

Abstract
Lentiviral vectors (LV) have seen considerably increase in use as gene therapy vectors for the treatment of acquired and inherited diseases. This review presents the state of the art of the production of these vectors with particular emphasis on their large-scale production for clinical purposes. In contrast to oncoretroviral vectors, which are produced using stable producer cell lines, clinical-grade LV are in most of the cases produced by transient transfection of 293 or 293T cells grown in cell factories. However, more recent developments, also, tend to use hollow fiber reactor, suspension culture processes, and the implementation of stable producer cell lines. As is customary for the biotech industry, rather sophisticated downstream processing protocols have been established to remove any undesirable process-derived contaminant, such as plasmid or host cell DNA or host cell proteins. This review compares published large-scale production and purification processes of LV and presents their process performances. Furthermore, developments in the domain of stable cell lines and their way to the use of production vehicles of clinical material will be presented.
Introduction
With the first marketing authorization of an AAV1 vector for the treatment of lipoprotein lipase deficiency (Glybera®) in Europe,1 viral vector-based gene therapy is more and more rapidly evolving towards the routine treatment of rare and acquired diseases for which different viral vectors systems are available. Depending on the purpose of the treatment as well as the target cells or tissues to be treated, one or the other vector system is preferable. In case of dividing, tissues or cells integrating vectors are required for the long-term expression of the transgene. Traditionally, retroviral vectors (in a large sense) are the vectors of choice because they lead to a stable integration of the transgene to be expressed. Mainly two different retroviral vector systems have been developed: γ-retroviral vectors derived from murine leukemia viruses (MLV)2 and lentiviral vectors (LV) mainly derived from HIV-1.3 In the past, many clinical trials based on the use of MLV vectors were successful4 and although these vectors are still used, the general tendency is towards the use of LV vectors. Different reasons can be quoted for this shift: (i) in contrast to γ-retroviral vectors, LVs are able to transduce nondividing cells because they can translocate across the nuclear membrane5; (ii) their integration patterns are different from MLV vectors and seem to be less risky with respect to insertional mutagenesis6; and (iii) they can be produced at high vector titer.
These are the main reasons why there is a clear transition from the use of MLV to LV vectors though the overall manufacturing conditions for LV vectors have not yet reached their maximal potential and the level of those used for MLV vectors.
LV vectors have been used successfully in clinical trials, in a first instance for the treatment of rare diseases, in particular, of primary immunodeficiencies7,8 and in neurodegenerative storage diseases.9,10 However, their application for the treatment of more frequent genetic and acquired diseases, including treatment of β-thalassemia,11 Parkinson’s disease,12 and chimeric antigen receptor-based immunotherapy of cancer,13 has been assessed in clinics with exciting outcomes. This means that manufacturing technology becomes a critical issue in view of the implementation of these novel therapies for routine use.
Thus this review, based on publically available sources, presents the actual state of the art of production means for LV vectors, providing information on advantages and short comings of actual protocols (or methods) and devices as well as on maximal manufacturing levels achievable (titer, total vector quantity) and finishing with a perspective of what should come next.
LV Vector System(S)
The prototype LV vector system is based on HIV-1, a very well-studied human pathogen virus. Besides HIV-1, other lentiviruses have also been developed as gene transfer vectors (TVs) but most of them have not yet reached the clinical study stage, such as HIV-2 (ref. 14) simian immunodeficiency viruses,15 or nonprimate lentiviruses including feline immunodeficiency virus,16 bovine immunodeficiency virus17 or caprine arthritis-encephalitis virus.18 Only equine infectious anemia virus (EIAV)-based vectors19 have been developed up to clinical use.
In the following, this review article will focus on HIV-1-based LV vector system.
Four-plasmid systems
Essentially guided by safety considerations due to the pathogenicity of HIV-1 in humans, different generations of LV vector systems20 have been developed of which the third generation is widely used for R&D and clinical purposes today. It is a four-plasmid system (Figure 1), consisting of three helper plasmids and one TV plasmid. The choice of the helper plasmids was dictated by the principle of the rationale design of a split genome conditional packaging system described by Dull et al.21 This production system is associated with all required features necessary for a safe use in clinics (use of nonoverlapping split-genome packaging constructs to maximally reducing the potential recombination events, which could lead to the generation of replication-competent lentivirus (RCL)).21 All accessory genes of HIV-1 (vif, vpr, vpu, and nef),22 present only in the very first generation of LV, have been removed because they are not necessary. Similarly, the regulatory tat gene, present in the second-generation LV, has been eliminated because its transacting function is dispensable as the U3 promoter of the 5′ long terminal repeat (LTR) in the TV has been replaced by a constitutively active promoter sequence, such as cytomegalovirus (promoter)22–24 or Rous Sarcoma Virus (promoter)21 plus an optional enhancer25 or an inducible/repressible promoter sequence, such as 7tetO.26 This is commonly referred as the pRRL (lentivirus transfer vector construct containing chimeric Rous sarcoma virus (RSV)-HIV 5’ LTRs) design or the pCCL design ((CMV)-HIV 5′LTR). These modifications lead to a LV vector system with the helper functions based on the use of gag-pol (encoding for the structural proteins and viral enzymes) and rev (encoding for a post-transcriptional regulator) derived from HIV-1 and env. Although LV vectors can be pseudotyped with different heterologous envelope glycoproteins,27 all large-scale (clinical scale) vector preparations have made use of the glycoprotein of the vesicular stomatitis virus (VSV-g) envelope, due to improved stability during downstream processing and its large transduction spectrum.
Figure 1.
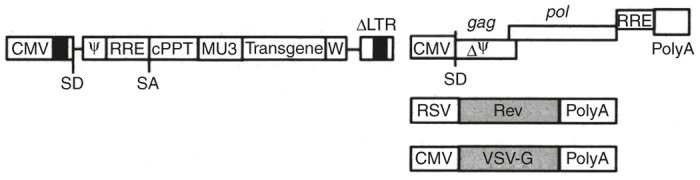
One example of the third generation Tat-independent HIV-1 vector system. The shown SIN transfer vector contains the cPPT for efficient nuclear import and uses the MSCV LTR promoter (MU3) as internal promoter for driving the expression of the transgene, as well as the WPRE (W) element for high-level transgene expression. The three other packaging constructs encode for the HIV-1 gag-pol and rev proteins as well as for the VSV-g envelope glycoprotein.20 SIN, self-inactivating; VSV-g, glycoprotein of the vesicular stomatitis virus.
