

Abstract
Objective:
To evaluate the association between the genetic variants in CACNA1C, which encodes the α1 subunit of the L-type voltage-sensitive calcium channel (LVSCC) and Parkinson disease (PD) while accounting for interactions with vitamin D concentration.
Methods:
Two independent case-control data sets (478 cases and 431 controls; 482 cases and 412 controls) were used. Joint effects of single nucleotide polymorphisms (SNPs) and SNP-vitamin D interaction were analyzed by comparing models containing vitamin D deficiency, SNP genotypes, SNP-vitamin D interaction, and covariates to a restricted model with only vitamin D deficiency and covariates. Meta-analysis was used to combine the joint effects in the 2 data sets. Analysis was stratified by vitamin D deficiency to demonstrate the pattern of SNP-vitamin D interaction.
Results:
Vitamin D deficiency was associated with PD in both data sets (odds ratio [OR] = 1.9–2.7, p ≤ 0.009). SNP rs34621387 demonstrated a significant joint effect (meta-analysis, p = 7.5 × 10−5; Bonferroni corrected, p = 0.02). The G allele at rs34621387 is associated with PD in vitamin D-deficient individuals in both data sets (OR = 2.0–2.1, confidence interval = 1.3–3.5, p = 0.002) but is not associated with PD in vitamin D–nondeficient individuals (p > 0.8 in both data sets).
Conclusions:
Previous studies suggest that vitamin D deficiency is associated with PD and sustained opening of LVSCC contributes to the selective vulnerability of dopaminergic neurons in PD. Our data demonstrate that the association between genetic variations in CACNA1C and PD depends on vitamin D deficiency, providing one potential mechanism underlying the association between vitamin D deficiency and PD.
Parkinson disease (PD) is characterized by the loss of dopaminergic neurons in the substantia nigra (SN). Sustained opening of L-type voltage-sensitive calcium channels (LVSCC) in adult dopaminergic neurons in the SN has been suggested to cause excessive Ca2+ entry, leading to excessive mitochondrial stress and production of reactive oxygen species, rendering SN neurons vulnerable to cell death.1 In the brain, Cav1.2, encoded by CACNA1C, accounts for approximately 90% of LVSCC activity, with Cav1.3, encoded by CACNA1D, accounting for the remaining activity.2 Cav1.2 and Cav1.3 are antagonized by dihydropyridine (DHP) derivatives. Both animal and retrospective human studies support a protective role of DHP derivatives in PD development.3–9 Nonetheless, genetic variations in CACNA1C or CACNA1D have not been studied with PD risk.
As a complex disease, PD has both genetic and environmental risk factors that act individually or through complex interactions to influence disease pathogenesis.10–18 Recently, several studies, including ours,19 have reported that lower circulating vitamin D concentration is associated with the risk of PD.20–22 Vitamin D is known to exert its biological actions by regulating the expression of many genes, providing a plausible molecular mechanism for gene-environment interactions. Previous studies have shown that vitamin D treatment decreases CACNA1C messenger RNA expression through the vitamin D receptor (VDR) in primary neuronal cultures.23,24 We hypothesize that CACNA1C and CACNA1D would be good candidate genes for PD and that the association of SNPs in these genes with PD might be modulated by circulating vitamin D concentrations.
METHODS
Subjects.
Subjects from 2 genome-wide association studies (GWAS) were included in the study. The first GWAS was conducted by the Morris K Udall Parkinson Disease Research Center of Excellence (UDALL) at the University of Miami,25 and the second GWAS was conducted as part of the NeuroGenetics Research Consortium (NGRC).26 Participant enrollment and clinical assessment were described previously in detail.25,26 In brief, cases were ascertained by neurology clinics associated with UDALL (from 1997 to 2003) or NGRC (from 2003 to 2009). Controls were ascertained from community outreach efforts or were spouses of individuals with PD or Alzheimer disease (M.A.P.-V., Principal Investigator). All participants with stored plasma samples available for vitamin D assessment were included in the study. Participants in this study are non-Hispanic whites by self-report and confirmed by principal component analysis of population structure in the GWAS.
