occasional forward head movements were noted at 8 years. She had only subtle finger movements and intermittent upward shrug of the right shoulder. Her school performance was always poor, and at 8 years, she was unable to read or calculate. Neurologic examination was otherwise normal.
Patient II-8 was an 18-month-old girl at the time of writing. The pregnancy and delivery were uneventful, and her gross motor development was normal; she could sit at 8 months and walked independently at 18 months. However, at that age, there were no words at all. Abnormal movements were not seen.
Routine laboratory investigations including a battery of tests for inborn errors of metabolism were normal in patient II-1. This patient also underwent brain MRI at 14 years of age, which was normal.
The other children in the family (individuals II-2, II-3, II-6, and II-7) aged 3.5–13.5 years had normal motor development and no involuntary movements. All had age-appropriate fluent speech and average learning performance.
Exome analysis.
Exome analysis was performed as previously described3 in the DNA samples of patients II-1 and II-4.
Standard protocol approvals, registrations, and patient consents.
Parental consent was given for DNA studies. The study was performed with the approval of the ethics committees of Hadassah Medical Center and the Ministry of Health.
RESULTS
The exome analyses of the DNA of patients II-1 and II-4 yielded 47.9 and 61.0 million reads, respectively (mean coverage X72.1 and X91.9). After alignment to the reference genome (Hg19) and variant calling, we removed variants that were called less than X8; were off-target, synonymous, or heterozygous; those with minor allele frequency (MAF) >1% in the Exome Aggregation Consortium (ExAC), Cambridge, MA (http://exac.broadinstitute.org); and those with MAF >2% in the Hadassah in-house database. Four homozygous variants were shared by the 2 sisters, but using Sanger sequencing we found that only 1 of the variants, Chr1:101005395C>A, NM_022049:c.C873A, p.C291X in the GPR88 gene, was carried by the parents and was found to be homozygous in patients II-5 and II-8. None of the other siblings, whose development was age appropriate at 3.5–13.5 years, was homozygous for the variant (figure, A and B). The variant was not carried by any of the ∼60,000 individuals whose exome analyses were deposited in ExAC (accessed June 2015).
DISCUSSION
The 4 female patients, ages 1.5–15 years, presented in infancy with developmental delay, marked speech retardation, and learning disability. The older 3, when reaching 8–9 years of age, developed movement disorder, which consisted of choreiform movements involving mainly the face and hands with involuntary movements of the shoulders, pelvis, and thighs. Molecular investigation revealed a homozygous nonsense mutation, p.C291X, in the GPR88 gene.
GPR88 encodes a 384–amino acid protein, an orphan G protein–coupled receptor that is abundantly expressed in the striatal medium spiny neurons (MSNs). The MSNs play a crucial role in movement control. The motor cortex receives input from other cortical areas, the cerebellum, and the basal ganglia, always through the thalamus. The thalamus is under inhibitory activity of the globus pallidus pars interna (GPint); this inhibition can be released when the striatal GABAergic MSN transmits an inhibitory signal to the GPint. The inhibition is further relieved by stimulation of the MSN dopamine (D1) receptors. This signaling is the “direct pathway,” which promotes movement. The opposing “indirect pathway” aims at preventing movement by using another striatal subpopulation of MSNs, which transmit an inhibitory signal to the globus pallidus pars externa (GPext). The inhibition is reinforced by stimulation of the MSN dopamine (D2) receptors. The GPext transmits inhibitory signal to the subthalamic nucleus, which in turn makes excitatory connections to cells in the GPint, maintaining the GPint inhibitory effect on the thalamus.4 The abundant expression of GPR88 in both D1 and D2 receptor–expressing MSNs5,6 suggests that GPR88 has an instrumental role in movement control. In agreement, our patients who were homozygous for a deleterious GPR88 mutation had a hyperkinetic movement disorder and the GPR88 knockout mice had increased locomotion, nonhabitual hyperactivity in novel environment (which is a primary landmark of striatal dysfunction7), increased circling, and prolonged duration of burying episodes, suggesting stereotypic behavior.6,8 In the mutant animals, the MSNs had higher average firing, which was attributed to decreased tonic GABAergic inhibition resulting from low level of β3 protein, a GABAA receptor subunit, and to an increased glutamatergic excitatory synaptic transmission proposed to result from enhanced phosphorylation of the AMPA-type glutamate receptor subunit GluR1.6
Speech delay and poor school performance in our patients recapitulated the marked impairment in motor skill learning of the mutant mice despite normal coordination and strength. This deficit was confined to striatal learning and memory and did not affect hippocampal learning behavior.8
GPR88 has been the center of scientific attention because administration of antidepressants, lithium, and valproate alters its expression, which likely mediates some of the adverse effects of these drugs. In addition, association studies proposed GPR88 as a candidate gene in major psychoses.9 With the clear reservation that our oldest patient is currently 15 years of age, we cannot lend support to these findings. Furthermore, the extended family has no known cases of psychiatric illness and the parents who carry the mutation have normal mental health.