The TV plasmid is the only genetic material transferred to the target cells and consists of the LV backbone containing the transgene expression cassette flanked by cis-acting elements required for encapsidation, reverse transcription, and integration. In view of biosafety improvement, self-inactivating (SIN)-LV vectors have been developed, for which a deletion has been introduced into the U3 element of the 3′LTR.23,24 This type of vector loses the transcriptional capacity of the viral LTR once transferred to the target cells minimizing the risk of emergence of replication competent recombinants and avoiding problems linked to promoter interference.
Considering vector production, HEK293 or HEK293T cells (see below) are transfected with four plasmids encoding the gag-pol genes, the rev gene, the VSV-g envelope gene, as well as a SIN LV TV plasmid with an internal promoter for transgene expression (Figure 1).
Further safety improvements of the LV vector system are related to the removal of residual sequence homologies existing between the vector genome because the packaging signal (Ψ) that extends into the first bp of the gag gene28 and the gag-pol construct via codon optimization. Given that codon optimization renders the translation of gag-pol proteins rev-independent29,30 the Rev-Responsive-Element (RRE) sequence can be removed from the packaging construct. Since the TV contains a portion of the gag sequence codon optimization of the gag-pol sequence renders the so-called ψ-gag recombination (for instance, reported by Sastry et al.31) impossible.32 An additional advantage of codon optimization is about 100-fold reduced frequency of potential recombination events.30
A complete rev-independence of the LV vector system characterized by the absence of the RRE sequence in the TV has not been shown for the moment. The function of RRE (which is part of the unspliced vector transcript) consists in fact in the assembly of multiple molecules of Rev to form an oligomeric complex stabilizing and mediating the export of the vector transcripts from the nucleus to the cytoplasm during the production of the vector, but not after integration of the TV in target cells.33 Alternative export sequences, such as the constitutive transport elements or the RNA transport element, were shown to be functional in the context of the gag-pol production,34–36 but they have not yet been shown to be as efficient as RRE sequence in context of the TV.3,22,35 Thus, today, LV vector systems still make use of Rev/RRE for efficient vector genome export to the cytoplasm.
Three-plasmid systems
A rev-independent vector technology was only successfully implemented for the EIAV system in which only three plasmids (two helper plasmids coding for the gag-pol and the env functions and the TV plasmid) are required37 (Table 1).
Table 1. Large-scale cell culture GMP productions of LV vectors.
| Company/institution | Cell line, vector system | Culture system (number of culture units) | Number of plasmids | Maximal production scale (l) | Titer (before purification)b |
|---|---|---|---|---|---|
| Virxsys52 | 293, HIV-1 | NC | 2 | 36–52 | 2.02 × 107 TU/ml |
| Généthon42/MolMed105 | 293T, HIV-1 | CF-10 (12–24) | 4 | 24–50 | 1–5 × 107 IG/ml |
| Beckman Research In-stitut (City of Hope/CA)43,61 | 293T, HIV-1 | CF-10 (12 × 10 = 120) | 4 | 120 | 0.5–1 × 106 TU/ml |
| Oxford Biomedica/Henogen[55, Mitrophanous, personal communication] | 293T, EIAV | CF-10 (24 per run, 3 campaigns) | 3 | 72 | 0.2–2 × 106 TU/ml |
| Généthon63,123 | 293T, HIV-1 | STR (50/ 200 l) | 4 | 50/200 | 5 × 107 IG/ml |
| St. Jude Children’s Hospital56 | Stable 293T, tet-off, HIV-1 | 50-l WAVE reactor with HEK293T cells immobilized on Fibra-cel | Induction by removal of doxycycline | About 138 per batch | 0.5–1 × 107 TU/ml |
| University of California Davis School of Medicine62 | 293T, HIV-1 | Hollow fiber systema | 4 | NC | 1.0–2.8 × 108 vg/ml |
LV, lentiviral vectors; NC, not communicated; STR, stirred tank reactor.
Equivalent with three CF-10.
It has to be kept in mind that the titers between the different laboratories cannot be compared because different transgenes, different promoters, and non standardized analytical methods have been used.
In the past, Virxsys has developed a two-plasmid system for the treatment of HIV infection using a conditionally replicating LV vector.38 This system is characterized by the assembly of all helper functions (gag-pol, rev, tat, and VSV-g) on one plasmid.39 Due to the use of full LTRs, this system is tat-dependent and therefore by definition a second-generation LV vector system. The maintenance of full LTRs is retained to allow the transcription of the antisense against the HIV envelope gene39 only in the HIV-infected target cells that express Tat. Though such a production system is easier to produce and less expensive in its application and leads to higher vector titer than the three or four plasmid systems,39 the presence of all helper genes located on one plasmid might be a concern with respect to the formation of RCLs. However, none of the vector lots or of the transduced cell products were shown to contain RCLs.40
Cell Lines Used for Vector Production
Most of the current LV vector production methods involve cotransfection of, preferentially, HEK293T or HEK293 cells. The reason for the preference of HEK293T cells is that the presence of the SV40 T-antigen in the producer cells renders them more efficient for vector production. In addition, HEK293T cells show increased cell growth and transfection efficiency in comparison to HEK293 cells.41–43 A thorough explanation for the improvement of the “cellular context” cannot be given today, however, Gama-Norton et al.41 could show that, in the context of stable HEK293-based producer cell lines, clones containing the SV40 T-antigen showed higher vector production rates than those not expressing the SV40 T-antigen. The only difference between the clones was the expression/nonexpression of this antigen. The positive effect of the presence of SV40 T-antigen during LV vector production was also shown by Smith and Shioda44 in the context of CV-1 (SV40 T-antigen negative) – COS-1 (SV40 T-antigen positive) cells. The latter cells are of high interest for screening purposes related to LV vectors as well as for automated manufacturing at small scale because of improved vector quality (improved infectious to total vector ratio; reduced level of contaminating cellular proteins) in comparison to HEK293T cells.44 However, the main drawback when using COS-1 cells is that these cells are obligatory adherent cells, whereas HEK293T cells can be adapted to suspension growth in serum-free medium which is of particular interest for large-scale vector productions.