Standard protocol approvals, registrations, and patient consents.
All participants gave informed consent prior to these studies. All ascertainment protocols were approved by each contributing center's institutional review board.
Genetic markers.
The UDALL data set was genotyped using Illumina 610-quad BeadChip (Illumina, San Diego, CA).25 The NGRC data set was genotyped using Illumina HumanOmni1-Quad_v1-0_B BeadChip (Illumina).26 Final marker sets after quality control (described in detail in these previous publications) were used for the imputation of untyped SNPs separately in each of the 2 data sets. Imputation allowed us to obtain genetic information on additional variants through the genome for a more thorough investigation. Both data sets were imputed up to 38 million SNPs using IMPUTE2 and the 1000 Genomes reference panel (phase 1, March 2012).27 During quality control, SNPs that had low imputation quality (info score <0.4) and low minor allele frequency (less than 1%) were removed from further analysis. SNPs within the 5-kb flanking regions of the start and end of CACNA1C and CACNA1D were examined for interaction with vitamin D status and association with PD.
Plasma vitamin D measurement.
Stored plasma samples were analyzed using liquid chromatography tandem mass spectrometry at the Emory Clinical Translational Research Laboratory as described previously.19 In brief, the samples were analyzed using the methods outlined before.28 Three quality control samples (approximately 6, 21, and 62 ng/mL) were included in duplicate at the beginning and end of each run. The assay was linear up to 200 ng/mL and had a limit of detection of 1 ng/mL. Total imprecision ranged from 10.8% to 17.1%. The assay was validated using the NIST 972a standards. The laboratory participates in the Vitamin D External Quality Assessment Scheme proficiency scheme. For both the discovery and replication data sets, the samples were organized into multiple batches for vitamin D measurement: each batch included ∼220 samples with a balanced number of cases and controls, analyzed in random order with respect to affection status. Laboratory technicians were blinded to affection status.
Statistical analysis.
Logistic regression analysis was used to evaluate the association between vitamin D status and PD, adjusting for age at sampling, sex, and sampling season in each data set separately using the R software package. Vitamin D deficiency was defined as having total 25-hydroxyvitamin D (25[OH]D) <20 ng/mL, and vitamin D insufficiency was defined as having total 25(OH)D <30 ng/mL. A joint test of SNP main effect and SNP-vitamin D interaction effect was conducted using a 2 degrees-of-freedom likelihood-ratio test.29 This joint test compares a full model containing vitamin D deficiency, SNP dosage (additive genetic model), an SNP-vitamin D deficiency interaction term, and covariates (age at sampling, sex, and sampling season) to a restricted model with only vitamin D deficiency and covariates. Such an analysis will detect loci missed by main-effects analysis conducted in previous GWAS if the combined effect of the SNP effect and environmental interaction is stronger than the main effect of the SNP alone.
Besides a different genotyping platform, the 2 GWAS data sets are also different in distribution of vitamin D concentration, age, sex, and sampling season covariates (table 1). Therefore, joint tests were conducted in each data set separately. To achieve better statistical power while accommodating the heterogeneity between the 2 data sets, the joint test results in the 2 data sets were then combined using a meta-analysis approach. This meta-analysis used the joint meta-analysis extension of METAL.30 To adjust for multiple testing, simpleM was used to estimate the effective number of independent tests taking into account linkage disequilibrium (LD) between SNPs31; this estimate of independent tests was used to determine a significant threshold for each gene, adjusting for the number of independent tests. To unravel the nature of the interactions between SNP dosage and vitamin D deficiency, stratification analysis was performed to evaluate the associations between SNPs and PD risk in vitamin D-deficient and vitamin D-nondeficient strata separately. The use of prevalent cases could lead to potential survival bias. Sensitivity analysis was carried out by duration of disease to test the effect of considering disease duration in the case definition on the identified association, i.e., cases with shorter duration (“near-incident” cases with less than 3 years of duration) or longer duration (3 years or longer). Additional sensitivity analyses were performed by age at onset and sex to test whether the identified association is restricted to early or later age at onset of PD and whether it is sex specific.
