

Abstract
Selective inhibition of cancer cells remains a challenge in chemotherapy. Here we report the molecular and cellular validation of enzyme-instructed self-assembly (EISA) as a multiple step process for selectively killing cancer cells that overexpress alkaline phosphatases (ALPs). We design and synthesize two kinds of d-tetrapeptide containing one or two phosphotyrosine residues and with the N-terminal capped by a naphthyl group. Upon enzymatic dephosphorylation, these d-tetrapeptides turn into self-assembling molecules to form nanofibers in water. Incubating these d-tetrapeptides with several cancer cell lines and one normal cell line, the unphosphorylated d-tetrapeptides are innocuous to all the cell lines, the mono- and diphosphorylated d-tetrapeptides selectively inhibit the cancer cells, but not the normal cell. The monophosphorylated d-tetrapeptides exhibit more potent inhibitory activity than the diphosphorylated d-tetrapeptides do; the cancer cell lines express higher level of ALPs are more susceptible to inhibition by the phosphorylated d-tetrapeptides; the precursors of d-tetrapeptides that possess higher self-assembling abilities exhibit higher inhibitory activities. These results confirm the important role of enzymatic reaction and self-assembly. Using uncompetitive inhibitors of ALPs and fluorescent d-tetrapeptides, we delineate that the enzyme catalyzed dephosphorylation and the self-assembly steps, together, result in the localization of the nanofibers of d-tetrapeptides for killing the cancer cells. We find that the cell death modality likely associates with the cell type and prove the interactions between nanofibers and the death receptors. This work illustrates a paradigm-shifting and biomimetic approach and contributes useful molecular insights for the development of spatiotemporal defined supramolecular processes/assemblies as potential anticancer therapeutics.
Introduction
Being a ubiquitous process used by cells, self-assembly (or aggregation, or clustering) to form oligomeric or supramolecular protein assemblies are critical to cell functions and fates. For example, the energy dissipation self-assembly (or self-organization) of actins or tubulins maintain the actin filaments and microtubules as the cytoskeletons for cell migration1 and mitosis.2 The oligomerization of the extrinsic cell death receptors (e.g., TRAIL-R1/R2, TNFR1, and CD95) initiates the downstream signaling of apoptosis.3 The self-assembly of Apaf-1 and cytochrome c results in apoptosomes.4 The nucleation of adaptor protein ASC triggers the formation of inflammasomes that are critical for host defense.5 Recently, prion-like proteins, such as the cytoplasmic polyadenylation element-binding protein,6 the mitochondrial antiviral signaling protein,7 the T-cell-restricted intracellular antigen 1,8 are reported to be beneficial to cells.9 One reason for nature to select oligomeric or higher-ordered protein structures is to achieve quantitative aspects of signaling transduction, such as location, duration, thresholds, amplitude, and amplification, even in the case of promiscuous binding.10
Notably, enzymatic reactions11 and molecular self-assembly are the most utilized processes by nature to achieve the oligomeric or higher-ordered structures with precise spatiotemporal control. For example, enzyme-catalyzed conversion of guanosine diphosphates (GDP) to guanosine triphosphates (GTP) on β-tubulin powers the self-assembly of α- and β-tubulin onto the (+) end of microtubules.12 Despite the prevalence of enzyme-instructed self-assembly (EISA) in nature,13 the application of this concept in supramolecular chemistry and chemical biology is just beginning. Recently, we and other researchers are exploring this concept for developing a biomimetic, multiple-step process for cancer therapy, especially in the studies and applications of small molecular self-assembly controlled by enzymatic transformation.14 For example, besides observing that intracellular EISA of small peptides, instructed by esterase, selectively inhibit cervical cancer cells,15 we found that pericellular EISA of small d-peptides16 or nanoparticles,17 catalyzed by placental alkaline phosphatases (PLAP18), selectively inhibit cancer cells, including drug-resistance uterine cancer cells.16 Most recently, we found that intracellular EISA can boost the activity of cisplatin against cisplatin-resistant ovarian cancer cells.19 Moreover, Maruyama et al. demonstrated the use of extracellular enzymes (e.g., MMP-7) to instruct the self-assembly of peptide lipids on cell membrane. They found that the internalization of the resulted assemblies leads to the death of multiple cancer cell lines.20 Using a phosphorylated carbohydrate derivative as the substrate of membrane phosphatases (likely the tissue nonspecific alkaline phosphatase (TNAP)21), Pires and Ulijn et al. also achieved pericellular EISA to inhibit the osteosarcoma cells without affecting related prechondrocyte cells with low expression of phosphatases.19 By applying phosphatase-based EISA, Gao and co-workers recently demonstrated that EISA of a tetrapeptide derivative22 and a clinical used dye is able to form tumor-specific nanofibers for cancer theranostics in animal model bearing tumor of HeLa cells.23 These results suggest that EISA is emerging as a new strategy, which consists of enzymatic reaction and self-assembly steps, for selectively targeting cancer cells.
As an emerging biomimetic approach for developing anticancer therapeutics, EISA is fundamentally different from the well-established prodrug approach.24 In a prodrug approach, enzymatic reaction in vivo releases the active parent drug that usually functions as a monomeric agonist or antagonist. In EISA, only the assemblies, not the unassembled products of the enzymatic conversion, are inhibitory to cancer cells.16 In other word, EISA, combining biotransformation (i.e., enzyme catalysis) and molecular self-assembly, mainly acts as a multiple-step process to inhibit cancer cells. Since it relies on localized molecular assemblies rather than on an individual molecule, EISA is able to directly disrupt multiple cellular processes16a and to create a global change (e.g., viscosity increase)16a in cellular environment of the cancer cells. Thus, EISA promises a new way for targeting evolutionary redundancy that results in drug resistance in cancer therapy.14
Despite its promises, the use of EISA for cancer therapy represents a venture into a previously unknown and unexplored intersection of chemical and biological spaces. Many challenges remain to be met in order to develop EISA of small molecules for selectively killing cancer. For example, achieving selectivity requires the knowledge of biomarkers (e.g., enzymes or proteins) that differentiate cancer cells from normal ones. Unfortunately, although the information accumulated in proteomics research is filling this gap,25 the information we currently have is far from sufficient. Besides, there is considerable difference between the methods and techniques used for studying the different approaches of cancer therapy. Those used in conventional chemotherapy and recently advanced immunotherapy may be inadequate for EISA. Moreover, lack of understanding of the protein targets of the assemblies or aggregates, as well as the limited techniques26 to identify and characterize the interactions between nanoscale assemblies of small molecules27 and proteins, remains an obstacle for further advances of EISA. Thus, a reliable molecular and cellular validation of EISA is the first essential step for progressing both into and through this unexplored space, as well as for generating the most reliable conclusions. Following the updated view on target validation through molecular design,28 we choose to design and synthesize a series of structural analogues of the substrates and products of EISA. By using these molecules to treat the same set of cells, we aim to answer the following questions: (1) how does the molecular structure modulate the EISA properties of the precursors, and thus regulate the biological activities of the resulting supramolecular assemblies? (2) Our previous studies have validated that the pericellular hydrogel/nanofibers formed by EISA of a small d-peptide containing a phosphotyrosine can selectively inhibit cancer cells over normal ones due to the overexpression of ALPs by cancer cells. How does the number of phosphotyrosine on a single molecule affect the cellular response even though the final products (i.e., hydrogelators) remain the same? This question is valid and relevant because it is common for a protein to have multiple post-translational modification sites (including phosphorylation). (3) What is the relationship between the levels of enzyme expression on different cell lines and the inhibitory activities of the precursors? (4) What is the role of different isoenzymes of ALPs in this multistep process for selectively inhibiting cancer cells?