Lentiviral Vector Production
The following section compares the production processes that have been described in the literature from bench scale to industrial manufacturing. The comparison focuses mainly on the technical methods developed by the producers. It would not be appropriate to discuss the performance and productivity of each process since the differences in lentivirus systems, transgenes of interest and titration methods do not allow objective comparisons. Nevertheless, the titers that were reported by the authors are given as an indication in this review.
Vector production at small scale
Small-scale productions for R&D purposes are performed using surface adherent cells grown in Petri dishes, T-flasks, multitray systems (Cell Factories, Cell Stacks), or HYPERFlask. At optimal confluence (<50%)45 cells are transfected using either the traditional Ca-phosphate protocol46 or the more recently developed polyethylenimine (PEI) method47 characterized by several advantages (independence of the culture conditions, no requirement for medium change after transfection, applicability to transfection in suspension culture).48 In principle, for small-scale applications, other very efficient cationic transfection agents including lipofectamine, fugene or 293fectin, have also been used resulting in high expression levels.49
By comparing the different cell culture systems, Ausubel et al.43 did not observe any differences with respect to vector titers produced among the systems evaluated (T-flasks, one- or ten-layer CFs). Concerning the use of HYPERFlasks, Kutner et al.50 observed that probably due to the improved oxygen availability, cultures in HYPERFlasks were able to produce about 10 times more LV vectors per surface unit than when using traditional culture devices. In addition, the crude vector product was less contaminated by cellular proteins and nucleic acid compared with supernatants produced in traditional dish cultures.
Since attachment of HEK293T cells to culture surfaces is not very strong, vector production is more difficult to perform in roller bottles and transfection conditions in these devices have to be carefully optimized for keeping cells attached. In this context, Patel et al.51 could show that the overexpression of alpha-v and beta-3 integrin by HEK293 led to an increased cell adhesion allowing efficient LV vector production in roller bottles. However, this approach requires the use of recombinant HEK293 cells overexpressing integrin.
Vector production at large scale
When moving towards clinical trials larger vector lots are required signifying that the production method has to be scaled up. This can either be performed by using a scale-out approach (addition of supplementary production units) or by the use of suspension cultures which are characterized by much better scalability than when using adherent cell cultures. Both approaches will be presented in the following:
Large-scale vector production using adherently grown cells
Large-scale productions of LV are mostly a direct scale-up (scale-out) of the small-scale production methods by augmenting the culture surface via addition of supplementary culture/production units. The productions are essentially performed in parallel cultures using large numbers of multitray systems (Cell Factories (CF) (CF-10 (Figure 2)) or Cell Stacks (CS)). Because of easier manipulation, 10 stack devices (CS-10) are preferable, though, in principle, 40 stack devices could equally be used, requiring nevertheless a specific handling system due to the elevated weight of the CF-40 stacks. Furthermore, gas exchange as well as medium layers may not be identical for all plates and growth control using microscopy is very difficult when using 40 stack devices. Productions are performed either in an “open mode” under a laminar air flow bench42 or in a semiclosed mode43 providing improved safety for the operator, the environment as well as for the final product.
Figure 2.

Ten stack Cell Factory from Nunc (CF-10).
Harvesting is performed via one simple medium exchange and in some cases the number of harvests has been increased to augment the final vector quantity. However, in the case of preclinical and clinical large-scale productions frequent harvest cycles are not practical, wherefore in most of the cases one to at most three harvests are performed.42,43,52–54
Based on the number of 10-stack culture devices and the number of harvests, traditionally, the harvest volumes range between 20 and 52 l42,52,54 using 12–24 CF-10 devices for a single campaign production schedule.
In the case of multicampaign production protocols (multiple sub-batch system), larger bulk volumes of a drug product (purified and vialed product) can be produced. Ausubel et al.43 presented such a protocol for the production of large volumes of lentiviral material (>100 l) based on the pooling of multiple 10-l sub-batches produced over several weeks. A similar approach was also used by Dupont.55 The harvests of single runs are treated separately leading to the purified bulk product per run which is quality controlled separately. For generating a lot of drug product, the necessary number of aliquots of purified bulk is thawed, pooled, sterile filtered and vialed.
In the case of stable inducible producer cell lines (tet-off induction system) 50-l WAVE reactor cultures (working volume: 25 l) could produce about 138 l of vector-containing cell culture supernatant in a continuous process (harvest period: 3 to 6–8 days postinduction)(56 and see below).
Unprocessed supernatants often contain vector titers ranging from 1 to 5 × 107 infectious particles (ip)/ml42,52 being thus similar to titers reported for small-scale productions.57–60 However, titers in the range of 0.5–1 × 107 and of 0.5–2 × 106 transducing units (TU)/ml have also been reported for large-scale production runs by Greene et al.56 and by Ausubel et al.43, respectively. These differences are mainly influenced by vector components such as the gene of interest, the promoter used, and any additional regulatory elements, but also by the titration methods which are not standardized for the moment.
All large-scale production protocols were developed for HIV-142,52,54,61 and EIAV37,55-based LV vectors.
Very recently a semi-“large-scale” LV vector production system based on the use of hollow fibers has been presented.62 The hollow fibers are seeded with HEK293T cells, which are then transfected with three plasmids after attachment for 24 hours. The advantage consists in the fact that it is a closed, fully automated culture system with an LV yield equivalent to three CF-10 stacks. This, however, requires that several parallel systems have to be set up for real larger scale productions.
Table 1 presents details of the available large-scale vector productions.
Vector production using suspension cultures
Although transfection of adherent cells is the gold standard methodology for producing LV, this system is rather limited in scalability. For industrial manufacturing, cell culture in large bioreactors is usually the most convenient approach.
Production in bioreactors requires the expansion of the producer cells in suspension. Several cell lines used for lentivirus production (293T, 293FT, and 293SF-3F6) have been described to be prone to adaptation to suspension culture in chemically defined media (Freestyle 293 and F17, Invitrogen, Carlsbad, CA; HyQSFM4TransFx293, Hyclone, Logan, UT).63–65 These cells grow readily in suspension with no need for microcarriers rendering thus their culture and expansion much easier than for adherently growing cells. In addition, the absence of bovine serum and animal origin components in the culture media is the most suitable situation for clinical manufacturing as this decreases the risk of contamination by adventitious agents.