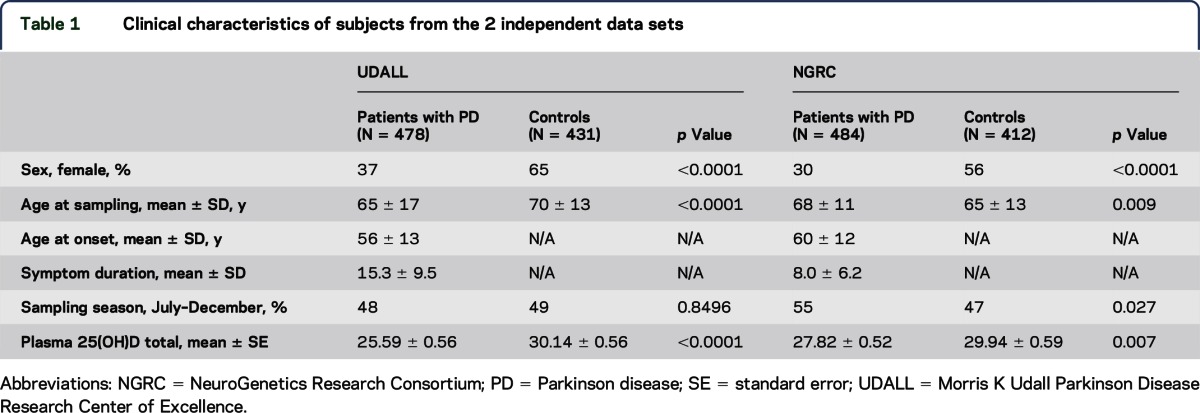
Table 1.
Clinical characteristics of subjects from the 2 independent data sets
To investigate the potential biological basis for the gene-vitamin D interaction at the most significant SNP, 1000 Genomes phase 3 data were used to identify all SNPs that are in LD within a 1 million base pair region surrounding the significant SNP. In addition, publicly available data from CHIP-Seq experiments with VDR32,33 were also used to evaluate the functional relevance of SNPs.
RESULTS
Characteristics of subjects in 2 data sets.
Table 1 displays the characteristics of participants from the 2 data sets. In both data sets, the percentage of women is lower in the PD cases than controls, consistent with prior observations of increased prevalence of PD in men. In the UDALL data set, the controls have older age at sampling than cases because of the intentional recruitment of controls at older ages to reduce the possibility of including controls who would develop PD after ascertainment. In the NGRC data set, the controls have younger age at sampling than cases. There is no difference in sampling season between cases and controls in UDALL; however, in NGRC, more PD cases were sampled between July and December (when higher vitamin D levels are usually observed for the same individual) than in controls. Owing to these differences in clinical characteristics and genotyping platforms between the 2 data sets, we elected to perform individual analyses in each data set separately.
Lower vitamin D concentrations are associated with PD in both data sets.
