

Abstract
Background:
Coronary heart disease (CHD) is a multifactorial disease and is thought to have a polygenic basis. Apolipoprotein E (APOE) gene is one such candidate with its common ε2/ε3/ε4 polymorphism in CHD. In recent years, numerous case-control studies have investigated the relationship of APOE polymorphism with CHD risk. However, the results are confusing.
Methods:
To clarify this point, we undertook a meta-analysis based on 14 published studies including 5746 CHD cases and 19,120 controls. Crude odds ratios (ORs) with 95% confidence intervals (CIs) were assessed for association using a random-effects or fixed-effects model using STATA version 10 (StataCorp LP, College Station, TX, USA).
Results:
Overall, the analysis showed that carriers of APOE ε2 allele decreased risk for CHD (ε2 allele vs. ε3 allele: OR = 0.82, 95% CI: 0.75–0.90, P < 0.001; ε2 carriers vs. ε3 carriers: OR = 0.81, 95% CI: 0.73–0.89, P < 0.001), compared with those carrying ε3 allele, especially in Caucasian population. However, those with ε4 allele had a significant increased risk for CHD (ε4 allele vs. ε3 allele: OR = 1.34, 95% CI: 1.15–1.57, P < 0.001), especially in Mongoloid population. Potential publication bias was observed in the genetic model of ε4 versus ε3, but the results might not be affected deeply by the publication bias. When we accounted for publication bias using the trim and fill method, the results were not materially alerted, suggesting the stability of our results.
Conclusions:
Taken together, our meta-analysis supported a genetic association between APOE gene and CHD. ε4 increased the risk of CHD, whereas ε2 decreased the risk of CHD.
Keywords: Apolipoprotein E, Coronary Heart Disease, Meta-analysis, Polymorphism
INTRODUCTION
Coronary heart disease (CHD), a complex disease, is the most important cause of mortality and morbidity worldwide. CHD accounts for 7.3 million death in 2008.[1] CHD is often initially detected from clinical manifestations such as myocardial infarction, angina, or sudden death due to artery occlusion.
The exact etiology underlying CHD is still unclear. Many environmental and genetic factors are identified to implicate the development of CHD. The environmental factors, such as tobacco smoking, physical activity and alcohol intake, and dietary fat consumption, in particular, the amount of saturated fatty acids, have been considered as the main risk factors for CHD.[2] In recent years, several susceptibility genes have been proposed to manifest an association with CHD.
Among the genetic determinants that have been more widely studied related to CHD risk is the common polymorphism in the apolipoprotein E (APOE) gene.[3] APOE is located on chromosome 19q13.2, with 3 common isoforms (ε2, ε3 and ε4) and 6 genotypes (E2E2, E2E3, E3E3, E2E4, E3E4, and E4E4).[4] The alleles differ at two single-nucleotide polymorphisms, giving rise to different amino acids at protein positions 112 and 158; cysteine at 112 and arginine at 158 for ε3, two cysteine for ε2 and two arginine for ε4. ε3 accounting for approximately 50–90% in all population is considered to be the wild type allele whereas alleles ε2 and ε4 are considered variants which vary between 5-35% and 1-15%, respectively.[5,6] APOE is highly polymorphic and plays an imperative role in endogenous lipoprotein metabolism and tissue distribution. It is present in lipoprotein particles and mediates lipoprotein through binding to the low-density lipoprotein (LDL) and lipoprotein remnant receptors. Defects in the APOE protein could diminish its ability to bind to the receptors, which may lead to an elevated blood cholesterol level.[7] It has been well-established that the presence of the APOE ε4 allele is associated with higher levels of total and LDL serum cholesterol, whereas the presence of the APOE ε2 allele is associated with the lower effect with reference to cholesterol effects from the ε3 allele.[8] These results suggest that APOE is associated with CHD.
To date, numerous epidemiological studies[2,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24] have recently focused on the association between APOE ε2/ε3/ε4 polymorphism and CHD risk. However, results in different studies are inconsistent. For example, Singh et al. found a significant association of ε4 polymorphism, especially E3/E4 genotype with CHD, along with high-density lipoprotein (HDL) and LDL concentrations. However, Marrzoq et al. suggested APOE genotypes were not associated with CHD. Moreover, there was no significant difference between the mean of triglyceride and HDL levels among different APOE genotypes. Therefore, the present meta-analysis is designed to derive a more precise estimation.
METHODS
Identification of studies
Relevant studies were identified from the following electronic databases: PubMed, EMBASE, Web of Science, CNKI, CBM, Wanfang, VIP and hand searching of relevant journals and the reference lists of retrieved articles, with the last updated search performed before September 2012. The search was performed using various combinations of keywords including (“APOE”) and (“polymorphism” or “variant” or “mutation”) and (“CHD”) without language limitation. The references of the selected papers were also checked by hand-search for other potential articles that possibly have been missed in the initial search. To be eligible for inclusion in this meta-analysis, a study must meet the following criteria: (1) APOE gene polymorphism in CHD; (2) sufficient data for calculating an odds ratio (OR) with its 95% confidence interval (CI). When duplicate articles were published, only the newest or most informative single article was selected. Study was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement.[25]
Data extraction
All the data were extracted independently by two reviewers with the standard protocol. Potential disagreements were documented and resolved by discussion with a third reviewer. The following characteristics were extracted from each study: First author, year of publication, ethnicity of the study population, sample size, genotype method; source of case and control subjects.
Quality assessment
The quality of the included studies was assessed using the following criteria modified from the previous report, and studies with four or five adequate items were defined as high quality.