As a suspension culture, the cells can be expanded in different kinds of vessels: shake flasks, glass bioreactors, stainless steel bioreactors, wave bags, and disposable stirred tank. In the case of HEK293T cells, expansion, transfection, and lentivirus production have been demonstrated at 50-l scale in single-use bioreactors.63
To transfect suspension cells, DNA precipitation using calcium phosphate is expected to be less effective because of continuous culture stirring. Therefore, other transfection agents like cationic polymers are used most of the time. Linear 25-kDa PEI appears to induce the highest transfection efficiency in 293T and 293-EBNA1 cells, leading to 75% of transfected cells using a green fluorescent protein (GFP) reporter plasmid.66 The same form of PEI is used in the patent from Marceau and Gasmi63 for the production of LV and leads to 90% of GFP-positive cells. However, in this example, the transfection efficiency was measured 48 hours post-transfection. Therefore, the GFP signal might also result from cell transduction by the neo-synthesized lentiviral vector.
For optimal transfection of several HEK293-derived cell lines, the cell density appears to be a critical parameter. Many papers converge towards values around 1 million cells/ml, more precisely between 8 × 105 cells/ml and 1.5 × 106 cells/ml, to proceed to the addition of the PEI/DNA polyplexes.63,66,67
One drawback of PEI-mediated transfection is the amount of plasmid DNA required to reach high transfection efficiency. Ansorge et al.65 used 1 µg/106 293SF-3F9 cells while Marceau and Gasmi63 reported optimal amounts at 2.5 µg/106 293T cells, which would lead to enormous amounts of plasmids at larger scale (e.g., 750 mg of total plasmid DNA for a potential 200-l bioreactor). This large amount of DNA represents a high cost of raw material and a substantial residue to be eliminated by downstream processing.
Another issue with PEI is the absence of analytical method to detect and quantify this molecule in process or in the purified vector preparation. Therefore, it is unclear whether PEI remains copurified with the vector, to which extent and if this can be detrimental or not for the vector infectivity and stability.
Alternatively, electroporation has been described as a method for transfection of eukaryotic cells. The production of lentiviral vector by electroporation has been shown possible and effective by Witting et al.64 However, this method appears difficult to translate to industrial scale as the electroporation step requires concentrating the cell culture to 108 cells/ml. As a matter of fact, the current protocol implies a centrifugation step before electroporation, then dilution back to the volume of the original vessel. At larger scale, such a manipulation would require specific equipment like continuous centrifuge. In general, technical operations during cell-culture phase should be limited as much as possible since they considerably increase the risk of microbial contamination. In addition, centrifugation may lead to substantial cell damage or cell stress that may correlate with lower productivity. Therefore, although promising, the electroporation method needs further development and simplification before being implemented at industrial scales.
In previous examples of lentivirus production by transient transfection in suspension cells, sodium butyrate was added to the culture at a final concentration between 2 and 10 mmol/l. Sodium butyrate, as other inhibitors of histone deacetylases like trichostatin A or valproic acid, has been reported to prevent DNA compaction thus providing more accessibility to promoters. This in turn improves RNA transcription and consequently vector production.68 However, the effect of sodium butyrate remains controversial as the gain in vector productivity is not consistent from one author to another one. For instance, Ansorge et al.65 reported a titer increase of more than 10-fold using 5-mmol/l sodium butyrate for the production of VSV-g-pseudotyped LV.65 At the opposite, Sena-Esteves et al.69 did not observe any gain of productivity for LV-bearing VSV-g protein at 10-mmol/l sodium butyrate while a positive effect was obtained for other pseudotypes.69 Therefore, it is difficult to conclude on the impact of histone deacetylase inhibitors. The major differences in these experiments may come from the plasmid constructs themselves since their possible interactions with histones should theoretically depend on the DNA sequences and the types of promoters. Hence, for each DNA construct the effect of sodium butyrate should be assessed.
In terms of productivity in suspension culture, the published data indicate that LV titers are similar to the values obtained in adherent cells. The titers achieved in the bulk harvest or culture supernatants are in the range of 107 to 108 infectious genomes (IG)/ml or TU/ml63–65 while the productivity of adherent cell systems varies from 106 to 108 TU/ml69 (Table 1). However, the comparison is rather difficult since a limited number of comparative studies has been reported using the same vector and because of differing analytical procedures.
Finally, in adherent cells, the LV culture supernatant can be harvested at least twice at one-day interval (see previous section) which is a good way to increase the cost effectiveness of the process. In suspension, such an approach is much more difficult to implement as the culture medium cannot be collected independently from the cells. A perfusion system has been evaluated by Ansorge et al.65 to allow medium replacement and successive harvests during 7 days. Their results showed that LV titers peaked between 48 and 96 hours post-transfection, suggesting that multiple harvests should be appropriate to suspension culture, too. Although perfusion systems can be complex and costly at large scale, they should have an enormous interest for transient transfection processes since they would allow multiplying the harvest volume by 2 or 3 while keeping constant the plasmid quantities.
In conclusion, large-scale production of LV using transient transfection of suspension cells is feasible and shows promising productivity. However, further developments are required before transferring this technology to industrial manufacturing. The major bottleneck in urge of optimization is the transfection procedure itself that requires massive amounts of plasmid DNA and consequently makes the production process extremely costly. An industrial-friendly transfection technology consuming less DNA and increasing the percentage of producing cells will be the key element to render such a process profitable for industries in the future.
Without those perspectives of improvement, stable producer cell lines will represent a much more affordable production system once they will be available for LV production.
Development of Stable LV Producer Cell Lines
Compared with transient production, stable LV manufacturing is the best option for gene therapy to reduce the production costs and increase the overall safety and reproducibility, which are, currently, the major limitations discouraging the application of the transient method to the large scale. The implementation of stable systems for clinic trials first and ultimately for gene therapy product commercialization represents, therefore, a mandatory milestone to reduce the manufacturing cost and to enhance quality and safety of LV. Since the early 1990s, when the first packaging cells were generated, various construction strategies have been developed differing, essentially, for the method chosen to introduce the vector encoding genes, i.e., plasmids versus integrating vectors, and the nature of the pseudotyping envelopes utilized. The latter has been one of the most important elements guiding towards the development of a larger number of inducible rather than constitutive packaging cells. As the heterologous VSV-g envelope, the most widely utilized envelope for LV, is highly cytotoxic, and its transcription must be induced only at the time of LV production to prevent packaging cells death. Furthermore, the expression of gag and pol genes of HIV is associated to cytotoxic and cytostatic effects and therefore high expression of these genes must also be regulated only during the production phase. The first and more frequently utilized inducible systems are the Tet-on and Tet-off systems, which are based on the addition or removal, respectively, of the tetracycline/doxycycline antibiotic in the culture medium to trigger gene transcription through the tetracycline response element (TRE). When alternative nontoxic envelopes were found suitable for LV pseudotyping, constitutive packaging cells were later generated. This section will treat separately inducible and constitutive packaging cells derived from both HIV-1 and EIAV lentiviruses,3,19 whose main features are summarized in Table 2.