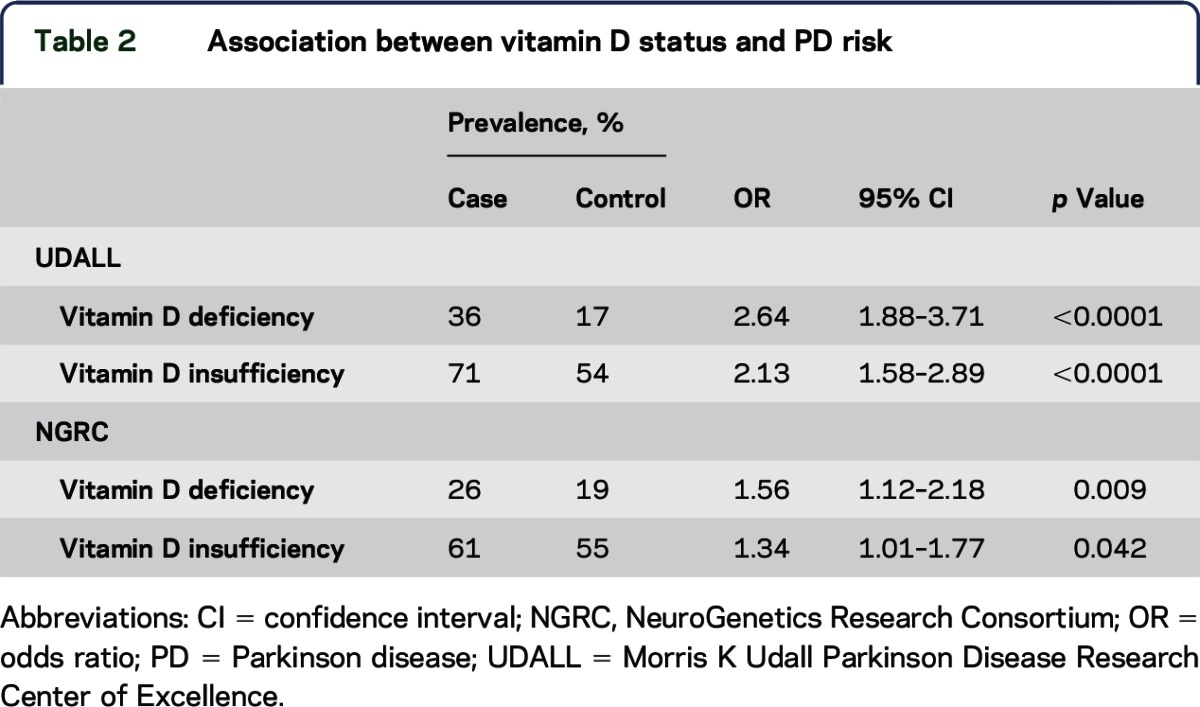
Total plasma 25(OH)D concentrations are significantly lower in PD cases than in controls in both data sets (table 1). Total 25(OH)D was also analyzed categorically, using the established clinical criteria for vitamin D deficiency (25[OH]D < 20 ng/mL) and vitamin D insufficiency (25[OH]D < 30 ng/mL). Cases with PD are significantly more likely than controls having vitamin D deficiency (25[OH]D < 20 ng/mL) and vitamin D insufficiency (25[OH]D < 30 ng/mL) in both data sets (table 2). In addition, a dosage effect was consistently observed in both data sets, with vitamin D deficiency more strongly associated with PD than was vitamin D insufficiency (odds ratio [OR] = 2.63 vs 2.13 in UDALL, OR = 1.56 vs 1.34 in NGRC).
Table 2.
Association between vitamin D status and PD risk
Analysis of interaction between vitamin D deficiency and SNPs in CACNA1C and CACNA1D.
Since the strongest association was found between vitamin D deficiency and PD, the initial gene-environment interaction analysis was focused on analyzing the association of PD with the joint effects of SNP dosage, and its interaction with vitamin D deficiency. In total, 1,244 SNPs in CACNA1C and 504 SNPs in CACNA1D were available for analysis. The effective number of independent tests after adjusting for LD is 274 and 175 in CACNA1C and CACNA1D, respectively. To maximize statistical power, we performed a meta-analysis across both data sets. The strongest evidence for a joint effect was found at SNP rs34621387 (meta-joint test p = 7.5 × 10−5, Bonferroni-adjusted p = 0.02) in the third intron of CACNA1C (figure, A). Within CACNA1D, the lowest meta-joint test p value is 0.02, which would not remain significant after adjusting for multiple testing (figure, B). To further evaluate the nature of the interaction, associations between rs34621387 and PD were evaluated in individuals who were vitamin D deficient or vitamin D nondeficient separately in each data set. This analysis revealed that the association of rs34621387 and PD depends on vitamin D concentration: allele G is associated with PD in the vitamin D-deficient stratum in both data sets (OR = 2.0, p = 0.002 and OR = 2.1, p = 0.002) but is not associated with PD in vitamin D-nondeficient stratum in both data sets (p > 0.8) (table 3). Sensitivity analysis suggested that this pattern of association (restricted to vitamin D-deficient individuals) is not affected by age at onset, disease duration, or sex (table e-1 at Neurology.org/ng).