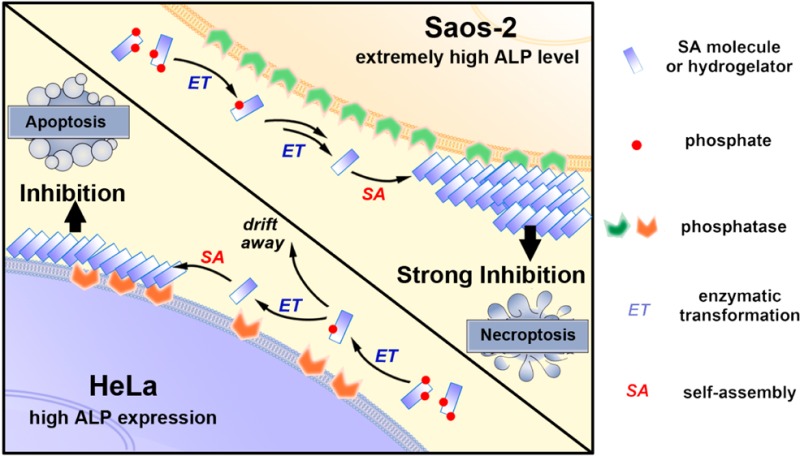
To answer the above basic and important questions, we systematically synthesize two kinds of d-tetrapeptides containing one or two phosphotyrosine residues and with the N-terminal capped by a naphthyl group. By mutating the sequence of amino acids, we obtain six precursors (a-1p, a-2p, b-1p, b-2p, c-1p, and c-2p) containing one (1p) or two phosphotyrosines (2p). All of the precursors are able to turn into hydrogelators (a, b, and c) that self-assemble in water to form nanofibers upon enzyme-instructed dephosphorylation (Figure 1). Despite that they all contain the same motifs (i.e., Phe, Tyr, and naphthyl), the resulting molecules exhibit different states of self-assembly (or aggregation) both before and after the action of ALP in vitro, according to several complementary methods (e.g., transition electron microscopy (TEM), static light scattering (SLS), and rheology) used for characterizing molecular self-assembly in water. Being incubated with the same cell lines, these precursors become self-assembling hydrogelators, and result in distinct cellular responses. The precursors exhibit more potent inhibitory activities when the d-tetrapeptides possess sufficiently high self-assembling abilities. Incubating these precursors or hydrogelators with several cancer cell lines and one normal cell line, the cell viability assay indicates that, while the dephosphorylated hydrogelators are innocuous to all the cell lines, the mono- and diphosphorylated precursors selectively inhibit the cancer cells, but are innocuous to the normal cells. This result agrees with that EISA localizes the assemblies of the hydrogelators at the cancer cells,16 which is further confirmed by imaging of fluorescent d-tetrapeptides. Besides, the monophosphorylated precursors exhibit more potent inhibitory activity than the diphosphorylated precursors; the cancer cell lines express higher level of ALPs are more susceptible to the inhibition by the precursors (Scheme 1). Using different uncompetitive inhibitors of ALPs (e.g., l-Phe for PLAP29 and tetramisole for TNAP30), we delineate that the enzyme catalyzed dephosphorylation and the self-assembly steps, together, result in the inhibition of the cancer cells. Moreover, our experiments confirm that EISA of these structurally different precursors/hydrogelators to inhibit cancer cells selectively according to the different levels of the isozymes of ALPs expressed in different cell lines. In addition, we find that zVAD-fmk,31 an antagonist of apoptosis, and Nec-1,32 an antagonist of necroptosis, counter the effect of EISA on HeLa and Saos-2 cells, respectively, implying that the modality of cell death likely associates with the types of the cancer cells. As the first comprehensive validation of a multistep process for selective inhibiting multiple cancer cell lines, this work contributes new insights for answering the fundamental question that how different cells respond to the EISA of structurally different molecules, and illustrates a new and biomimetic approach for the development of spatiotemporal controlled supramolecular processes/assemblies as potential anticancer drugs.
Figure 1.

Molecular structures of the precursors (a-2p, b-2p, c-2p, a-1p, b-1p, and c-1p) that have one or two phosphotyrosine residues and the corresponding self-assembling d-peptides (i.e., hydrogelators a, b, and c).
Scheme 1. Enzyme-Instructed Self-Assembly of Small Molecules to Inhibit Cancer Cells.

Results and Discussions
Molecular Design
In our exploration of EISA of small molecules for making supramolecular hydrogels, we unexpectedly found that EISA of a small d-peptide derivative (e.g., Nap-d-Phe-d-Phe-d-pTyr), with the tyrosine being phosphorylated, selectively inhibits cancer cells.16 A recent tissue-based map of human proteome has also validated PLAP as a generic difference between cancer and normal cells.25 This generic and rare difference between cancer and normal cells presents an unprecedented opportunity for targeting cancer cells selectively so it is worthwhile and necessary to conduct a comprehensive molecular and structural validation of EISA for potential cancer therapy. Thus, we designed and synthesized a series of structural analogues of the substrates with one or two tyrosine phosphorylated (a-1p, a-2p, b-1p, b-2p, c-1p, and c-2p) and their corresponding products (a, b, and c) of enzymatic dephosphorylation (Figure 1). We choose to use backbone of 2-(naphthalen-2-yl)acetic-d-Phe-d-Phe-d-Tyr-d-Tyr (NapDFDFDYDY) or its analogues (by simply varying the amino acid sequence) for the following reasons: (1) 2-(Naphthalen-2-yl)acetic-Phe-Phe (NapFF) and 2-(naphthalen-2-yl)acetic-Phe (NapF) are excellent motifs for promoting molecular self-assembly in water due to the extensive aromatic–aromatic interactions33 and hydrogen bonding among those molecules.34 (2) Besides being part of the molecular backbone, the incorporation of Tyr motifs provides sites for mono- or diphosphorylation, which can elucidate whether increasing the number of enzymatic triggers on a single molecule enhances its solubility and selectivity for targeting cancer cells. (3) Varying the amino acid sequence provides related yet different derivatives for evaluating the relationship between the molecular structures and self-assembly properties, as well as its corresponding cellular activities. (4) We prefer the self-assembling molecules to be hydrogelators because hydrogelation provides a facile assay to report molecular self-assembly in water.35 (5) The self-assembling molecules are exclusively composed of d-amino acids because d-peptides, as the enantiomers of naturally occurring l-peptides, usually resist endogenous proteases and barely to have strong interactions with cellular proteins.
Synthesis
Figure 1 shows the structures of a series of precursors and their corresponding hydrogelators resulted from ALP treatment. According to Alewoods’ report,36 we made tyrosine phosphate in 90% yield, followed by the conjugation of Fmoc protecting group to the N-terminal. The resulting Fmoc-phosphotyrosine can be directly used in the solid phase synthesis (SPPS) (Figure S1).37 Utilizing Fmoc-protected d-amino acids, we prepared all the compounds by SPPS with 2-chlorotrityl chloride resin, and obtained these molecules in 70% yields after high-performance liquid chromatography (HPLC) purification. LC–MS and 1H NMR confirm the purity and structures of these designed molecules (Figure S2).
Self-Assembly of the Designed Molecules
We evaluate the enzyme-instructed hydrogelation/self-assembly of the hydrogelators in water at pH = 7.4 by using ALP to treat the precursors and using transmission electron microscopy (TEM) to examine the formed nanostructures. As shown in Figure 2, either the mono- or diphosphorylated precursors dissolve well in water to make clear solutions, while TEM images reveal subtle difference between them after water evaporates. The diphosphorylated precursors generally tend to form amorphous aggregates, whereas the monophosphorylated precursors start to form relative short nanofibers. This observation agrees with that two phosphotyrosine residues enhance the aqueous solubility of the precursors. Moreover, the structure difference of these precursors leads to different self-assembly behaviors even when they have same number of phosphates. For example, in the solutions of a-1p and c-1p, there are sporadic nanofibers interwoven into aggregates, and for b-1p, only short nanofibers with diameters of around 7 ± 2 nm appear with relatively high density. Similarly, a-2p and c-2p form the aggregates with similar morphologies, but b-2p results in larger aggregates than those of a-2p or c-2p. These results suggest that the backbone of DFDYDFDY has higher tendency to self-assemble to form nanoscale structures among these analogues. Adding ALP into the six precursor solutions afford the hydrogelators, which self-assemble in water to form nanofibers that act as the matrices of the hydrogels (Figure 2). TEM images reveal that the hydrogel networks, formed by the hydrogelator resulted from enzymatic dephosphorylation of the precursors, are all composed of uniformed nanofibers with similar diameters, as summarized in Table 1.
Figure 2.

TEM images of aggregates/nanofibers in the solutions of different precursors (a-2p, a-1p, b-2p, b-1p, c-2p, and c-1p) or nanofibers in the hydrogels formed by treating the solutions of the precursors with alkaline phosphatase (ALP). C = 0.5 wt %, pH = 7.4, [ALP] = 1 U/mL. Insets are optical images of the solutions of the precursors and the hydrogels formed after enzymatic dephosphorylation. The scale bar is 100 nm.