Description of the case and control groups (not described, inadequate, adequate).
Assessment and validation of CHD in the patients (not described, inadequate, adequate). Adequate validation would include confirmation by coronary angiography; inadequate validation would include recollection of the patient as the only clinical evidence without examination.
Description of the laboratory procedures for the genotyping (not described, inadequate, adequate).
Confirmation of Hardy-Weinberg equilibrium (HWE) in the genotype distribution (not described, inadequate, adequate). Since HWE is a surrogate to assess study quality, and the effect of HWE is associated with problems in the design and conduct of genetic association studies, studies with departures from HWE were defined as low-quality studies.
Equal assessment for confounding factors in the case and control groups (not described, inadequate, adequate).
Statistical analyses
Allele frequencies at genetic polymorphisms from each study were determined by the allele counting method. HWE for the controls in each study was assessed by Chi-square interval or Fisher exact test. The combined OR and 95% CI was used to compare contrasts of genotypes and alleles between cases and controls. Statistical heterogeneity was measured using the Chi-square based Q statistic (P < 0.10 was considered statistically significant heterogeneity). We also quantified the effect of heterogeneity using I2 statistic:[26]
I2=100% × (Q – df)/Q
I2 values of 25%, 50%, and 75% were defined as low, moderate, and high estimates, respectively. Either a random-effects model (DerSimonian-Laird method)[27] or fixed-effects model (Mantel-Haenszel method)[28] was used to calculate pooled effect estimates in the presence or absence of heterogeneity, respectively.
Finally, potential publication bias was evaluated through funnel plot visual analysis and with the Begg's and Egger's tests.[29,30] A P < 0.05 was considered statistically significant. For the possible publication bias, we used trim and fill method to evaluate the influence to the results. All statistical analyses were performed by STATA version 10 (StataCorp LP, College Station, TX, USA).
RESULTS
Studies included in the meta-analysis
The flow chart of literatures selection was provided in Figure 1. There were 161 papers relevant to the search words. Through screening the title and reading the abstract, 135 papers were excluded. Of these, the type of 26 papers was review. Twenty-two papers were not related to the genetic polymorphism. Twenty-seven papers were not about APOE gene polymorphism. Fifty-four papers were not about CHD. Six papers were published in other language (Russian: 2; Spanish 2; Portuguese: 1; Norwegian: 1). Twenty-six studies were left for full publication review. Of the 26 studies, 2 studies did not have a control group, 5 did not provide enough information on genotype distribution, leaving 19 studies for more detailed assessment. Two studies[31,32] were excluded because of overlap data. Four studies[13,15,19,24] excluded were deviated from HWE. One study[17] contained data on two different groups (Whites and African Americans). Thus, in accordance with the objectives of the present study, we analyzed them independently. As a result, a total of 14 studies from 13 independent articles including 5746 CHD cases and 19,120 controls were subjected to the final analysis. The gene discussed in all included studies was consistent with target gene. Of the 14 studies, 6 studies were conducted in Mongoloid population, 5 were conducted in Caucasians population, and 3 in other population. Characteristics of studies included in the current meta-analysis are shown in Table 1.
Figure 1.
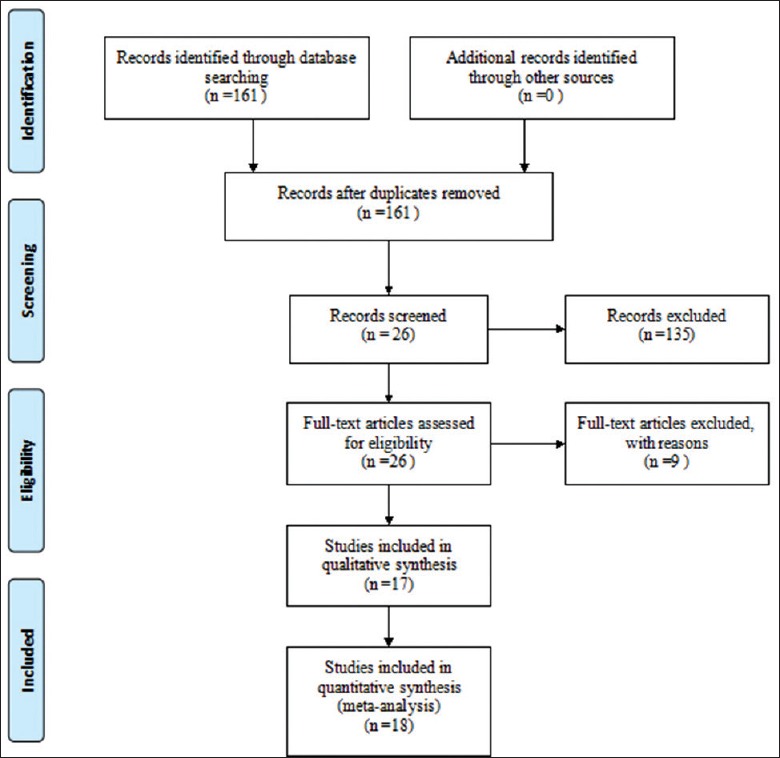
The flow diagram of study selection.
Table 1.