Figure. Joint tests of SNPs and vitamin D deficiency in CACNA1C and CACNA1D.

Each point represents a common variant (minor allele frequency >5%) in CACNA1C (A) and CACNA1D (B). In total, 1,244 SNPs in CACNA1C and 504 SNPs in CACNA1D were analyzed. A joint test of SNP main effect and SNP-vitamin D interaction effect was conducted using a 2 degrees-of-freedom likelihood-ratio test that compares a full model containing vitamin D deficiency, SNP dosage, an SNP-vitamin D deficiency interaction term, and covariates to a restricted model with only vitamin D deficiency and covariates. Each circle represents the p value of the meta-analysis of the joint tests using both UDALL and NGRC data sets. The most significant SNP in each gene is displayed as a purple diamond. The color of the circles indicates LD (r2) between each SNP and the top SNP. The blue line displays the recombination rates estimated from phase 2 HapMap. NGRC = NeuroGenetics Research Consortium; SNP = single nucleotide polymorphism; UDALL = Morris K Udall Parkinson Disease Research Center of Excellence.
Table 3.
Association between rs34621387 and PD in the presence or absence of vitamin D deficiency

Functional relevance of the intronic SNP rs34621387 in CACNA1C.
No SNPs in substantial LD (r2 > 0.2) with rs34621387 were found to be located in a known VDR binding site, as delimited by reported Chip-Seq experiments.32,33 We used RegulomeDB34 and HaploReg35 to determine whether rs34621387 or its proxies are located within any other regulatory elements. This analysis found that rs34992881 (r2 = 0.76 with rs34621387) is located within a region displaying epigenomic signatures for chromatin enhancer, which include histone H3 lysine 4 trimethylation (H3K4me3), H3K27 acetylation (H3K27ac), and H3K9ac. In addition, the enhancer signatures are only detected in some of the tissues or cell types surveyed by the Roadmap Epigenome project, suggesting the possibility of tissue- or cell-specific regulation of gene expression of CACNA1C.
DISCUSSION
It is well established that vitamin D exerts its main biological actions by regulating the expression of a large array of genes and one of them is CACNA1C.23,24,32,33 Using 2 case-control data sets where vitamin D deficiency is significantly associated with PD, we found that vitamin D status significantly modified the association between SNPs in CACNA1C and PD: genetic effects of CACNA1C only manifest in individuals with vitamin D deficiency, but not in individuals who are not vitamin D deficient. As such, the effect of CACNA1C in PD would have become difficult to detect in a sample where a substantial portion of individuals have sufficient vitamin D levels, if only SNP main effects were examined. Our study demonstrates the importance of incorporating environmental exposure to understand the genetic underpinning for interindividual phenotypic variance in PD.
Using existing databases, no definitive evidence for regulatory potential at rs34621387 or its proxies was observed. Therefore, the biological mechanism underlying the association of rs34621387 and PD in the presence of vitamin D deficiency is currently not understood. Interestingly, SNP rs1006737, in the same intron (intron 3) as rs34621387 in CACNA1C, has been widely replicated in association with schizophrenia and other neuropsychiatric conditions, making it one of the most robust findings in psychiatric genetics.36–38 Similar to rs34621387, no compelling evidence for regulatory function was initially found for rs1006737 or proxies, using existing databases. Subsequent studies revealed that SNPs in LD with rs1006737 interact with the proximal promoter region and direct allelic-specific expression of CACNA1C in human induced pluripotent stem cell-derived neurons, providing a molecular basis for the SNP-phenotype association.39,40 SNP rs1006737 and rs34621387 are not in strong LD with each other (r2 < 0.1). Whether rs34621387 confers genetic risk to PD by directing allelic-specific expression of CACNA1C, in a similar fashion as rs1006737 in schizophrenia, will require further, more in-depth functional studies.