Table 2. Stable packaging cell lines.
| Packaging/cell line | LV generation/virus | Induction system | Envelope | Titer |
|---|---|---|---|---|
|
Inducible | ||||
| HtTA-1/HeLa71 | First/HIV | Tet-off | gp120/gp41 | 7.3 × 103 cfu/ml |
| B16 clone/HeLa72 | First/HIV | Tet-off | gp120/gp41 | 2.9 × 104 IU/ml |
| SODk1CG1/293124 | Second/HIV | Tet-off | VSV-g | 3.0 × 106 TU/ml |
| LVG/293125 | Second/HIV | Tet-off | VSV-g | 3.5 × 106 TU/ml |
| 293G/293126 | Second/HIV | Tet-off | VSV-g | 5.0 × 106 TU/ml |
| SODK1cSCG/29373 | Third/HIV | Tet-off | VSV-g | 2.0 × 106 TU/ml |
| SODk3/29374 | Third/HIV | Tet-off | VSV-g | 1.0 × 107 TU/ml |
| 17B-5/29375 | Second/HIV | Tet-off | VSV-g | 3.5 × 107 TU/ml |
| GPRT-CL204i-EF1α-hγc-OPT/293T/1726,56 | Third/HIV | Tet-off | VSV-g | 5.0 × 107 TU/ml |
| 650MNDhWASp1/293T/1777 | Third/HIV | Tet-off | VSV-g | >1.0 × 107 TU/ml |
| REr1.35/293T83 | Second/HIV | Ecdysone | VSV-g | 1.2 × 105 TU/ml |
| 293-Rev/Gag/poli/29384 | Second and third /HIV | Ecdysone | VSV-g | 3.0 × 105 TU/ml |
| PS5.8 and PS46.2/293T80,81 | Third/EIAV | Tet-on | VSV-g | <1.0 × 106 TU/ml |
| 293SF-PacLV/293SF82 | Third/HIV | Tet-on/Cum. | VSV-g | 3.4 × 107 TU/ml |
|
Constitutive/continuous | ||||
| STAR/293T76 | Second/HIV | NA | RD114-PR, GALV, MLV 4070A | 1.0 × 107 TU/ml |
| WinPac/293T85 | Third/HIV | NA | RD114-PR | 1.0 × 106 TU/ml |
| RD2-MolPack-Chim3/293T | Second/HIV | NA | RD114-TR | 1.0 × 107 TU/ml |
| RD3-MolPack-GFP/293T 89,92 | Third/HIV | NA | RD114-TR | 1.0 × 107 TU/ml |
Cum., cumate; LV, lentiviral vectors.
Inducible packaging cells
Tet-off inducible system.
The well-defined and characterized tetracycline-inducible expression system70 was used in the first-generation packaging cells in which the HIV gp120/gp41 envelope was not substituted yet by the VSV-g envelope and the regulatory and accessory genes were still maintained in the packaging construct.71,72 The HeLa-derived HtTA-1 (i.e., HeLa-derived cell line having been stably transfected with a tetracycline-controlled transactivator (tTA)) cells constitutively expressed tTA, the chimeric protein created by fusing the Escherichia coli tetracycline repressor (TetR) with the activation domain of the herpes simplex virus HSV-VP16 transactivator. The tTA controlled either the expression of the regulatory Rev protein, which in turn stimulated the expression of gag and pol genes,71 or directly the expression of all vector packaging genes.72 Later, the Tet-off system was applied to regulate the expression of the VSV-g envelope and first-generation packaging construct in a human embryonic kidney (HEK) 293-based stable packaging cell line, named SODk1, which could produce virus particles at titers greater than 106 TU/ml for at least 3–4 days. A similar approach was then followed by two independent groups to construct two second-generation stable cells based on the doxycycline-repressible expression of HIV-1 Rev/Gag/Pol and VSV-g envelope in 293G cells, whose LV titers were in the range of 3.5–5 × 106 TU/ml. Yet, the Tet-off system was also exploited to express the conditional SIN (cSIN) TV in SODk1 cells73 and later in the SODk3 third-generation packaging cell line which was devoid of the HIV-1 Tat transactivator and the remaining accessory genes, but Rev.74 In the cSIN TV, the U3 transcription regulatory element is replaced with the Tet-responsive element TRE. This vector design retains, therefore, the SIN properties and, at the same time, allows LV production exclusively in cells expressing the synthetic Tet-regulated trans-activator tTA. Contrary to the standard SIN TV, which must be integrated into stable packaging cells by plasmid transfection to avoid 5′LTR inactivation after reverse transcription, the cSIN TV can be introduced by transduction. In fact, after that the reverse transcription process takes place, the TRE in the presence of Dox guides the full-length vector proviral genome transcription that contains the packaging signal (Ψ) for encapsidation. Following transduction of Tet-regulated SODk1 and SODk3 packaging cell lines with the cSIN TV, high titers of cSIN recombinant vector (>106 TU/ml)73 and (1 × 107 TU/ml),74 respectively, could be generated.