Table 1. Summary of the EISA of the Precursors.
| compounda | a-2p | b-2p | c-2p | a-1p | b-1p | c-1p |
|---|---|---|---|---|---|---|
| in pbs (pH 7.4) | solution | solution | solution | solution | solution | solution |
| + ALP (1 U/mL) | gel | gel | gel | gel | gel | gel |
| critical strain Y0 (%) | 1.0% | 1.8% | 1.0% | 1.5% | 2.5% | 1.5% |
| storage modulus G′b (Pa) | 29 | 79 | 60 | 54 | 123 | 100 |
| loss modulus G″b (Pa) | 8 | 30 | 23 | 12 | 39 | 63 |
| morphology before ALP treatment (dc (nm)) | aggregate | aggregate | aggregate | aggregate | nanofibers (7 ± 2) | aggregate |
| morphology after ALP treatment (dc (nm)) | nanofiber (7 ± 2) | nanofiber (5 ± 2) | nanofiber (6 ± 2) | nanofiber (7 ± 2) | nanofiber (5 ± 2) | nanofiber (6 ± 2) |
The concentration is 0.5 wt %.
The modulus is taken at the frequency of 6.28 rad/s.
Diameter of nanofibers.
To further determine the self-assembly properties of the precursors and hydrogelators, we also examine them by rheology and static light scattering. Figure 3A shows the strain- and frequency-dependence of dynamic storage moduli (G′) and loss moduli (G″) of the hydrogels aforementioned. The values of G′ of all six hydrogels are larger than those of their G″, indicating that all the samples are viscoelastic materials. The values of G′ of the hydrogels change little during the frequency sweep (from 0.1 to 200 rad/s), suggesting that these hydrogels have good tolerance to external shearing force. Overall, both strain- and frequency-dependent G′ values of the hydrogels formed by monophosphorylated precursors after ALP treatment are larger than those of gels formed by diphosphorylated precursors (Figure 3A). Since precursors with two phosphates likely need longer time to turn into the hydrogelators completely, the lower values of G′ suggest incomplete dephosphorylation of the precursors consisting of two phosphotyrosine residues (vide infra). In agreement with TEM image results, the rheology of the hydrogel formed by dephosphorylating b-1p shows stronger mechanical strength than those of the hydrogels formed by dephosphorylating c-1p or a-1p. The similar trend appears in the hydrogels resulted from the treatment of a-2p, b-2p, or c-2p by ALP. These trends indicate that the self-assembling property largely depends on the molecular structures of the hydrogelators, and the backbone DFDYDFDY favors self-assembly in water. Gelation test by rheometer shows that precursors with one phosphate form hydrogels much faster than their corresponding analogues that possess two phosphates (Figure 3B). Due to that the backbone of DFDYDFDY exhibits higher tendency to self-assemble, the gelation point of b-1p emerges quickly—0.2 h after the addition of ALP, followed by c-1p (0.3 h) and then a-1p (0.7 h). The dephosphorylation of a-2p, b-2p, and c-2p forms hydrogels in the same order, taking 3.7, 2, and 2.5 h, respectively. In general, the precursors with two phosphates, requiring two dephosphorylation steps, take considerably longer time to turn into hydrogels than those of monophosphorylated precursors do. These results also agree with static light scattering (SLS) data (Figure 3C), which shows that the solution of b-1p exhibits strongest signal after ALP treatment for 24 h, followed by c-1p, b-2p, a-1p, c-2p and a-2p.
Figure 3.
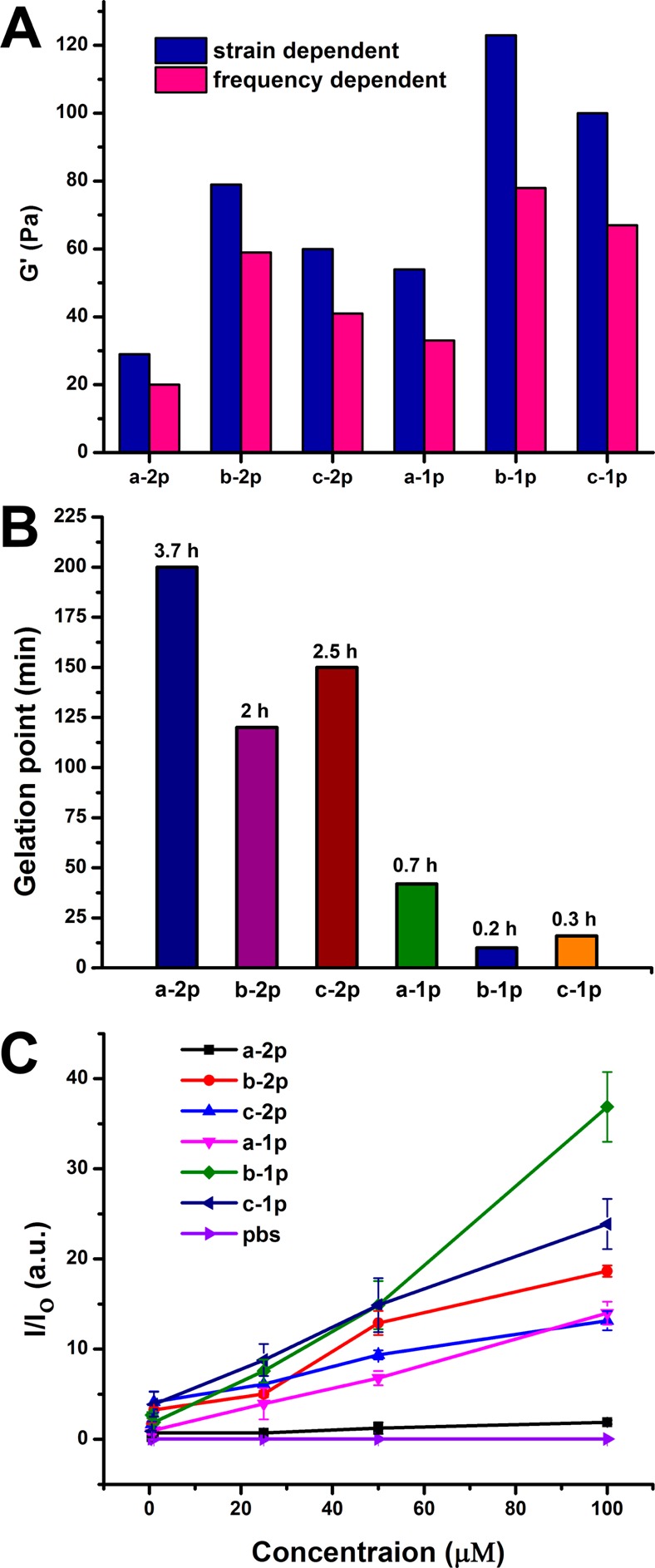
(A) Rheological characterization of hydrogels formed by different precursors (a-2p, b-2p, c-2p, a-1p, b-1p, and c-1p) treated with ALP (1.0 U/ml). C = 0.5 wt %. The strain-dependent dynamic storage (G′) is taken at a frequency equal to 6.28 rad/s, and the frequency-dependent dynamic storage (G′) is taken at a strain equal to 1.00%. (B) Time-dependent rheometry to show the gelation points (that is, at the gel state, where storage modulus (G′) dominates loss modulus (G″)) of different precursors treated with ALP (0.05 U/ml). C = 0.5 wt %. (C) Static light scattering (SLS) shows the scattering signals after the addition of ALP (1 U/mL) into the solution of different precursors at different concentrations, pH = 7.4, detecting angle = 30°.