Basic information of studies included in the meta-analysis
| Author | Year | Population | Sample size (n) | Characteristics of the included studies | Genotype method | HWE for controls (P) | Case source | Control source | Quality assessment | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | CHD group | Control group | ||||||||
| Ou et al. | 1998 | Japanese | 214 | 310 | 86.5% male, age: 55.4 ± 7.4 years BMI: 23.2 ± 2.6 | 86% male, Age: 54.5 ± 5.9 years BMI: 23.9 ± 2.8 | PCR-RFLP | 0.095 | Hospital (A hospital in Tsuchiura city) | Hospital (A hospital in Tsuchiura city) | High |
| Cao et al. | 1999 | Chinese | 78 | 85 | NA | NA | PCR-RFLP | 0.789 | Hospital (Han population in northeast China) | NA (Han population in northeast China) | Low |
| Yang et al. | 2001 | Chinese | 204 | 136 | CHD1 age: 45.4 ± 6.8 years, BMI: 23.8 ± 2.6 CHD2 age: 70.1 ± 6.4 years, BMI: 24.2 ± 2.3 | Age: 52.5 ± 12.8 years BMI: 34.1 ± 2.4 | PCR-RFLP | 0.491 | Hospital | Population (A local company) | High |
| Freitas et al. | 2002 | Caucasian | 640 | 624 | Age: 44 ± 4 years 88% Male | Age: 40 ± 6 years 52% Male | PCR-RFLP | 0.767 | Hospital | Population (local community) | High |
| Wang et al. | 2004 | Chinese | 186 | 350 | Age: 65.0 ± 10.6 years | Age: 63.6 ± 8.3 years | Multi- ARMS | 0.431 | Hospital (Han population in Hubei) | Hospital (Han population in Hubei) | High |
| Ouyang et al. | 2005 | Chinese | 200 | 100 | Male/female: 105/95 Age: 63.6 ± 5.8 years BMI: 15.1 - 34.3kg/m2 | Male/female: 55/45 Age: 62.1 ± 7.8 years | PCR-RFLP | 0.399 | Hospital (Han population in Beijing and Hebei) | Volunteers (Han population in Beijing and Hebei) | High |
| Volcik et al. | 2006 | Whites/African Americans | 1017/534 | 8287/5840 | NA | NA | TaqMan | 0.807/0.235 | Population (four US communities) | Population (four US communities) | High |
| Akanji et al. | 2007 | Kuwait | 50 | 65 | Age: 54.0 (40.0-76.0) years BMI: 28.1 (22.5-38.1) | Age: 39.0 (25.0-60.0) years BMI: 29.8 (21.0-41.7) | PCR-RFLP | 0.441 | Hospital (Kuwaiti Arab) | Volunteers (Kuwaiti Arab) | High |
| Singh et al. | 2008 | Indians | 193 | 150 | Age: 54.94 ± 11.43 years Male/female: 74.6%/25.4% | Age: 53.42 ± 12.47 years Male/female: 70%/30% | PCR | 0.327 | Hospital (northwest India) | Volunteers (northwest India) | High |
| Kolovou et al. | 2009 | Athens | 359 | 248 | Normoweight (98): Age: 61 ± 11 years, BMI: 24 ± 1 Overweight (189): Age: 59 ± 10 years, BMI: 27 ± 1 Obese (72): Age: 59 ± 10 years, BMI: 32 ± 2 | Age: 30 ± 12 years BMI: 22 ± 3 | PCR-RFLP | 0.133 | Hospital (Greek Caucasian) | Volunteers (Greek Caucasian) | High |
| Marrzoq et al. | 2011 | Asians | 69 | 68 | Male/female: 45/24 | Male/female: 33/35 | PCR-RFLP | 0.708 | Hospital (Gaza Strip) | NA (Gaza Strip) | High |
| Corella et al. | 2011 | Spanish | 534 | 1123 | Male/female: 424/110 Age: 54.0 ± 7.3 years BMI: 29.2 ± 3.5 | Male/female: 884/239 Age: 53.7 ± 7.2 years BMI: 28.7 ± 3.7 | Taqman | 0.169 | Volunteers (Spanish EPIC cohort) | Volunteers (Spanish EPIC cohort) | High |
| Gustavsson et al. | 2012 | Sweden | 1735 | 4654 | NA | 2642 women | PCR-SPM | 0.766 | Population (INTERGENE and SHEEP) | Population (INTERGENE and SHEEP) | High |
NA: Not available; HWE: Hardy-Weinberg equilibrium; PCR-RFLP: Polymerase chain reaction with restriction fragment length polymorphism; PCR-SPM: Polymerase chain reaction with solid-phase minisequencing; BMI: Body mass index; CHD: Coronary heart disease.
According to the quality criteria, there were thirteen studies with high quality, and only one with low quality.
Main results of meta-analysis
A summary of meta-analysis findings regarding the relation between the APOE ε2/ε3/ε4 polymorphism is presented in Table 2. Forest plots are shown in Figures 2–5.
Table 2.