Another strategy for the development of second-generation packaging cells, established by Virxsys in the context of anti-HIV gene therapy, was based on 293 cells operated by a three-level cascade gene regulation system to avoid the toxicity and the leaky expression of the packaging and envelope regulated genes, and the toxicity of the constitutive expression of the tTA itself. Furthermore, codon optimization of the Tat, Rev, and Gag-Pol genes were attained to reduce the risk of homologous recombination. This new approach reduced the overall poisoning effect of the packaging and VSV-g genes, but it was still unable to constrain the leakiness of p24Gag expression. The production of LV carrying the anti-HIV envelope antisense payload VRX496 was allowed for over a period greater than 11 days with the highest titer corresponding to 3.5 × 107 TU/ml and p24Gag 300 ng/ml. No RCL was detected, but long-term analysis demonstrated that partial gene silencing occurred after 2–3 months in culture.75 The short-term stability can be explained by the fact that the Virxsys’ as well as the other packaging cells described so far, were built using plasmids which are frequently silenced over time. To overcome this problem Ikeda et al.76 in 2003 at UCL, London (see below) and later Throm et al.26 in 2009 at St. Jude Children’s Research Hospital, Memphis described the use of integrating MLV vectors as delivery vehicles for integrating the vector genes. The St. Jude’s approach for the treatment of severe combined immunodeficiency X-linked grounded on the construction of inducible third-generation 293T-derived GPR (gag-pol-rev) and second-generation GPRT (gag-pol-rev and tat) packaging cells, in which Rev and Tat expression was tightly regulated by a Tet-off system. High level of Rev induced, in turn, the expression of the remaining packaging genes, the VSV-g gene and the SIN TV, CL204i-EF1α-hγc-OPT, expressing human IL-2Rγc codon-optimized cDNA, and flanked at both extremities by the 400-bp version of the chicken β-globin HS4 insulator. In contrast to the original method described by Ikeda et al.76, in which LTR-driven MLVs were used, Throm and collaborators used SIN-MLV vectors to avoid crossencapsidation of the LTR-MLV packaging genome into the LV particles. Another innovative aspect introduced in this approach was a novel concatemeric array transfection technique for the integration of SIN vector genomes linked to the antibiotic resistance gene. Producer cells expressing either GFP or IL2RG transgene generated supernatants with titers greater than 5 × 107 TU/ml.26 As previously mentioned, the CL204i-EF1α-hγc-OPT vector was produced at scale supporting clinical trial for severe combined immunodeficiency X-linked of ~280 l in two productions obtaining after concentration final titers of 4.5 and 7.2 × 108 TU/ml.56
The St. Jude Children’s Research Hospital took advantage of the GPRGT packaging cells to generate also the 650MNDhWASp1 producer cells expressing the Wiskott–Aldrich syndrome protein for clinical grade production of Wiskott–Aldrich syndrome LV-pseudotyped VSV-g.77 To obtain producer cells, the TV, containing HS4 chromatin insulator in the deleted U3 region, was introduced by a further improvement of the method used by Throm and collaborators.26 The hWASp TV monomers ligated to the bleomycin resistance cassette, instead of the longer concatemeric array, were stably transfected in GPRGT cells. The clone produced unconcentrated vector titers ≥1 × 107 TU/ml and remained stable for up to 8 weeks of continued passaging in culture.77
To the best of our knowledge, up to now, only the GPRG-EF1α-hγcOPT packaging cells have already been used, whereas the 650MNDhWASp1 cells are planned to be used shortly in clinical trials conducted at the St. Jude Children’s Research Hospital.56,77,78
Very recently, the first stable packaging cell line for the production of integration defective lentiviral vectors has been developed by Tal Kafri’s group.80 The packaging cells were obtained by transfecting the tetracycline-regulated packaging construct pTK1574, carrying a D64E integrase mutant, and the VSV-g envelope into PVG3 cells that constitutively express the trans-activator tTA. Producer cells were then established by either the transduction of cSIN-TV (titer: 107 TU/ml) or stable transfection of a novel polypurine tract-deleted TV (titer: 108 TU/ml). Both types of integration defective lentiviral vectors were suitable for transducing in vivo neurons of rat striatum.
Tet-on inducible system.
Oxford BioMedica (Oxford) developed a stable packaging cells based on EIAV lentivirus expressed in 293T cells and a Tet-on inducible system in which the Tet repressor (TetR) tightly regulates the expression of VSV-g and Gag/Pol genes after addition of Dox to the culture medium. This system showed reduced leakiness as compared with the Tet-off system. The EIAV TV encoded “ProSavin”, a therapeutic gene therapy product for Parkinson’s disease.80 Two ProSavin producer cell lines, PS5.8 and PS46.2, were characterized in details showing culture stability for at least 49 days, even in the absence of selective pressure. Although the titers were comparable to the established transient production system, they were on average <106 TU/ml.81
Tet-on/cumate inducible system.
As the Tet-off inducible system could not abolish completely some leakiness for the expression of VSV-g leading to toxicity and poor stability of the packaging cells,8 a double switch system was developed to obtain the 293SFPacLV, a cell line derived from 293SF cells that produced LVs in serum-free suspension cultures. The 293SFPacLV cells expressed the repressor (CymR) of the cumate switch system and the reverse transactivator (rtTA2S-M2) of the tetracycline (Tet) switch system. Gene induction was promoted by the addition of the dox and cumate inducers to the growth medium. The producer cells were able to generate up to 3.4 × 107 TU/ml after transducing the packaging cells with a cSIN-LV expressing GFP transgene.82 Nevertheless, although the very encouraging results obtained with the GFP marker gene, these cells never reached the clinical study stage.
Ecdysone-inducible system.
The ecdysone-inducible system, based on the insect hormone ecdysone analog ponasterone A, was employed in alternative to the tetracycline-regulated technology.83 A ponasterone A-responsive 293T-based cell line was generated in which the expression of gag, pol, rev and VSV-g genes was placed under control of an inducible ecdysone promoter. This cell line consistently produced second-generation pseudotyped lentiviral vector stocks that after concentration gave titers up to 108 TU/ml.83
Similarly, the 293-Rev/Gag/Poli cell line was obtained by introducing HIV rev and gag/pol genes, each under the control of separate ecdysone inducible promoters, into 293 cells in the continuous presence, during selection, of the specific HIV protease inhibitor, Saquinavir, to further control the cytotoxic effect of HIV protease. The 293-Rev/Gag/Poli cells released within 48 hours post-induction, high amounts of HIV Gag/Pol particles (about 10 µg p24/ml) that could package and transduce third-generation HIV vectors to high titers.84
Constitutive packaging cells
Constitutive (or continuous, as also referred) packaging cells with high productivity rate are no doubts more difficult than inducible cells to be obtained. In fact, the toxicity of the vector genes renders unfeasible either the use of the pantropic VSV-g envelope or the selection of p24Gag highly expressing cells. Therefore, this category includes packaging cells carrying only envelopes different from VSV-g and usually producing lower amount of p24Gag as compared with inducible cells.