Previous studies have shown that vitamin D inhibits the expression of CACNA1C.23,24 One possible mechanism underlying the observed interaction is that high vitamin D levels effectively suppress LVSCC expression and thus reduce the activity of LVSCC in the dopaminergic neurons in SN, masking the effect of rs34621387.
It has been proposed that Cav1.3, encoded by CACNA1D, might be particularly responsible for the selective death of dopaminergic neurons in the SN based on the observation that Cav1.3 channels open at relatively hyperpolarized membrane potentials, which makes them more suitable for pacemaking.1 We did not find any evidence for association between genetic variants in CACNA1D and PD, even after taking into account vitamin D status, which is consistent with the lack of evidence suggesting that CACNA1D is regulated by vitamin D.23,24 Our study, however, does not exclude the possibility that genetic variations in CACNA1D are involved in PD pathogenesis as our study is limited by a modest sample size, a focus on common variants, and interactions with vitamin D concentration. In addition, we used an ethnically homogeneous population (i.e., non-Hispanic whites) to minimize confounding by population substructure. Additional studies are needed to assess generalizability to other populations.
As in other case-control association studies of vitamin D concentration and PD, our results could reflect “reverse causation,” i.e., lower vitamin D concentrations in patients with PD are a consequence of the disease (lower levels of sun exposure due to reduced mobility and less time spent outdoors) rather than a cause. However, a previous longitudinal study demonstrated that lower vitamin D levels at baseline are associated with increased risk of PD during a 29-year follow-up20; a second study showed that patients with early, nondisabling PD and relatively normal mobility have lower vitamin D levels21; and our own work demonstrated that lower vitamin D2, a form of vitamin D that is obtained independently from sunlight exposure, is associated with PD, as is vitamin D3 (the form of vitamin D that is mainly obtained from sunlight exposure).19
Our data demonstrate that genetic variations in CACNA1C and vitamin D deficiency act together to increase PD risk. This study also highlights the importance of incorporating environmental exposure to identify genetic factors influencing interindividual phenotypic variance in PD.
Supplementary Material
ACKNOWLEDGMENT
Some of the samples used in this study were collected while the Udall Parkinson Disease Research Center of Excellence was at Duke University. The authors are thankful to Dr. D. James Surmeier for insightful discussion of the manuscript. The authors are grateful for the individuals and families who participated in the study.
GLOSSARY
- 25(OH)D
25-hydroxyvitamin D
- DHP
dihydropyridine
- GWAS
genome-wide association studies
- LD
linkage disequilibrium
- LVSCC
L-type voltage-sensitive calcium channel
- NGRC
NeuroGenetics Research Consortium
- PD
Parkinson disease
- SN
substantia nigra
- UDALL
Morris K Udall Parkinson Disease Research Center of Excellence
- VDR
vitamin D receptor
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
L.W. and W.K.S. conceived and designed the experiments. J.L.H., C.P.Z., H.P., M.A.P.-V., and J.M.V. were responsible for collecting biosamples and obtaining genotype data. M.L.E. and J.C.R. were responsible for measuring vitamin D concentrations. L.M., G.W.B., and E.R.M. imputed genotype data and performed statistical analyses. L.W., W.K.S., and J.M.V. interpreted the data. L.W. wrote the manuscript. W.K.S. and J.M.V. edited the manuscript. All authors read and approved the final manuscript.
STUDY FUNDING
The majority of this work was supported by NIH P50 NS071674 (J.M.V., W.K.S., L.W.). This work was also supported by grants from NIH P50 NS062684, R01NS065070, R01 NS036960, R01 NS067469, and the Department of Veterans Affairs 1I01BX000531.