Summary estimates of the association between APOE gene polymorphism and CHD: Overall analysis and subgroup analysis
| Polymorphism | Subgroup | Number of studies | Test of association | Test of heterogeneity | ||||
|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | PZa | Modelb | I2 (%) | PQc | |||
| ε2 allele versus ε3 allele | All | 14 | 0.82 | 0.75–0.90 | <0.001 | F | 27.1 | 0.164 |
| High quality studies | 13 | 0.84 | 0.77–0.91 | <0.001 | F | 33.9 | 0.111 | |
| Race | ||||||||
| Mongoloid | 6 | 1.07 | 0.83–1.37 | 0.611 | F | 0.0 | 0.848 | |
| Caucasian | 5 | 0.71 | 0.63–0.81 | <0.001 | F | 0.0 | 0.893 | |
| Others | 3 | 0.96 | 0.84–1.09 | 0.488 | F | 0.0 | 0.739 | |
| ε4 allele versus ε3 allele | All | 14 | 1.34 | 1.15–1.57 | <0.001 | R | 71.3 | <0.001 |
| High quality studies | 13 | 1.28 | 1.12–1.46 | <0.001 | R | 73.1 | <0.001 | |
| Race | ||||||||
| Mongoloid | 6 | 1.97 | 1.61–2.41 | <0.001 | F | 0.0 | 0.951 | |
| Caucasian | 5 | 1.04 | 0.97–1.12 | 0.271 | F | 0.0 | 0.939 | |
| Others | 4 | 1.11 | 1.02–1.21 | 0.018 | R | 77.7 | 0.011 | |
| ε2 carriers versus ε3 carriers | All | 14 | 0.81 | 0.73–0.89 | <0.001 | F | 12.8 | 0.313 |
| High quality studies | 13 | 0.83 | 0.76–0.90 | <0.001 | F | 21.8 | 0.223 | |
| Race | ||||||||
| Mongoloid | 6 | 1.01 | 0.79–1.29 | 0.927 | F | 0.0 | 0.877 | |
| Caucasian | 5 | 0.72 | 0.64–0.81 | <0.001 | F | 0.0 | 0.838 | |
| Others | 3 | 0.94 | 0.82–1.07 | 0.348 | F | 0.0 | 0.630 | |
| ε4 carriers versus ε3 carriers | All | 14 | 1.29 | 1.12–1.48 | <0.001 | R | 60.0 | 0.002 |
| High quality studies | 13 | 1.21 | 1.08–1.35 | 0.0001 | R | 62.3 | 0.001 | |
| Race | ||||||||
| Mongoloid | 6 | 1.76 | 1.44–2.15 | <0.001 | F | 0.0 | 0.933 | |
| Caucasian | 5 | 1.05 | 0.98–1.13 | 0.147 | F | 0.0 | 0.978 | |
| Others | 3 | 1.08 | 0.99–1.18 | 0.065 | R | 73.1 | 0.024 | |
aZ test used to determine the significance of the overall OR. PZ < 0.05 was considered to be significant; bF: Fixed-effects model; R: Random-effects model; cChi-square Q statistic test used to assess the heterogeneity in each group. PQ > 0.1, Fixed-effects model was used, PQ < 0.1, Random-effects model was used. OR: Odds ratio; CI: Confidence interval; CHD: Coronary heart disease; APOE: Apolipoprotein E.
Figure 2.
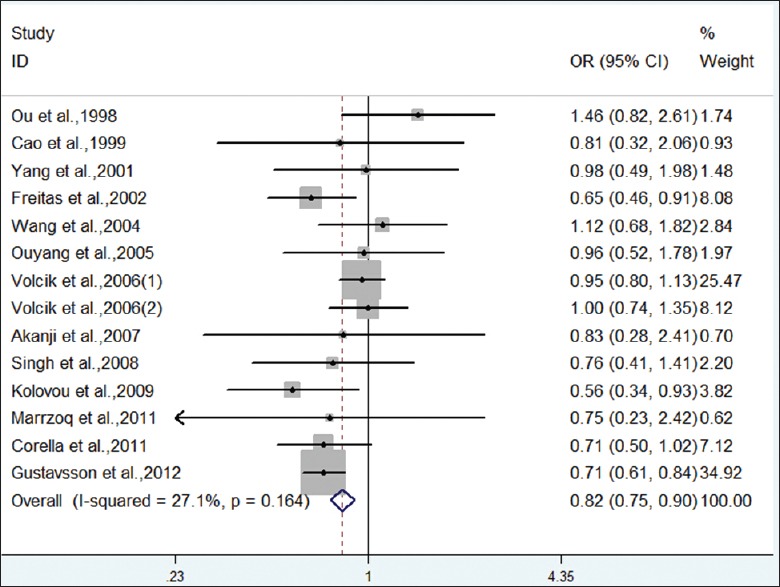
Forest plot on the association between ε2 allele (vs. ε3 allele) and CHD.
Figure 5.
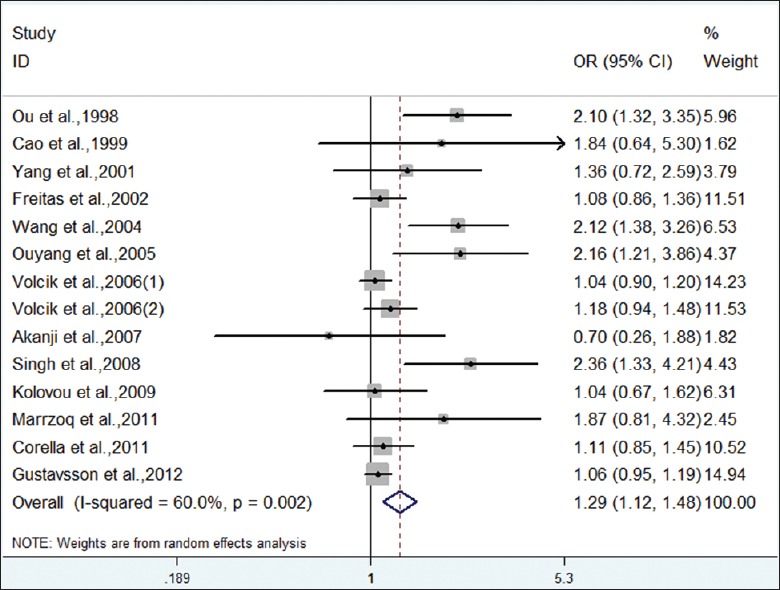
Forest plot on the association between ε4 carriers (vs. ε3 carriers) and CHD.