The first continuous packaging cell line, named STAR cells, were developed by Ikeda et al.76, in 2003. Several innovative aspects were introduced in the STAR’s approach: (i) testing three gamma retroviral envelopes: a derivative of the feline endogenous gamma retrovirus RD114 envelope with an HIV protease site introduced at the R-peptide cleavage site (RD114-Pro), the gibbon ape leukemia virus with an MLV cytoplasmic tail (GALV+) and the MLV 4070A (Ampho) envelopes; (ii) the introduction of the packaging genes not by stable plasmid transfection, but by the use of MLV integrating vector; and (iii) testing HeLa and HT1080, in addition to 293T cells as starting cells. Gag-pol genes were under the transcriptional control of the MLV LTR, whereas the remaining vector genes were expressed by standard plasmids. The STAR technology was suitable for the production of both second- and third-generation LV with a continuous production of up to 850 ng/ml p24 for 3 months in culture with a titer of 107 TU/ml. However, no STAR-derived producer cells reached clinic application for the possibility of crosspackaging the MLV genome encoding Gag-Pol and Rev within LV particles. This risk was later reduced by the use of SIN-MLV vector combined to the recombinase-mediated cassette exchange (RMCE) technology in the development of the WinPac cells.85 The RMCE was first described as a useful technology to exchange different transgenes using the integrated TV in stable gamma retroviral packaging cells,86–88 whereas in the WinPac cells, the RMCE technology was applied to the integration of the packaging genes. A “tagging” SIN-MLV vector expressing GFP was utilized for the screening of the best expressing locus in traced 293FT cells and a “targeting” vector for the exchange of the GFP cassette with a cassette expressing the codon-optimized HIV-1 Gag-Pol genes. Although the RMCE is a very elegant and useful technology for screening transcriptionally open genomic loci, in this case its use seems superfluous because cells selected to support high level of GFP could not support continuous high levels of cytotoxic Gag-Pol gene products and, more importantly, regardless the method of delivery, the best Gag-Pol expressing locus can be easily and directly screened by p24Gag ELISA test of the packaging cell supernatant. The nontoxic RD114-PR envelope, the rev gene, and the vector genome plasmids were serially transfected and producer cells were screened by antibiotic selection. Vector titers in excess of 106 TU/ml, which could be increased to a concentration of 108 TU/ml were obtained. Scale up and reduction of FCS concentration down to 1% achieved a continued production of >5 × 106 TU/ml up to the sixth harvest in HYPERFlask.85
Another model of constitutive packaging technology is represented by the RD-MolPack cells that have been developed by MolMed S.p.A, Milan. The RD-MolPack system is based on traced HEK-293T cells that, similarly to the STAR and WinPac cells, express a derivative of the RD114 envelope. Unlike the WinPac pseudotyped with the RD114-PR, RD-MolPack cells carry the RD114-TR envelope that contains the extracellular and transmembrane domain of RD114 fused to the cytoplasmic tail (TR) of MLV-Ampho 4070 envelope that facilitates its incorporation into the lentiviral vector particles. A unique feature of the RD-MolPack cells is that the HIV-1 gag, pol, rev and hygro-resistance genes were introduced by infecting 293T cells with a chimeric baculo-AAV vector. The cells were first transfected with a plasmid expressing the AAV Rep78 to guide genome integration of the baculo-AAV vector. The resulting intermediate PK-7 clone, expressing two copies of gag-pol-rev genes, showed an extremely high genetic stability for a year of continuous culture either in the absence or presence of hygromycin selection by producing, on average, p24Gag level of 6.7 ± 3.5 and 15.3 ± 8.4 ng/106 cells/day, respectively.89 From PK-7 cells, the RD2-MolPack and RD3-MolPack were then independently derived for the production of second and third-generation LV. Similarly to the approach of the St. Jude Children’s Research Hospital also in MolMed’s strategy, the envelope and, only for RD2-MolPack cells, the Tat gene, were delivered by VSV-g-pseudotyped SIN-LV rather than SIN-MLV integrating vectors. A major safety concern for using either MLV or HIV SIN vectors in the construction of packaging cells is the remote, but real possibility of mobilization of the vector genes (i.e., env or gag-pol) in the newly produced LV particles. It was shown, in fact, either 0.1–0.03% mobilization frequency of true vector integrations for SIN-LV or some residual 3′LTR promoter activity for SIN-MLV.90,91 This safety concern together with that related to RCL formation were ruled out in both packaging cells by specific tests.89,92 The producer RD2-MolPack-Chim3 cells, expressing the anti-HIV chim3 therapeutic gene, a dominant negative HIV Vif,89,93,94 generated LVs that outperformed VSV-g pseudo-typed transiently derived LVs in transducing human cord blood-derived hematopoietic stem cells.95 While the SIN-LV expressing the GFP marker from RD3-MolPack-GFP producer cells transduced human peripheral blood T lymphocytes at 90% with MOI = 3 and at level comparable to that of VSV-g-pseudotyped transiently derived LV.92 Of interest, the titer of both RD2- and RD3-MolPack cells (106 TU/ml and after concentration 108 TU/ml) was higher than that obtained by transient transfection using the RD114-TR envelope.96,97
The different systems presented here are not easily comparable because in most cases cellular productivity, measured as TU or ng p24/cell/day is not reported or nonstandardized assays have been used wherefore reported titers are not comparable. For stable packaging cells, cellular productivity must be considered, with no doubts, one of the most important parameters that makes worth or not further development of a stable system. Low cellular productivity can however be compensated by high infectivity of the LV (TU/ng p24) that generally correlates with high transduction efficiency.
Lentiviral Vector Purification
A downstream recovery process should provide a product with the desired concentration, purity, and the other quality attributes at minimal costs.98 This statement is true for all large-scale manufacturing processes, but often not for small-scale purification protocols destined for research purposes.
Small-scale purification
Most of the research grade LV vector batches have been used as concentrated but rather crude preparations essentially generated via two step centrifugation methods. After concentration of LV particles by ultracentrifugation at about 70,000g (pelleting) the vectors are purified through a sucrose cushion (50,000g) and taken up in a formulation buffer99. An improvement, in particular, with respect to purity represents the combined centrifugation/chromatography-based purification method. For instance, Kutner et al.50 assessed the combination of both purification/concentration methods. The combination of ultracentrifugation through a sucrose cushion followed by an anion exchange chromatography (e.g., Mustang Q membrane cartridge) led to a yield of (TU) 88.2%, whereas the inverted combination yielding 77.6%. In both cases, concentration factors beyond 100× and vector titers >1010 TU/mL were obtained (VSV-g-pseudotyped LV vectors).