Cellular Responses
To investigate how molecular modification and the corresponding EISA affects the cellular response, we use MTT cell viability assay to examine the cytotoxicity of the precursors toward HeLa cells and Saos-2 cells, two cell lines known to allow EISA to form pericellular nanofibers.16,19 Our results (Figure 4A) show the following: (1) All the six precursors inhibit both HeLa and Saos-2 cells in a dose-dependent manner at the concentrations above certain thresholds, indicating that enzymatic dephosphorylation leads to the self-assembly of these d-tetrapeptidic derivatives on the cell surface to kill HeLa and Saos-2 cells. (2) b-2p or b-1p exhibits significantly higher cytotoxicity to both HeLa and Saos-2 cell than their analogues (i.e., a-2p, c-2p or a-1p, c-1p). These results match with the TEM and rheological results that the Nap-DFDYDFDY motif has the highest self-assembling ability. (3) In general, Saos-2 cells are more vulnerable to the six precursors compared with HeLa cells, largely because of the significantly higher expression of ALPs on Saos-2 (vide infra, Figure 6). (4) The precursors with one phosphotyrosine residue (i.e., a-1p, b-1p, and c-1p) are more cytotoxic to HeLa cells than those with two phosphotyrosine residues (i.e., a-2p, b-2p, and c-2p), while the precursors with same backbones (i.e., b-1p and b-2p; c-1p and c-2p) show similar inhibitory activities to Saos-2 cells, except that a-1p is more potent than a-2p. These observations agree with the different levels of expression of ALPs on the surface of HeLa and Saos-2 cells (Figure 6). To be specific (Scheme 1), HeLa cells express less ALPs on cell surface than Saos-2. For the precursors with two phosphotyrosines, when one of the phosphates is removed by ALP, the resulting intermediates can drift away due to relatively good solubility rendered by the remaining phosphotyrosine group before continuously interacting with ALP to lose another phosphate and to result in self-assembly. For the monophosphorylated precursors, self-assembly immediately occurs on cell surface upon dephosphorylation by ALP, and then kills cells. So it is reasonable that the precursors with two phosphotyrosine exhibit reduced cytotoxicity, comparing to monophosphorylated precursors, on HeLa cells. In the case of Saos-2 cells, there is considerably high expression level of ALPs on the cell surface, which quickly dephosphorylates the two phosphotyrosine residues on a single molecule before they diffuse away (Scheme 1). So mono- and diphosphorylated precursors show almost same cytotoxicity toward Saos-2 cells.
Figure 4.
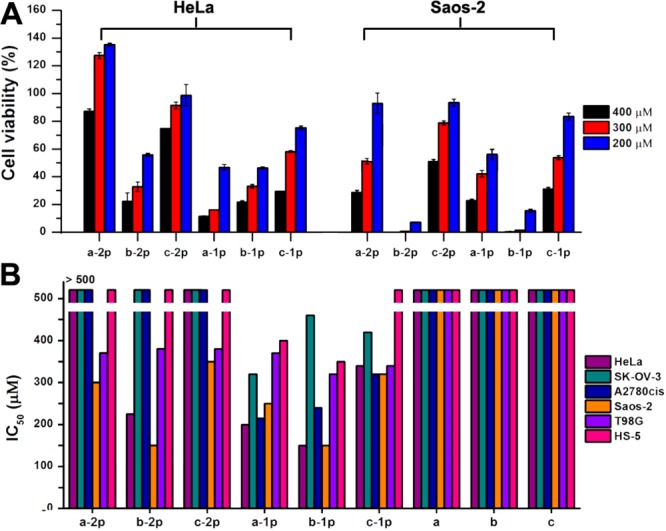
(A) 48-h cell viability (determined by MTT assay) of HeLa and Saos-2 cells incubated with different precursors at the concentrations of 200, 300, and 400 μM in culture medium. The initial cell numbers are 1 × 104 cells/well. (B) IC50 of different precursors/hydrogelators against different cell lines after 48-h incubation.
Figure 6.
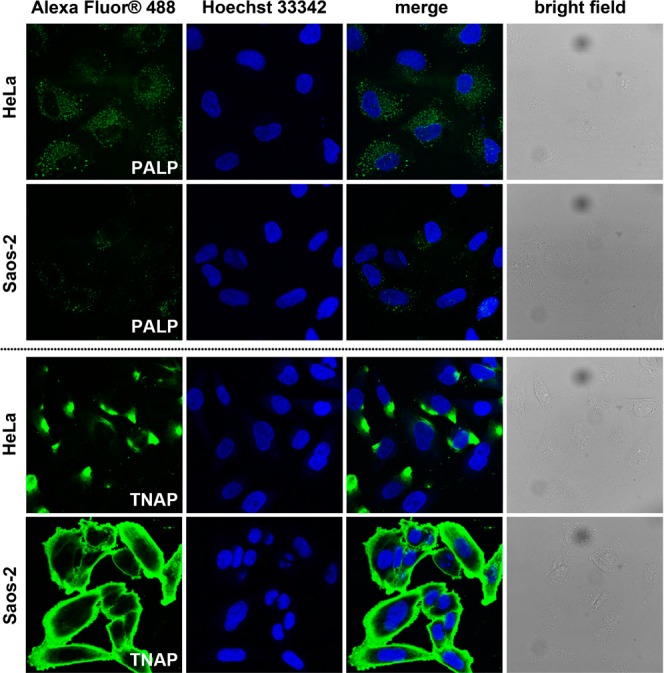
Confocal microscopy images of HeLa and Saos-2 cells after PLAP or TNAP antibody staining. Nuclei are stained by Hoechst 33342.
To evaluate the generality that the EISA can selectively inhibit cancer cells, we treated SK-OV-3, A2780cis (two drug-resistant ovarian carcinoma cell lines), T98G cells (a glioblas-toma multiforma tumor cell line) with the six precursors, using the same procedure as that for the HeLa and Saos-2 cells. Our results reveal that all the six precursors inhibit these cancer cells above certain concentrations (Figure S3). As a control, we also examined the cellular response of HS-5 cell (an immortalized normal stromal cell38) upon the treatment of precursors, and found that all of precursors hardly show any cytotoxicity (Figure S3) to the HS-5 cells at the concentrations that they exhibit significant cytotoxicities to the cancer cells. These results confirm that EISA of these precursors can selectively inhibit cancer cells over normal ones due to overexpression of ALPs on the cancer cells.39 According to Figure 4B and Table 2, which summarizes the 48-h IC50 of these precursors on different cell lines, the IC50 values of monophosphorylated precursors are generally lower than those of diphosphorylated ones on HeLa, SK-OV-3, and A2780cis cells. This result can be explained by that the intermediates with one phosphotyrosine would drift away before self-assembling (Scheme 1, upper left). The monophosphorylated precursors are less inhibitory to SK-OV-3 cells than to A2780cis, indicating that different cells exhibit different response to the precursors. Similar to the case of Saos-2, T98G exhibits almost same response to the mono- and diphosphorylated precursors, suggesting that the ALP level on T98G surface is sufficient to dephosphorylate the second phosphotyrosine before the intermediate diffusing way. However, T98G is less vulnerable than Saos-2 to the six precursors, suggesting higher level of ALP expression on Saos-2 than on T98G cells. The lower left panel of Scheme 1 illustrates the case of Saos-2. By measuring the membrane-associated ALP activities with pNPP assay (Figure S4), we found that the cells with higher membrane ALP abundance are generally more vulnerable to these precursors. These results not only reveal the relationship among cellular responses, molecular structures, and ALP levels, but also indicate that increasing the numbers of enzyme active sites helps to amplify the selectivity in a situation where molecules need to bypass some cells with relatively high ALP expression to reach their targets.
Table 2. 48-h IC50 (μg/mL) of Different Precursors/Hydrogelators against Different Cell Lines.
| compound | a-2p | b-2p | c-2p | a-1p | b-1p | c-1p | a | b | c |
|---|---|---|---|---|---|---|---|---|---|
| HeLa | >483 | 217 | >483 | 177 | 132 | 301 | >500 | >500 | >500 |
| Saos-2 | 326 | 144 | 338 | 170 | 132 | 283 | >500 | >500 | >500 |
| A2780cis | >483 | >483 | >483 | 189 | 212 | 283 | >500 | >500 | >500 |
| SK-OV-3 | >483 | >483 | >483 | 283 | 407 | 372 | >500 | >500 | >500 |
| T98G | 357 | 367 | 360 | 327 | 283 | 301 | >500 | >500 | >500 |
| HS-5 | >483 | >483 | >483 | 354 | 310 | >443 | >500 | >500 | >500 |
Although the different sequences of the tetrapeptides may result in different self-assembled nanostructures to interact with the cells, the cell viabilities are largely inverse proportional to the intensity of the signal of SLS (Figure S5), further supporting that the degree of self-assembly indeed correlates with the effect of EISA on cell fate.
Notably, the corresponding hydrogelators (a, b, and c), hardly inhibit any of the cell lines tested even at the concentration as high as 500 μM, which completely differ from the cytotoxicities of the six precursors. This result agrees with that the nanofibers generated by EISA result in inhibition. Moreover, the results in Figure 4 indicate that cellular responses of the six precursors are closely associated with molecular structure, self-assembly ability, dephosphorylation rate, and cell difference.