Figure 3.
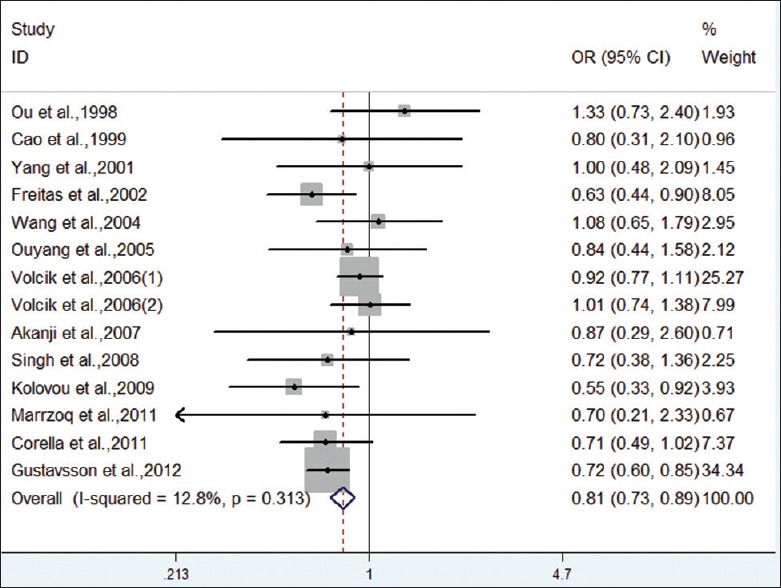
Forest plot on the association between ε2 carriers (vs. ε3 carriers) and CHD.
Figure 4.

Forest plot on the association between ε4 allele (vs. ε3 allele) and CHD.
Meta-analysis showed a decreased risk for the ε2 allele compared with the ε3 allele in overall population (OR = 0.82, 95% CI: 0.75–0.90, P < 0.001), without heterogeneity among studies (I2 = 27.1%, P = 0.164). When stratified by race, the pooled OR was just statistically significant in Caucasian population, not in Mongoloid and others. We also compared the model of ε2 carriers versus ε3 carriers. Under a fixed-effect model, the pooled OR for ε2 carriers versus ε3 carriers was 0.81 (95% CI: 0.73–0.89, P < 0.001). Analysis after stratification by race indicated significant association with CHD in Caucasian population. Therefore, carriers of APOE ε2 allele had a decreased risk for CHD, especially in Caucasians.
Meta-analysis showed an increased risk for the ε4 allele compared with the ε3 allele in overall population (OR = 1.34, 95% CI: 1.15–1.57, P < 0.001) with significant inter-study heterogeneity (I2 = 71.3%, P < 0.001). When stratified by race, heterogeneity was not detected. The combined OR obtained using a fixed-effect model was significant in Mongoloid population. Caucasians failed to find an association. Similarly, for genotype contrast of ε4 carriers versus ε3 carriers, a significant association was noticed in overall population (OR = 1.29, 95% CI: 1.12–1.48, P < 0.001). Analysis after stratification by race indicated a significant association with CHD risk in Mongoloid population (OR = 1.76, 95% CI: 1.44–2.15, P < 0.001). Therefore, carriers of APOE ε4 allele had an increased risk for CHD, especially in Mongoloid population.
Publication bias
Both Begg's test and Egger's test were performed to assess the publication bias of the literature. Funnel plots on the distribution of the ORs from individual studies in relation to their respective standard deviation are shown in Figures 6 and 7. No evidence of publication bias was observed in the genetic model of overall ε2 allele versus ε3 allele (PBegg = 0.583, PEgger = 0.820). However, the shape of the funnel plot for the genetic model of overall ε4 allele versus ε3 allele seemed to be asymmetry, suggesting potential publication bias (PBegg = 0.002). This might be a limitation for our analysis because the studies with null findings, especially those with small sample size, were less likely to be published. By using the trim and fill method, we showed that if the publication bias was the only source of the funnel plot asymmetry, it needed seven more studies to balance the funnel plot. The adjusted risk estimate was attenuated but remained significant (ε4 allele vs. ε3 allele: OR = 1.08, 95% CI: 1.02–1.15, P = 0.009; ε4 carriers vs. ε3 carriers: OR = 1.08, 95% CI: 1.02–1.16, P = 0.016), indicating the stability of our results.
Figure 6.
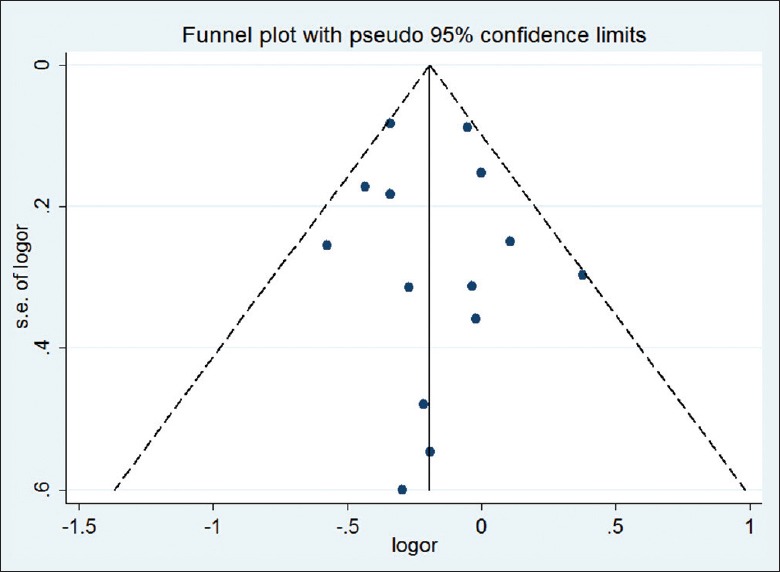
Funnel plot for the results of the meta-analysis of the ε2 allele vs. ε3 allele.