DISCLOSURE
Dr. Wang has received research support from National Institute of Neurological Disorders and Stroke (2P50NS071674-02, 1R01NS065114-01, 2R01NS040807-09) and NHLBI (1R01HL102487-01A1). Ms. Maldonado has received research support from the NIH (NS071674) and the National Parkinson Foundation. Dr. Beecham has received research support from and receives funding from the NIH (R01HL102487, P50NS071674, 5454NS065712, R011NS075764, U01AG032984, R01AG027944) and the Department of Defense (W81XWH-14-1-0097). Dr. Martin serves on the editorial board of Frontiers in Statistical Genetics and Methodology and holds a patent Test for Linkage and Association in General Pedigrees: The Pedigree Disequilibrium Test (US Patent No. 6697739). Dr. Evatt has served on the scientific advisory boards for Cynapsus Therapeutics Inc. and on the editorial boards of The American Journal of the Medical Sciences (AJMS) and Advances in Neuroscience. Dr. Ritchie has served on the scientific advisory boards for Beckman Coulter, Inc. and Sebia, Inc. and routinely performs vitamin D assays in his laboratory. Dr. Haines has received funding for travel or speaker honoraria from Novartis Vision Research Day; serves on the editorial boards of NeuroGenetics, Current Protocols in Human Genetics, and Human Molecular Genetics; has received publishing royalties from Current Protocols in Human Genetics (John Wiley & Sons); has received research support from NIH/NIA Consortium for Alzheimer's Sequence Analysis (CASA) (R01EY023164) NIH/NEI (R01EY020928, R01EY012118, R01EY022310, R01EY022305); and receives royalty payments for the use of APOE in clinical diagnostics from Athena Diagnostics. Dr. Zabetian has served on the Scientific Advisory Council for the Lewy Body Dementia Association; and has received research support from NIH/National Institute of Neurological Disorders and Stroke (P50 NS062684, U01 NS082137, R01NS065070, P50 NS062684, R01 NS036960, R01 NS057567, R01 AG033398), the Department of Veterans Affairs (1I01BX000531), Parkinson's Disease Foundation, and American Parkinson Disease Association. Dr. Payami has received research support from the NIH (R01 NS036960, R01 NS067469). Dr. Pericak-Vance reports no disclosures. Dr. Vance has received funding for travel or speaker honoraria from the University of Alaska, Fairbanks, and NETPR Department of Defense; serves on the editorial boards of American Journal of Neurodegenerative Diseases and Neurology Genetics; holds patents for the method of detecting Charcot-Marie-Tooth disease type 2A, TRPC6 involved in glomerulonephritis, and methods for identifying an individual at increased risk of developing coronary artery disease; has received research support from NIH/National Institute of Neurological Disorders and Stroke (5P50NS071674, 1R25NS090624) and the Hussman Foundation; and receives royalty payments from Duke University. Dr. Scott has served on the scientific advisory boards for the PSG Scientific Review Committee; serves on the editorial boards of Frontiers in the Genetics of Aging and Journal of Clinical Investigation; holds patent regarding the use of genetic data for risk assessment in age-related macular degeneration (Duke University); and has received research support from the NIH (R01 NS071674, R01 AI068804, R01 EY12118, R01 EY023164, T32 EY023194), Florida Biomedical Research Program, and American Health Assistance Foundation. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Surmeier DJ, Schumacker PT. Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J Biol Chem 2013;288:10736–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sinnegger-Brauns MJ, Huber IG, Koschak A, et al. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 2009;75:407–414. [DOI] [PubMed] [Google Scholar]
- 3.Chan CS, Guzman JN, Ilijic E, et al. “Rejuvenation” protects neurons in mouse models of Parkinson's disease. Nature 2007;447:1081–1086. [DOI] [PubMed] [Google Scholar]
- 4.Wang R, Ma Z, Wang J, Xie J. L-type Cav1.2 calcium channel is involved in 6-hydroxydopamine-induced neurotoxicity in rats. Neurotox Res 2012;21:266–270. [DOI] [PubMed] [Google Scholar]
- 5.Kupsch A, Sautter J, Schwarz J, Riederer P, Gerlach M, Oertel WH. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res 1996;741:185–196. [DOI] [PubMed] [Google Scholar]
- 6.Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis 2011;43:364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosharov EV, Larsen KE, Kanter E, et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009;62:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasternak B, Svanstrom H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol 2012;175:627–635. [DOI] [PubMed] [Google Scholar]