The Nanofibers Forming in Pericellular Space
The distribution of Hoechst 33342 (a nucleus dye), upon the formation of the pericellular hydrogel/nanonets on cells, further validates the assumption in Scheme 1. As shown in Figure 5A, after incubation with b-2p at 500 μM for 6 h and then the addition of Hoechst 33342 for 5 min, HeLa cells show the fluorescence in nuclei, behaving similarly to the control cells (i.e., untreated HeLa cells). Being treated with b-1p (500 μM), HeLa cells hardly show blue fluorescence in nuclei, agreeing with that the formation of pericellular nanofibers prevents or delays the nuclei dye entering the cells. This result further confirms that b-1p affords nanofibers faster than b-2p does on HeLa cells, which is consistent with the observed difference in the cytotoxicity of b-1p and b-2p. Contrasting to the case of HeLa cells, Hoechst 33342 is unable to enter the Saos-2 cells treated with either b-2p or b-1p at 500 μM for 6 h (Figure 5A). Moreover, after staining for 10 min, some nuclei dyes are even trapped in the pericellular nanofibers. This observation differs drastically from the staining of the untreated Saos-2 cells (showing fluorescence in the nuclei) and indicates that b-1p and b-2p result in almost the same density of self-assembled nanofibers due to high abundance of ALPs on Saos-2 cells. This result also agrees with the same toxicity of b-1p and b-2p on Saos-2 cells.
Figure 5.

(A) Nuclei staining (by Hoechst 33342) of HeLa and Saos-2 cells treated with b-2p and b-1p (500 μM) for 6 h. HeLa cells were stained for 5 min, and Saos-2 for 10 min to guarantee a clear contrast in control images. Accumulated nanofibers on cell surface trap the Hoechst 33342 and prevent this nuclei dye from entering cells. (B) Chemical structures of NBD-2p and NBD-1p (analogues of b-2p and b-1p), which turn into the same hydrogelator after dephosphorylation. (C) Confocal microscopy images of HeLa and Saos-2 cells treated with NBD-2p and NBD-1p for 12 h. Nuclei are stained by Hoechst 33342.
To directly visualize the pericellular nanofibers on cells, we designed and synthesized NBD-2p and NBD-1p by replacing the Nap motif with NBD (Figure 5B). The replacement of N-terminal-capped motifs results in reduced self-assembly ability and low cytotoxicity (Figure S6) of NBD-2p and NBD-1p, but still allows their corresponding hydrogelators to form nanofibers in pericellular space of cells. According to Figure 5C, treating HeLa cells with NBD-2p (500 μM) for 12 h only leads to faint yellow fluorescence inside the cells, likely due to endocytosis, while the addition of NBD-1p into HeLa cell culture results in strong fluorescence on the surface of the cells. Contrasting to the case of HeLa cells, the addition of NBD-2p or NBD-1p in Saos-2 cell culture leads to the same phenomenon—significant fluorescence appears on cell surface (Figure 5C), which is obviously more fluorescent than that on HeLa cell treated by NBD-1p. Because NBD-modified peptides fluoresce intensely in self-assembled nanofibers,40 the yellow fluorescence reflects the amount of nanofibers formed by EISA of NBD-2p or NBD-1p. These results, being consistent with Hoechst 333342 staining results and cellular response on HeLa and Saos-2, further verify the assumption illustrated in Scheme 1.
Two previous works visualized the self-assembled nanofibers formed by EISA on cell surface by electron microscopy (EM).16a,19 To provide direct evidence that the hydrogelators self-assemble in fibrous structures on cell membrane, we use TEM images of the freeze-drying sample to confirm the formation of nanofibrils of the tetrapeptides (Figure S7), using the method described in our previous work.16a
Expression of the Isozymes of ALPs on HeLa and Saos-2 Cells
As ectophosphatases, ALPs have different isotypes.41 Using antibody staining, we evaluated the expression level of two types of isozymes of ALPs on HeLa and Saos-2 cells. As shown in Figure 6, HeLa cells express more PLAP than Saos-2 cells while TNAP are more abundant on Saos-2 cells. According to the staining, Saos-2 cells, in overall, express significantly more ALPs on cell surface. This result, together with the Hoechst 33342 staining and imaging results of NBD-1p and NBD-2p on HeLa and Saos-2 cells, not only explains the different cellular response of HeLa and Saos-2 cells, but further supports the assumption illustrated in Scheme 1.
Cell Responses under ALP Inhibition
Since antibody staining is unable to reveal the activity of ALPs on cancer cells, we choose to inhibit ALPs and evaluate cell responses. On the basis of the fact that HeLa cells express more PLAP42 on their surface than Saos-2 do, we used l-phenylalanine (l-Phe, an uncompetitive inhibitor for PLAP43) to inhibit PLAP during cell culture. As shown in Figure 7A, the cytotoxicity of all six precursors against HeLa cell decrease significantly after the addition of l-Phe (3 mM). Since l-Phe is cell compatible at 3 mM, the decrease of the cytotoxicity likely originates from the inhibition of PLAP, which slows down the EISA process. This result confirms that PLAP catalyzes the formation of hydrogelators a, b, or c, and their self-assembly on cell surface for inhibiting the growth of HeLa cells. However, the addition of (−)-tetraimisole (an uncompetitive inhibitor of TNAP41) hardly rescues HeLa cells (Figure S8). This result indicates that PLAP catalyzed EISA largely contributes the death of HeLa cells. While the addition of (−)-tetramisole significantly increases the cell viability of Saos-2 cells (Figure 7B), the addition of l-Phe into Saos-2 culture hardly rescue the Saos-2 cells at all (Figure S8). This result agrees with that the majority of the phosphatases on the membrane of Saos-2 are TNAP (Figure 6). Notably, as shown in Figure 7B, (−)-tetramisole generally saves more Saos-2 cells treated by diphosphorylated precursors than monophosphorylated ones, which further confirms the inhibition mechanism illustrated in Scheme 1. Because the inhibition of ALPs on cell surface effectively reduces the numbers of active phosphatases, it is reasonable that there are more significant reductions of the cytotoxicity of diphosphorylated precursors than those of the monophosphorylated precursors.
Figure 7.
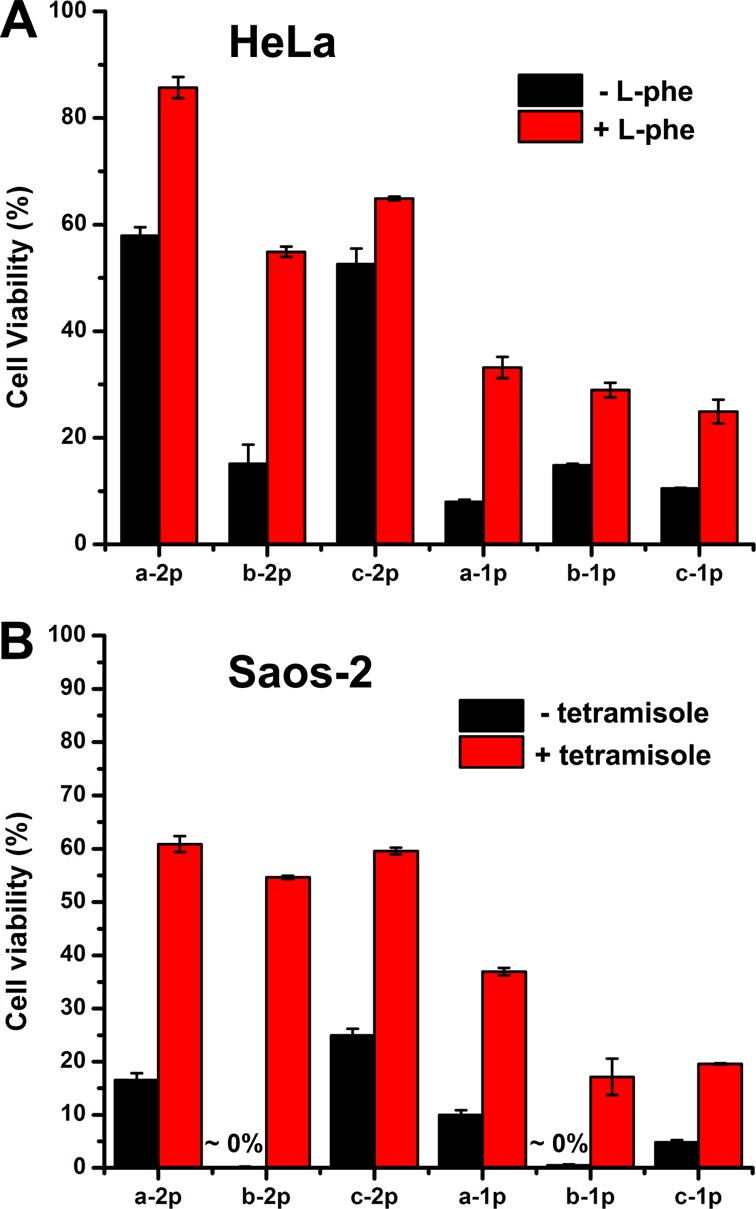
Viability of (A) HeLa and (B) Saos-2 cells incubated with six precursors (500 μM) with or without different phosphotase inhibitors for 48 h; [l-phe] = 3 mM; [levamisole] = 1 mM.