Figure 7.
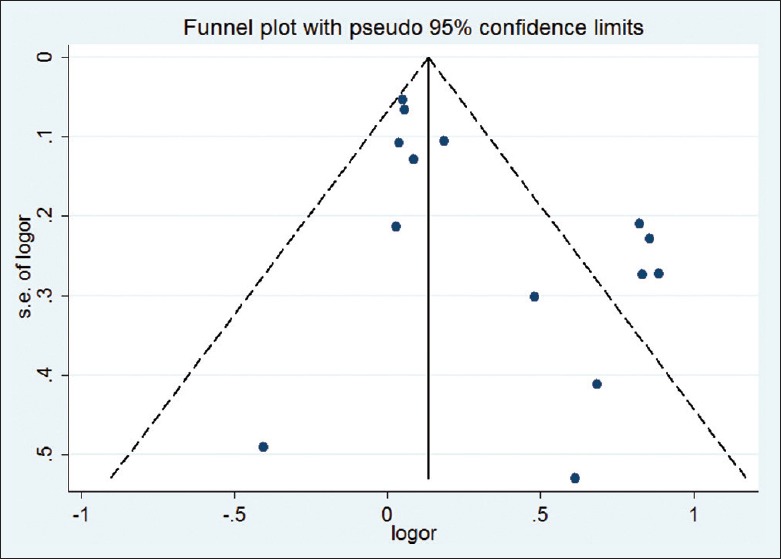
Funnel plot for the results of the meta-analysis of the ε4 allele vs. ε3 allele.
DISCUSSION
Many epidemiological studies have investigated the relationship between the APOE polymorphism and the risk of CHD, but because of small sample size and the low statistical power of individual studies, results have been contradictory. The main purpose of the present meta-analysis is to summarize the published literature and attempt to clarify the role of APOE ε2/ε3/ε4 polymorphism in CHD. We quantitatively combined 18 case–control studies including 6342 CHD cases and 19,688 controls, and found that carrying one or more ε4 allele was associated with an increased risk for CHD, whereas individuals with one or more ε2 allele had a significantly lower risk of developing CHD compared to those with ε3 allele. Therefore, it is safe to assume that ε4 allele is an independent factor for the development of CHD, however, ε2 allele tends to be a protective factor.
We also conducted subgroup analysis by race. As for the Caucasian population, significant association was observed in the contrast of the genetic model of ε2 allele vs. ε3 allele and ε2 carriers vs. ε3carriers. However, the genetic model ε4 allele versus ε3 allele and ε4 carriers versus ε3carriers remained significant difference only in Mongoloid population. Therefore, ethnic difference may contribute to this phenomenon.
Utermann et al.[33] first reported that the presence of APOE polymorphisms has inspired widespread interest in the genetic associations between these single-nucleotide polymorphisms and many complex diseases including Alzheimer's disease,[34] type 2 diabetes,[35] hypertension,[36] and others. Besides, several studies have provided evidence that APOE is functional. Structural defects in APOE could result in an impaired interaction between APOE-containing lipoproteins and their receptors and induce the development of atherogenic dyslipidemias and premature cardiovascular disease, including CHD. Several former meta-analyses had documented that ε4 was associated with increased risk for CHD.[37,38] In our study, we also found that ε2 carriers had a lower risk of CHD.
It is worth mentioning that testing for deviations from HWE in controls is an important requirement in population genetic studies. HWE is based on five basic assumptions: (1) The population is large (i.e., there is no genetic drift); (2) there is no gene flow between population due to migration or transfer of gametes; (3) mutations are negligible; (4) individuals are mating randomly; and (5) natural selection is not operating on the population. Deviation from HWE may point to genotyping error, racial heterogeneity, or selection bias. In our study, four of 18 studies were not conformed to HWE. We excluded the four studies in combined analysis.
Heterogeneity did not exist in overall comparisons of ε2 allele versus ε3 allele and ε2 carriers versus ε3carriers, but in the genetic model of ε4 allele versus ε3 allele and ε4 carriers versus ε3carriers, which may affect the results of the present meta-analysis. We adopted the random-effects model, as it is more conservative than the fixed-effects model and accounts for additional sources of inter-study variation when heterogeneity exists. This heterogeneity may attribute to the potential confounding due to case definition and sampling, sample size, genotyping method, difference of ethnicity, or it may be due to interaction with other risk factors. In order to understand the sources of heterogeneity, we conducted the subgroup analysis by race. The heterogeneity disappears in each subgroup. Hence, the heterogeneity might be resulted from the ethnicity difference.
We had to state the potential publication bias. Significant bias was observed in genetic model of ε4 allele versus ε3 allele and ε4 carriers versus ε3 carriers. The explanations might arise from some aspects. First, our meta-analysis took into consideration only fully published data, and the abstract and conference papers were excluded. Second, this meta-analysis only focused on papers published in English language, and some eligible studies which were reported in other languages might be missed. Third, positive results tend to be accepted by journals while negative results are often rejected or not even submitted. We should point out that the publication bias might partly account for the results, but which were not affected deeply. When we adjusted the results using the trim and fill method, the adjusted risk estimate was attenuated but remained significant, indicating the stability of our results. Hence, the summary statistics obtained may approximate the actual average.