We propose that GPR88 deficiency in humans manifests as developmental delay with pronounced speech acquisition impairment, learning disabilities, and hyperkinetic movement disorder at 8–9 years of age. Although further functional studies are needed, our clinical data corroborate those observed in the GPR88 knockout mice and underscore the role of GPR88 in movement control and learning.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients' family for their cooperation and Dr. David Arkadir for reviewing the clinical data.
GLOSSARY
- ExAC
Exome Aggregation Consortium
- GPext
globus pallidus pars externa
- GPint
globus pallidus pars interna
- MAF
minor allele frequency
- MSN
medium spiny neuron
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Dr. Fadi Alkufri and Dr. Bassam Abu-Libdeh designed and conceptualized the study and drafted and revised the manuscript. Dr. Avraham Shaag designed the study, analyzed and interpreted the data, and drafted the manuscript. Dr. Orly Elpeleg designed the study, analyzed and interpreted the data, and drafted and revised the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Hermann A, Walker RH. Diagnosis and treatment of chorea syndromes. Curr Neurol Neurosci Rep 2015;15:514–525. [DOI] [PubMed] [Google Scholar]
- 2.Breedveld GJ, van Dongen JWF, Danesino C, et al. Mutations in TITF-1 are associated with benign hereditary chorea. Hum Mol Genet 2002;11:971–979. [DOI] [PubMed] [Google Scholar]
- 3.Damseh N, Danson CM, Al-Ashhab M, et al. A defect in the retromer accessory protein, SNX27, manifests by infantile myoclonic epilepsy and neurodegeneration. Neurogenetics 2015;16:215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci 1990;13:281–285. [DOI] [PubMed] [Google Scholar]
- 5.Massart R, Guilloux JP, Mignon V, et al. Striatal GPR88 expression is confined to the whole projection neuron population and is regulated by dopaminergic and glutamatergic afferents. Eur J Neurosci 2009;30:397–414. [DOI] [PubMed] [Google Scholar]
- 6.Quintana A, Sanz E, Wang W, et al. Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat Neurosci 2012;15:1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Do J, Kim JI, Bakes J, Lee K, Kaang BK. Functional roles of neurotransmitters and neuromodulators in the dorsal striatum. Learn Mem 2012;20:21–28. [DOI] [PubMed] [Google Scholar]
- 8.Meirsman AC, Le Merrer J, Pellissier LP, et al. Mice lacking GPR88 show motor deficit, improved spatial learning, and low anxiety reversed by delta opioid antagonist. Biol Psychiatry Epub 2015 Jun 6. doi: 10.1016/j.biopsych.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Del Zompo M, Deleuze JF, Chillotti C, et al. Association study in three different populations between the GPR88 gene and major psychoses. Mol Genet Genomic Med 2014;2:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.