To further confirm the critical role of the membrane-bound ALPs (i.e., endogenous ectophosphatases), we add ALP (5 U/mL), as an exogenous, soluble enzyme, together with these precursors into the HeLa and Saos-2 cell culture. As shown in Figure S9, the addition of the soluble ALP abrogates the cytotoxicity of the precursors to a certain extent. This result therefore proves that the dephosphorylation of the precursors by the membrane-bound ALPs on the cell surface, indeed, localize the self-assembly of the hydrogelators on the cell surface to form nanofibers in the pericellular space to inhibit the cancer cells.
The Modality of Cell Death
To unravel the mechanism of cell death induced by pericellular nanofibers formed by EISA, we examine the effect of a pan-caspase inhibitor (i.e., zVAD-fmk (45 μM))31 and a necroptosis inhibitor (i.e., Nec-1 (50 μM)),32 respectively, on the cytotoxicity caused by b-1p (the most effective one) on HeLa and Saos-2 cells. zVAD-fmk, a cell permeable irreversible caspase inhibitor with no cytotoxic effects, obviously ameliorates the cytotoxicity of b-1p against HeLa cells but hardly shows any effect on Saos-2 cells. On the contrary, Nec-1, significantly suppresses the inhibition of b-1p on Saos-2 cells but aggravates the situation in case of HeLa cells (Figure 8). These results indicate that the nanofibers formed by b-1p after EISA largely result in apoptosis of HeLa cells and necroptosis of Saos-2 cells.
Figure 8.
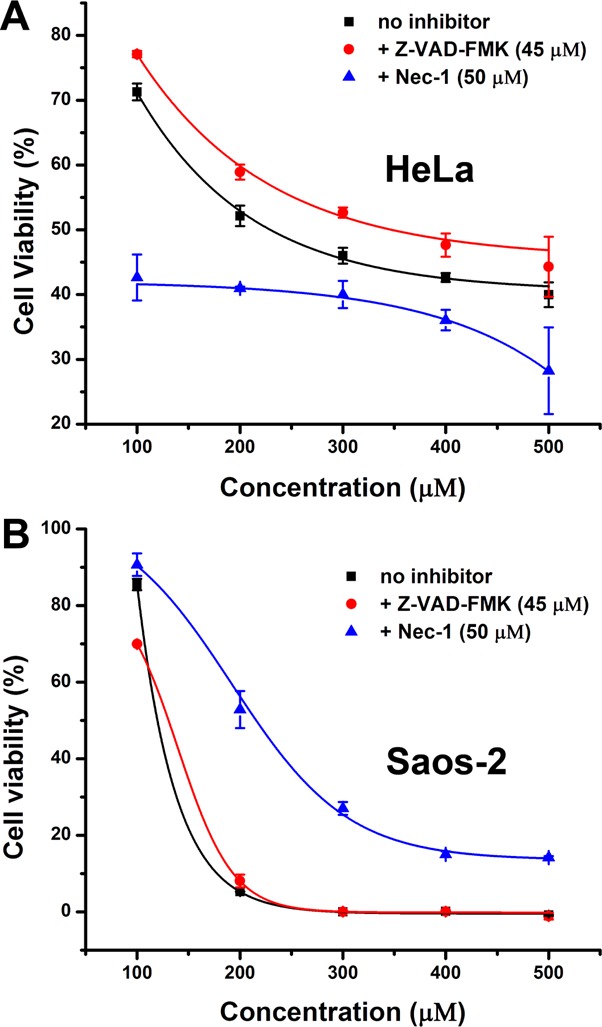
Dose-curves show that (A) pan-caspase inhibitor (Z-VAD-FMK) suppresses the cytotoxicity of b-1p on HeLa cells while necroptosis inhibitor (Nec-1) aggravates the inhibition and (B) Z-VAD-FMK has no effect on the toxicity of b-1p on Saos-2 cells but Nec-1 ameliorates the inhibition.
Rates of Dephosphorylation
To examine the relationship between enzymatic dephosphorylation rate and cellular response, we also evaluate the rate of enzymatic dephosphorylation process, using 31P NMR at the gelation concentration (i.e., 0.5 wt %, ALP = 0.02 U/mL). Unlike 31P NMR obtained in DMSO (Figure S2), which shows two peaks for the two phosphates on a single molecule when enlarging the scale, 31P NMR is unable to distinguish the two phosphates of a-2p, b-2p, and c-2p when the solvent is Tris buffer. According to time-dependent 31P NMR spectra (Figure S10) of the reaction mixture, the dephosphorylation of a-2p or a-1p almost completes within 24 h, though that of a-2p is slightly slower, likely due to two phosphates on a-2p. The dephosphorylation rates of c-2p and c-1p are slower than those of a-2p and a-1p, followed by those of b-2p and b-1p. The dephosphorylation rates of these precursors at gelation concentrations follow the opposite trend to their cytotoxicity against cancer cells, which likely originates from the strong tendency of self-assembly of b. Even a small portion of precursors, for example, of b-1p, are dephosphorylated, the mixture tends to self-assemble to make the solution viscous, which decreases the diffusion of molecules and enzymes and retards the dephosphorylation process. This observation, indeed, agrees with the strong self-assembling tendency of DFDYDFDY suggested by TEM and rheology data (vide supra).
The opposing trends between the results in Figure 4 and Figure 9 prompt us to evaluate the dephosphorylation process at the concentration used in cell culture (i.e., 500 μM) in PBS buffer for better understanding the different cytotoxicities exhibited by different precursors. For a-2p, b-2p and c-2p, the dephosphorylation of either of the two phosphotyrosines produces the intermediate P1 or P1′ (Figure S11). We would expect that enzyme shows different affinity to two phosphates at different positions on the precursors. However, according to Figure 9A, B, C, the curves of these intermediates, which goes up first and decays later, hardly show significant difference, suggesting that ALPs dephosphorylate the phosphotyrosine regardless its position on the precursors. In the case of monophosphorylated precursors (i.e., a-1p, b-1p, and c-1p), ALPs dephosphorylate these precursors at almost the same rate, indicating that the structure difference barely affects enzymatic dephosphorylation (Figure 9D, E, F). These results, on the other hand, suggest that the different cytotoxicity of a-1p, b-1p, and c-1p largely originates from the self-assembling abilities of a, b, and c, which depend on their molecular structures.
Figure 9.