In addition, heterogeneity and publication bias, some limitations of this meta-analysis should be acknowledged. Due to the relative small number of the eligible studies, we were unable to perform further subgroup analysis such as by gender, body mass index. Besides, CHD is a complex disease with various involved factors including environmental and genetic factors. However, many eligible studies included in this meta-analysis did not consider most of the important environmental factors, moreover, a single gene polymorphism is unlikely to provide the complete explanation of genetic risk for CHD. Therefore, gene-gene and gene-environment interactions should be considered in future studies.
Our study suggested a genetic association between APOE gene and CHD, ε4 increased the risk of CHD, whereas ε2 decreased the risk of CHD. Well-designed case-control studies with larger sample sizes regarding the association of APOE polymorphism and CHD need to be performed in the future.
ACKNOWLEDGMENTS
The authors express their sincere appreciation to the study participants.
Footnotes
Edited by: Li-Min Chen
Source of Support: This study was supported by a grant from Anhui Province Scientific Research Program (No. 12010402116).
Conflict of Interest: None declared.
REFERENCES
- 1.Zhou L, Cai J, Liu G, Wei Y, Tang H. Associations between interleukin-1 gene polymorphisms and coronary heart disease risk: Ameta-analysis. PLoS One. 2012;7:e45641. doi: 10.1371/journal.pone.0045641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corella D, Portolés O, Arriola L, Chirlaque MD, Barrricarte A, Francés F, et al. Saturated fat intake and alcohol consumption modulate the association between the APOE polymorphism and risk of future coronary heart disease: A nested case-control study in the Spanish EPIC cohort. J Nutr Biochem. 2011;22:487–94. doi: 10.1016/j.jnutbio.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298:1300–11. doi: 10.1001/jama.298.11.1300. [DOI] [PubMed] [Google Scholar]
- 4.Paik YK, Chang DJ, Reardon CA, Davies GE, Mahley RW, Taylor JM. Nucleotide sequence and structure of the human apolipoprotein E gene. Proc Natl Acad Sci U S A. 1985;82:3445–9. doi: 10.1073/pnas.82.10.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashavaid TF, Todur SP, Nair KG. Apolipoprotein E polymorphism and coronary heart disease. J Assoc Physicians India. 2003;51:784–8. [PubMed] [Google Scholar]
- 6.Bazrgar M, Karimi M, Fathzadeh M, Senemar S, Peiravian F, Shojaee A, et al. Apolipoprotein E polymorphism in Southern Iran: E4 allele in the lowest reported amounts. Mol Biol Rep. 2008;35:495–9. doi: 10.1007/s11033-007-9113-3. [DOI] [PubMed] [Google Scholar]
- 7.Ghebranious N, Ivacic L, Mallum J, Dokken C. Detection of ApoE2, E3 and E4 alleles using maldi-tof mass spectrometry and the homogeneous mass-extend technology. Mol Diagn. 2005;33:1–6. doi: 10.1093/nar/gni155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahley RW, Rall SC., Jr Apolipoprotein E: Far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 9.Ou T, Yamakawa-Kobayashi K, Arinami T, Amemiya H, Fujiwara H, Kawata K, et al. Methylenetetrahydrofolate reductase and apolipoprotein E polymorphisms are independent risk factors for coronary heart disease in Japanese: A case-control study. Atherosclerosis. 1998;137:23–8. doi: 10.1016/s0021-9150(97)00244-x. [DOI] [PubMed] [Google Scholar]
- 10.Cao W, Chen F, Teng L, Wang S, Fu S, Zhang G. The relationship between apolipoprotein E gene polymorphism and coronary heart disease and arteriosclerotic cerebral infarction. Chin J Med Genet. 1999;16:249–51. [PubMed] [Google Scholar]
- 11.Yang Z, Zhu T, Ma G, Yin H, Qian W, Zhang F, et al. Apolipoprotein E polymorphism in the early onset of coronary heart disease. Chin Med J. 2001;114:983–5. [PubMed] [Google Scholar]
- 12.Freitas EM, Phan TC, Herbison CE, Christiansen FT, Taylor RR, Van Bockxmeer FM. The poliovirus receptor related 2 (PRR2) and apolipoprotein E genes and coronary heart disease. J Cardiovasc Risk. 2002;9:59–65. doi: 10.1177/174182670200900109. [DOI] [PubMed] [Google Scholar]
- 13.Kumar P, Luthra K, Dwivedi M, Behl VK, Pandey RM, Misra A. Apolipoprotein E gene polymorphisms in patients with premature myocardial infarction: A case-controlled study in Asian Indians in North India. Ann Clin Biochem. 2003;40:382–7. doi: 10.1258/000456303766477020. [DOI] [PubMed] [Google Scholar]
- 14.Wang CH, Zhou X, Zhou GD, Tan XD, Han DF, Zheng F, et al. Genetic association of apoE and apoCI gene polymorphisms with coronary heart disease. Chin J Epidemiol. 2004;25:982–5. [PubMed] [Google Scholar]
- 15.Ranjith N, Pegoraro RJ, Rom L, Rajput MC, Naidoo DP. Lp (a) and apoE polymorphisms in young South African Indians with myocardial infarction. Cardiovasc J S Afr. 2004;15:111–7. [PubMed] [Google Scholar]
- 16.Ouyang T, Song JN, Miao Y, Lin Q, Niu XH, Jin H, et al. Study on relationship between polymorphism of apolipoprotein E gene and syndromes of phlegm and blood stasis in patients with coronary heart disease. J Chin Integr Med. 2005;3:438–42. doi: 10.3736/jcim20050605. [DOI] [PubMed] [Google Scholar]