- 9.Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 2010;67:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009;18:R48–R59. [DOI] [PubMed] [Google Scholar]
- 11.Nalls MA, Pankratz N, Lill CM, et al. Large scale meta analysis of genome-wide association data in Parkinson's disease reveals 6 novel risk loci. Nat Genet 2014;46:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elbaz A, Clavel J, Rathouz PJ, et al. Professional exposure to pesticides and Parkinson disease. Ann Neurol 2009;66:494–504. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Jacobs E, Schwarzschild MA, et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol 2005;58:963–967. [DOI] [PubMed] [Google Scholar]
- 14.Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol 2002;52:276–284. [DOI] [PubMed] [Google Scholar]
- 15.Scott WK, Zhang F, Stajich JM, Scott BL, Stacy MA, Vance JM. Family-based case-control study of cigarette smoking and Parkinson disease. Neurology 2005;64:442–447. [DOI] [PubMed] [Google Scholar]
- 16.Hancock DB, Martin ER, Stajich JM, et al. Smoking, caffeine, and nonsteroidal anti-inflammatory drugs in families with Parkinson's disease. Arch Neurol 2007;64:576–580. [DOI] [PubMed] [Google Scholar]
- 17.Hamza TH, Chen H, Hill-Burns EM, et al. Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson's disease modifier gene via interaction with coffee. PLoS Genet 2011;7:e1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hancock DB, Martin ER, Fujiwara K, et al. NOS2A and the modulating effect of cigarette smoking in Parkinson's disease. Ann Neurol 2006;60:366–373. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Evatt ML, Maldonado LG, et al. Vitamin D from different sources is inversely associated with Parkinson disease. Mov Disord 2015;30:560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knekt P, Kilkkinen A, Rissanen H, Marniemi J, Saaksjarvi K, Heliovaara M. Serum vitamin D and the risk of Parkinson disease. Arch Neurol 2010;67:808–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol 2008;65:1348–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding H, Dhima K, Lockhart KC, et al. Unrecognized vitamin D3 deficiency is common in Parkinson disease: Harvard biomarker study. Neurology 2013;81:1531–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci 2001;21:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gezen-Ak D, Dursun E, Yilmazer S. The effects of vitamin D receptor silencing on the expression of LVSCC-A1C and LVSCC-A1D and the release of NGF in cortical neurons. PLoS One 2011;6:e17553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards TL, Scott WK, Almonte C, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 2010;74:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet 2010;42:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lensmeyer G, Poquette M, Wiebe D, Binkley N. The C-3 epimer of 25-hydroxyvitamin D(3) is present in adult serum. J Clin Endocrinol Metab 2012;97:163–168. [DOI] [PubMed] [Google Scholar]
- 29.Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Hum Hered 2007;63:111–119. [DOI] [PubMed] [Google Scholar]
- 30.Manning AK, LaValley M, Liu CT, et al. Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP x environment regression coefficients. Genet Epidemiol 2011;35:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X, Starmer J, Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol 2008;32:361–369. [DOI] [PubMed] [Google Scholar]
- 32.Ramagopalan SV, Heger A, Berlanga AJ, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res 2010;20:1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer MB, Benkusky NA, Lee CH, Pike JW. Genomic determinants of gene regulation by 1,25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. J Biol Chem 2014;289:19539–19554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeates TO. Determination of the correct reference frame from an atomic coordinate list. Acta Crystallogr A 1990;46:625–626. [DOI] [PubMed] [Google Scholar]
- 36.Erk S, Meyer-Lindenberg A, Schnell K, et al. Brain function in carriers of a genome-wide supported bipolar disorder variant. Arch Gen Psychiatry 2010;67:803–811. [DOI] [PubMed] [Google Scholar]
- 37.Ripke S, O'Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 2013;45:1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ripke S, Sanders AR, Kendler KS, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011;43:969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roussos P, Mitchell AC, Voloudakis G, et al. A role for noncoding variation in schizophrenia. Cell Rep 2014;9:1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshimizu T, Pan JQ, Mungenast AE, et al. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol Psychiatry 2015;20:284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.