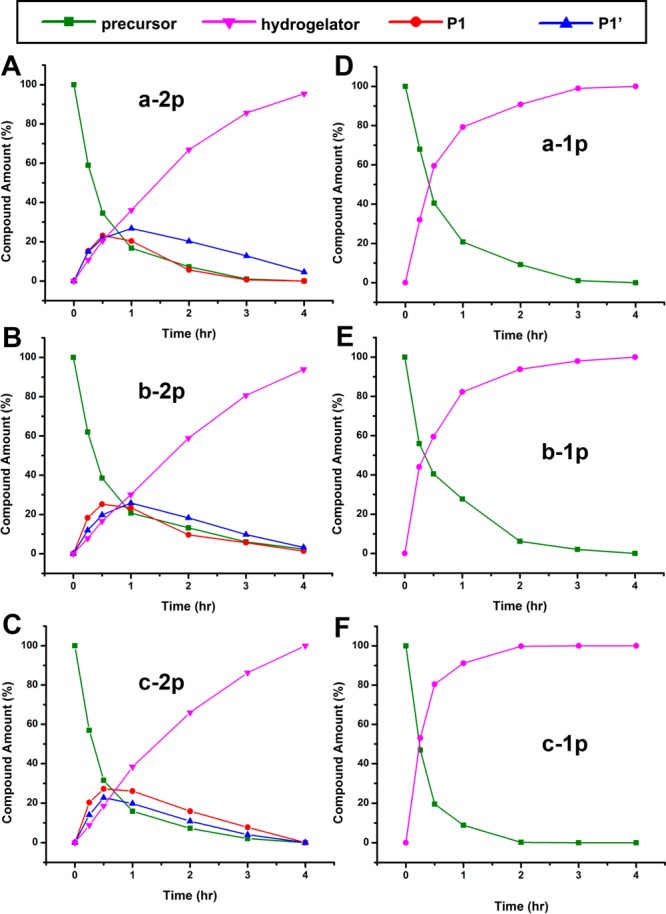
Time-dependent curves for dephosphorylation process of the precursors (A) a-2p, (B) b-2p, (C) c-2p, (D) a-1p, (E) b-1p, (F) c-1p, after incubation with ALP (0.1 U/mL) at 37 °C. The precursors gradually turn into hydrogelators after enzymatic dephosphorylation by ALP. The precursors dissolve in PBS (PBS) buffer at a concentration of 500 μM.
Conclusions
In summary, we designed and synthesized a series of d-tetrapeptide precursors containing one or two enzymatic dephosphorylation sites (i.e., phosphotyrosine) and evaluated their inhibitory activities on several representative cells lines. Besides establishing that EISA is a fundamentally new multiple-step process for selectively inhibiting cancer cells, our results reveal several key insights related to the structure–activity relationship (SAR) of EISA for killing cancer cells. (1) The increase of the number of phosphotyrosine, though improving the aqueous solubility of the precursors, hardly improves the inhibitory activity of EISA. This result suggests that it is critical to generate the self-assembling molecules quickly for inhibiting cancer cells. (2) The relatively high activities of b-1p and b-2p imply that the phenylalanine residues on the same side of peptidic backbone apparently result in more effective self-assembly. (3) The modality of cell death caused by EISA depends on the cell types, which implies it may depend on the isozymes of ALPs. However, the detailed cell death mechanism44 remains to be elucidated. (4) By varying the numbers of enzyme action sites on the peptides, one should be able to tune the cytotoxicity against different cancer cells by controlling the self-assembling process. (5) Most importantly, the observation of the apparently counterintuitive results—precursors significantly inhibit the cancer cells, while hydrogelators remain innocuous—confirms that EISA, as a multiple-step process, accounts for the selective inhibition of cancer cells. Since ALP may play a key role in cell survival via purinergic signaling pathway,45 this work illustrates a facile way to turn a cell survival signaling into a cell killing process by simply engineering the molecules for dephosphorylation and taking advantage of the ALPs overexpressed on cancer cells. In a more broad perspective, the approach illustrated in this work should be applicable for the exploration of supramolecular assemblies in cellular environment by using other enzymes46 and other well-established self-assembling molecules and systems.47
Acknowledgments
We thank Brandeis EM and Optical Imaging facilities for TEM and fluorescent imaging. JZ is an HHMI student fellow. This work was partially supported by NIH (R01CA142746) NSF (MRSEC-1420382) and Keck Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b13541.
The details of the synthesis, NMR and LC–MS data for all compounds, and rheological data. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ridley A. J.; Schwartz M. A.; Burridge K.; Firtel R. A.; Ginsberg M. H.; Borisy G.; Parsons J. T.; Horwitz A. R. Science 2003, 302, 1704. 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Heng Y.-W.; Koh C.-G. Int. J. Biochem. Cell Biol. 2010, 42, 1622. 10.1016/j.biocel.2010.04.007. [DOI] [PubMed] [Google Scholar]
- a Schulte M.; Reiss K.; Lettau M.; Maretzky T.; Ludwig A.; Hartmann D.; de Strooper B.; Janssen O.; Saftig P. Cell Death Differ. 2007, 14, 1040. 10.1038/sj.cdd.4402101. [DOI] [PubMed] [Google Scholar]; b Hanada K.-i.; Wang Q. J.; Inozume T.; Yang J. C. Blood 2011, 117, 4816. 10.1182/blood-2010-11-317743. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Black R. A.; Rauch C. T.; Kozlosky C. J.; Peschon J. J.; Slack J. L.; Wolfson M. F.; Castner B. J.; Stocking K. L.; Reddy P.; Srinivasan S.; Nelson N.; Boiani N.; Schooley K. A.; Gerhart M.; Davis R.; Fitzner J. N.; Johnson R. S.; Paxton R. J.; March C. J.; Cerretti D. P. Nature 1997, 385, 729. 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- a Hengartner M. O. Nature 2000, 407, 770. 10.1038/35037710. [DOI] [PubMed] [Google Scholar]; b Shi Y. Curr. Opin. Cell Biol. 2006, 18, 677. 10.1016/j.ceb.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Lu A.; Magupalli V. G.; Ruan J.; Yin Q.; Atianand M. K.; Vos M. R.; Schroeder G. F.; Fitzgerald K. A.; Wu H.; Egelman E. H. Cell 2014, 156, 1193. 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si K.; et al. Nat. Rev. Neurosci. 2004, 5, 81. 10.1038/nrn1341. [DOI] [Google Scholar]
- Huang C.; Yu H.; Miao Z.; Zhou J.; Wang S.; Fun H.-K.; Xu J.; Zhang Y. Org. Biomol. Chem. 2011, 9, 3629. 10.1039/c1ob05143a. [DOI] [PubMed] [Google Scholar]
- Gilks N.; Kedersha N.; Ayodele M.; Shen L.; Stoecklin G.; Dember L. M.; Anderson P. Mol. Biol. Cell 2004, 15, 5383. 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Colby D. W.; Prusiner S. B. Cold Spring Harbor Perspect. Biol. 2011, 3, 3. 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Prusiner S. B. Annu. Rev. Genet. 2013, 47, 601. 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru H.; Zhang P.; Wu H. Mol. Cell 2014, 54, 208. 10.1016/j.molcel.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R. J.; Smith A. M.; Collins R.; Hodson N.; Das A. K.; Ulijn R. V. Nat. Nanotechnol. 2009, 4, 19. 10.1038/nnano.2008.378. [DOI] [PubMed] [Google Scholar]
- Weisenberg R. C.; Deery W. J.; Dickinson P. J. Biochemistry 1976, 15, 4248. 10.1021/bi00664a018. [DOI] [PubMed] [Google Scholar]
- a Sigler P. B.; Xu Z. H.; Rye H. S.; Burston S. G.; Fenton W. A.; Horwich A. L. Annu. Rev. Biochem. 1998, 67, 581. 10.1146/annurev.biochem.67.1.581. [DOI] [PubMed] [Google Scholar]; b Higgs H. N.; Blanchoin L.; Pollard T. D. Biochemistry 1999, 38, 15212. 10.1021/bi991843+. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Xu B. Bioconjugate Chem. 2015, 26, 987. 10.1021/acs.bioconjchem.5b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. M.; Xu K. M.; Guo Z. F.; Guo Z. H.; Xu B. Adv. Mater. 2007, 19, 3152. 10.1002/adma.200701971. [DOI] [Google Scholar]
- a Kuang Y.; Shi J.; Li J.; Yuan D.; Alberti K. A.; Xu Q.; Xu B. Angew. Chem., Int. Ed. 2014, 53, 8104. 10.1002/anie.201402216. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shi J.; Du X.; Yuan D.; Zhou J.; Zhou N.; Huang Y.; Xu B. Biomacromolecules 2014, 15, 3559. 10.1021/bm5010355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Du X.; Zhou J.; Guvench O.; Sangiorgi F. O.; Li X.; Zhou N.; Xu B. Bioconjugate Chem. 2014, 25, 1031. 10.1021/bc500187m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Du X.; Zhou J.; Xu B. J. Colloid Interface Sci. 2015, 447, 273. 10.1016/j.jcis.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Millan J. L. J. Biol. Chem. 1986, 261, 3112. [PubMed] [Google Scholar]; b Lange P. H.; Millan J. L.; Stigbrand T.; Vessella R. L.; Ruoslahti E.; Fishman W. H. Cancer Res. 1982, 42, 3244. [PubMed] [Google Scholar]; c Doellgast G. J.; Fishman W. H. Nature 1976, 259, 49. 10.1038/259049a0. [DOI] [PubMed] [Google Scholar]; d Solomon C. G.; Gibson C. J.; Britton K. A.; Miller A. L.; Loscalzo J. N. Engl. J. Med. 2014, 370, 1742. 10.1056/NEJMcps1213589. [DOI] [PubMed] [Google Scholar]; e Stolbach L. L.; Krant M. J.; Fishman W. H. N. Engl. J. Med. 1969, 281, 757. 10.1056/NEJM196910022811403. [DOI] [PubMed] [Google Scholar]