- 17.Volcik KA, Barkley RA, Hutchinson RG, Mosley TH, Heiss G, Sharrett AR, et al. Apolipoprotein E polymorphisms predict low density lipoprotein cholesterol levels and carotid artery wall thickness but not incident coronary heart disease in 12,491 ARIC study participants. Am J Epidemiol. 2006;164:342–8. doi: 10.1093/aje/kwj202. [DOI] [PubMed] [Google Scholar]
- 18.Akanji AO, Suresh CG, Fatania HR, Al-Radwan R, Zubaid M. Associations of apolipoprotein E polymorphism with low-density lipoprotein size and subfraction profiles in Arab patients with coronary heart disease. Metabolism. 2007;56:484–90. doi: 10.1016/j.metabol.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Al-Bustan SA, Alkhalaf M, Al-Rashdan I, Al-Otaibi S, Al-Baker E, Balding D, et al. Apolipoprotein E, CI and B gene polymorphisms in a sample of patients with coronary heart disease in the Kuwaiti population. Med Princ Pract. 2009;18:294–9. doi: 10.1159/000215727. [DOI] [PubMed] [Google Scholar]
- 20.Singh PP, Singh M, Bhatnagar DP, Kaur TP, Gaur SK. Apolipoprotein E polymorphism and its relation to plasma lipids in coronary heart disease. Indian J Med Sci. 2008;62:105–12. [PubMed] [Google Scholar]
- 21.Kolovou GD, Anagnostopoulou KK, Kostakou P, Giannakopoulou V, Mihas C, Hatzigeorgiou G, et al. Apolipoprotein E gene polymorphism and obesity status in middle-aged men with coronary heart disease. In Vivo. 2009;23:33–9. [PubMed] [Google Scholar]
- 22.Marrzoq LF, Sharif FA, Abed AA. Relationship between ApoE gene polymorphism and coronary heart disease in Gaza Strip. J Cardiovasc Dis Res. 2011;2:29–35. doi: 10.4103/0975-3583.78584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gustavsson J, Mehlig K, Leander K, Strandhagen E, Björck L, Thelle DS, et al. Interaction of apolipoprotein E genotype with smoking and physical inactivity on coronary heart disease risk in men and women. Atherosclerosis. 2012;220:486–92. doi: 10.1016/j.atherosclerosis.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 24.Yilmaz-Aydogan H, Kucukhuseyin O, Kurnaz O, Akadam-Teker B, Kurt O, Tekeli A, et al. Investigation of polymorphic variants of PPARD and APOE genes in Turkish coronary heart disease patients. DNA Cell Biol. 2012;31:867–75. doi: 10.1089/dna.2011.1464. [DOI] [PubMed] [Google Scholar]
- 25.Moher D, Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. J Clin Epidemiol. 2009;62:1006–12. doi: 10.1016/j.jclinepi.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–58. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 27.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 28.Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst. 1959;22:719–48. [PubMed] [Google Scholar]
- 29.Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50:1088–101. [PubMed] [Google Scholar]
- 30.Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315:629–34. doi: 10.1136/bmj.315.7109.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Andrade M, Thandi I, Brown S, Gotto A, Jr, Patsch W, Boerwinkle E. Relationship of the apolipoprotein E polymorphism with carotid artery atherosclerosis. Am J Hum Genet. 1995;56:1379–90. [PMC free article] [PubMed] [Google Scholar]
- 32.Wang CH, Zhou X, Zhou GD, Han DF, Zheng F. Interaction of ApoE and LDL-R gene polymorphisms and alcohol drinking and smoking on coronary heart disease. Nat Med J China. 2004;84:554–8. [PubMed] [Google Scholar]
- 33.Utermann G, Hees M, Steinmetz A. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinaemia in man. Nature. 1977;269:604–7. doi: 10.1038/269604a0. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Q, Peng D, Yuan X, Lv Z, Pang S, Jiang W, et al. APOE and APOC1 gene polymorphisms are associated with cognitive impairment progression in Chinese patients with late-onset Alzheimer's disease. Neural Regen Res. 2014;9:653–60. doi: 10.4103/1673-5374.130117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaudhary R, Likidlilid A, Peerapatdit T, Tresukosol D, Srisuma S, Ratanamaneechat S, et al. Apolipoprotein E gene polymorphism: Effects on plasma lipids and risk of type 2 diabetes and coronary artery disease. Cardiovasc Diabetol. 2012;11:36. doi: 10.1186/1475-2840-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Xu JR, Liu XM, Zhou J, Yang B, Li M, et al. Polymorphisms of 2836 G>A in the apoE gene are strongly associated with the susceptibility to essential hypertension in the Chinese Hui population. Genet Mol Res. 2014;13:1212–9. doi: 10.4238/2014.February.27.6. [DOI] [PubMed] [Google Scholar]
- 37.Song Y, Stampfer MJ, Liu S. Meta-analysis: apolipoprotein E genotypes and risk for coronary heart disease. Ann Intern Med. 2004;141:137–47. doi: 10.7326/0003-4819-141-2-200407200-00013. [DOI] [PubMed] [Google Scholar]
- 38.Wilson PW, Schaefer EJ, Larson MG, Ordovas JM. Apolipoprotein E alleles and risk of coronary disease. A meta-analysis. Arterioscler Thromb Vasc Biol. 1996;16:1250–5. doi: 10.1161/01.atv.16.10.1250. [DOI] [PubMed] [Google Scholar]