- Pires R. A.; Abul-Haija Y. M.; Costa D. S.; Novoa-Carballal R.; Reis R. L.; Ulijn R. V.; Pashkuleva I. J. Am. Chem. Soc. 2015, 137, 576. 10.1021/ja5111893. [DOI] [PubMed] [Google Scholar]
- Tanaka A.; Fukuoka Y.; Morimoto Y.; Honjo T.; Koda D.; Goto M.; Maruyama T. J. Am. Chem. Soc. 2015, 137, 770. 10.1021/ja510156v. [DOI] [PubMed] [Google Scholar]
- a Orimo H.; Shimada T. Mol. Cell. Biochem. 2008, 315, 51. 10.1007/s11010-008-9788-3. [DOI] [PubMed] [Google Scholar]; b Fedde K. N.; Lane C. C.; Whyte M. P. Arch. Biochem. Biophys. 1988, 264, 400. 10.1016/0003-9861(88)90305-0. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Kuang Y.; Guo Z.-F.; Guo Z.; Krauss I. J.; Xu B. J. Am. Chem. Soc. 2009, 131, 13576. 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- Huang P.; Gao Y.; Lin J.; Hu H.; Liao H. S.; Yan X.; Tang Y.; Jin A.; Song J.; Niu G.; Zhang G.; Horkay F.; Chen X. ACS Nano 2015, 9, 9517. 10.1021/acsnano.5b03874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brunton L.; Blumenthal D.; Buxton I.; Parker K.. Goodman and Gilman’s Manual of Pharmacology and Therapeutics; McGraw-Hill Medical: 2008. [Google Scholar]; b Rautio J.; Kumpulainen H.; Heimbach T.; Oliyai R.; Oh D.; Jaervinen T.; Savolainen J. Nat. Rev. Drug Discovery 2008, 7, 255. 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- Uhlen M.; Fagerberg L.; Hallstroem B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson A.; Kampf C.; Sjoestedt E.; Asplund A.; Olsson I.; Edlund K.; Lundberg E.; Navani S.; Szigyarto C. A.-K.; Odeberg J.; Djureinovic D.; Takanen J. O.; Hober S.; Alm T.; Edqvist P.-H.; Berling H.; Tegel H.; Mulder J.; Rockberg J.; Nilsson P.; Schwenk J. M.; Hamsten M.; von Feilitzen K.; Forsberg M.; Persson L.; Johansson F.; Zwahlen M.; von Heijne G.; Nielsen J.; Ponten F. Science 2015, 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Long M. J. C.; Shi J.; Hedstrom L.; Xu B. Chem. Commun. 2012, 48, 8404. 10.1039/c2cc33631f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J. F.; Xu B. Nano Today 2015, 10, 615. 10.1016/j.nantod.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis R. F. ACS Med. Chem. Lett. 2015, 6, 618. 10.1021/acsmedchemlett.5b00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoylaerts M. F.; Manes T.; Millan J. L. Biochem. J. 1992, 286, 23. 10.1042/bj2860023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon M. D.; Whyte M. P.; Teitelbaum S. L. Lab. Invest. 1980, 43, 489. [PubMed] [Google Scholar]
- Julien O.; Kampmann M.; Bassik M. C.; Zorn J. A.; Venditto V. J.; Shimbo K.; Agard N. J.; Shimada K.; Rheingold A. L.; Stockwell B. R.; Weissman J. S.; Wells J. A. Nat. Chem. Biol. 2014, 10, 969. 10.1038/nchembio.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N.; Duprez L.; Grootjans S.; Cauwels A.; Nerinckx W.; DuHadaway J. B.; Goossens V.; Roelandt R.; Van Hauwermeiren F.; Libert C.; Declercq W.; Callewaert N.; Prendergast G. C.; Degterev A.; Yuan J.; Vandenabeele P. Cell Death Dis. 2012, 3, e437. 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S. K.; Petsko G. A. Science 1985, 229, 23. 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Li N.; Delgado J.; Gao Y.; Kuang Y.; Fraden S.; Epstein I. R.; Xu B. Langmuir 2012, 28, 3063. 10.1021/la203923d. [DOI] [PubMed] [Google Scholar]
- Du X.; Zhou J.; Shi J.; Xu B. Chem. Rev. 2015, 115, 13165. 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alewood P. F.; Johns R. B.; Valerio R. M.; Kemp B. E. Synthesis 1983, 1983, 30. 10.1055/s-1983-30206. [DOI] [Google Scholar]
- Chan W. C., White P. D., Eds.; Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford Univ. Press, 2000. [Google Scholar]
- Roecklein B. A.; Torok-Storb B. Blood 1995, 85, 997. [PubMed] [Google Scholar]
- Uhlén M.; Fagerberg L.; Hallström B. M.; Lindskog C.; Oksvold P.; Mardinoglu A.; Sivertsson Å.; Kampf C.; Sjöstedt E.; Asplund A. Science 2015, 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Shi J.; Yuan D.; Xu B. Nat. Commun. 2012, 3, 2040/1. 10.1038/ncomms2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan J. L.Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2006. [Google Scholar]
- Fishman W. H.; Inglis N. R.; Green S.; Anstiss C. L.; Gosh N. K.; Reif A. E.; Rustigian R.; Krant M. J.; Stolbach L. L. Nature 1968, 219, 697. 10.1038/219697a0. [DOI] [PubMed] [Google Scholar]
- Fishman W. H. Clin. Biochem. 1990, 23, 99. 10.1016/0009-9120(90)80019-F. [DOI] [PubMed] [Google Scholar]
- a Siegel R. M.; Frederiksen J. K.; Zacharias D. A.; Chan F. K. M.; Johnson M.; Lynch D.; Tsien R. Y.; Lenardo M. J. Science 2000, 288, 2354. 10.1126/science.288.5475.2354. [DOI] [PubMed] [Google Scholar]; b Chan F. K. M.; Chun H. J.; Zheng L. X.; Siegel R. M.; Bui K. L.; Lenardo M. J. Science 2000, 288, 2351. 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Nat. Rev. Drug Discovery 2008, 7, 575. 10.1038/nrd2605. [DOI] [PubMed] [Google Scholar]
- Liao S. W.; Yu T.-B.; Guan Z. J. Am. Chem. Soc. 2009, 131, 17638. 10.1021/ja907097t. [DOI] [PubMed] [Google Scholar]
- a Maeda Y.; Javid N.; Duncan K.; Birchall L.; Gibson K. F.; Cannon D.; Kanetsuki Y.; Knapp C.; Tuttle T.; Ulijn R. V.; Matsui H. J. Am. Chem. Soc. 2014, 136, 15893. 10.1021/ja509393p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yoshii T.; Mizusawa K.; Takaoka Y.; Hamachi I. J. Am. Chem. Soc. 2014, 136, 16635. 10.1021/ja508955y. [DOI] [PubMed] [Google Scholar]; c Anzini P.; Xu C.; Hughes S.; Magnotti E.; Jiang T.; Hemmingsen L.; Demeler B.; Conticello V. P. J. Am. Chem. Soc. 2013, 135, 10278. 10.1021/ja404677c. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Weiss R. G. J. Am. Chem. Soc. 2014, 136, 7519. 10.1021/ja503363v. [DOI] [PubMed] [Google Scholar]; e Cheetham A. G.; Zhang P.; Lin Y.-a.; Lock L. L.; Cui H. J. Am. Chem. Soc. 2013, 135, 2907. 10.1021/ja3115983. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Weingarten A. S.; Kazantsev R. V.; Palmer L. C.; Fairfield D. J.; Koltonow A. R.; Stupp S. I. J. Am. Chem. Soc. 2015, 137, 15241. 10.1021/jacs.5b10027. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Nagy K. J.; Giano M. C.; Jin A.; Pochan D. J.; Schneider J. P. J. Am. Chem. Soc. 2011, 133, 14975. 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.