The main drawbacks of these methods are the lack of scalability and the fact that the purity of the final vector preparation is often insufficient for an in vivo application due to the presence of residual cell and culture medium/process-derived contaminants. They are coconcentrated potentially leading to adverse effects and/or reduced transduction efficiency when used in vivo.100,101 Thus, purification methods based on chromatographic and membrane-based separation methods have been developed. The advantages are that such purification schemes can be developed in a way that they are scalable and can be implemented for the industrial-scale purification of LV vectors. In this context, several groups have shown that purification protocols based on chromatographic and membrane-based separation methods not only ensure vector safety but significantly improve vector efficacy.52,100–102
General overviews on downstream processing principles for the purification of γ-retroviral vectors and LV at small scale have been published by Segura et al.103 and Rodrigues et al.104
Large-scale downstream processing
In view of industrial application, downstream processing protocols have been developed using process steps traditionally used in the biotech industry. These are membrane (filtration/clarification, concentration/diafiltration using tangential flow filtration (TFF), membrane-based chromatography) and chromatography (ion-exchange chromatography (IEX), affinity chromatography, and size exclusion chromatography-based process steps. The combination of these different process steps is variable and in some cases, different purification principles can be used for the same purpose. Furthermore, a benzonase/DNase treatment for the degradation of contaminating DNA is either part of the downstream protocol or is already performed during vector production.
In principle, three general phases can be distinguished: (i) Capture is the initial purification of the target molecule from either crude or clarified cell culture and leads to elimination of major contaminants. (ii) Intermediate purification consists of steps performed on clarified feed between capture and polishing stages which results in removing specific impurities (proteins, DNA, and endotoxins). (iii) Polishing is the final step aiming at removing trace contaminants and impurities leaving an active and safe product in a form suitable for formulation or packaging. Contaminants at this stage are often «conformers» to the target molecule, trace amounts of other impurities or suspected leakage products. Practically any type of chromatography and ultrafiltration is used for the intermediate purification and the final polishing step(s).
The technologies that have been described for downstream processing LV are summarized in Table 3. The step that significantly differs from one team to the other is the capture step. Scherr et al.100, Yamada et al.102, Slepushkin et al.52, Bellintani et al.105, Merten et al.42 and Greene et al.56 showed that VSV-g-pseudotyped LV vectors could be purified using anion-exchange chromatography, whereby large-scale purification protocols are based on the use of low-pressure column chromatography42,105 or membrane-based ion exchange chromatography.52,55,56 Such membrane cartridges allow the purification of up to 1,500 l/day45,52 when used as first concentration step in a protocol. Scale-up is straightforward leading to pure vector preparations (e.g., removal of >98% of contaminating proteins and DNAs42); however, vector recovery is not as efficient as for ultracentrifugation. The low recovery from chromatography matrix is attributed to the restrictive process conditions that are imposed by LV fragility. As a matter of fact, LVs are very sensitive to pH variations and to high salt concentrations which are the two parameters that are normally altered to optimize binding and elution in ion-exchange chromatography.
Table 3. Standard downstream process technologies applicable to LV purification and recovery.
| Purpose of downstream process steps | Technologies used for LV downstream processing |
|---|---|
| Removal of cells and debris | Frontal filtration 0.45µm42 |
| Centrifugation99 | |
| Capture chromatography | Anion-exchange chromatography (Mustang Q42,52,100,102,105 or DEAE Sepharose52,55,56) |
| Affinity chromatography (heparin)106 | |
| Polishing | Size-exclusion chromatography42,52,105,110 |
| Concentration and buffer exchange | Tangential flow filtration42 |
| Ultracentrifugation99 | |
| DNA reduction | Benzonase42,52,54,55,61,105 |
| Sterilization | 0.2-µm filter113 |
LV, lentiviral vectors.
Alternatively, Segura et al.106 developed an affinity chromatography based on heparin column. Using this approach, up to 94% of proteins impurities and 56% of residual DNA were eliminated while 53% of LV was recovered. Despite these promising data, this kind of chromatography media is not a suitable option for the industry where components of animal origin such as heparin should be precluded.
In order to concentrate the bulk product (supernatant or an intermediate product), TFF systems are employed allowing also diafiltration for buffer exchange and formulation. At small scale or at later stages of an industrial downstream processing protocol centrifuge-based disposable devices42,45,69,100,102,105 and at a larger scale hollow fibre42,52,55,107 or flat membrane cartridges56,106,108 are used. TFF devices have also been applied for the concentration and diafiltration of LV vectors pseudotyped with other than VSV-g envelope proteins.59,69,109 Depending on the therapeutic indication (i.e., in vivo administration or ex vivo application), the concentration factor will vary significantly. For instance, in the case of intracerebral administration of LV, as exemplified by the Parkinson’s disease treatment ProSavin from Oxford Biomedica,12 the vector has to be highly concentrated as the injectable volume in brain is extremely small. In this specific case, a double TFF has been implemented to achieve titers above 108 TU/mL.81 As a consequence, the TFF was the ultimate step of the purification process since any additional procedure like polishing or 0.2-µm filtration would have resulted in significant loss or dilution of the final product.
Finally, size exclusion chromatography can be employed as polishing step because it efficiently removes all contaminants smaller than the pore size of the chromatographic matrix and thus to a further and efficient purification. 750 kDa (with an equivalence of 50 nm) is the maximal pore size which can be employed to preclude any potential retention of vector particles (size: 80–120 nm). However, the main drawback is the dilution of the vector preparation by at least three times. Many large-scale purification protocols have adopted size exclusion chromatography as a final polishing step.42,52,105,110
Figure 3 presents some large-scale purification protocols for which some details are available. It is possible to distinguish between protocols which are more or less transpositions from small scale and very probably research protocols without any chromatographic steps to a larger scale (those used by Beckman Research Institute) and protocols characterized by an assembly of different separation principles (including at least one chromatography step) as used in the biotech industry: protocols used by Virxsys,52 Généthon/MolMed,42,105,111,112 Oxford BioMedica/Henogen,55 and St. Jude Children’s Research Hospital.56
Figure 3.

Principle process steps of large-scale downstream processing protocols for the purification of VSV-g-pseudotyped LV vectors (for clinical purposes). The company/institution name indicated in red informs that their downstream processing protocol makes use of ion-exchange (IEX) chromatography. (?)—no details are available on the process step (e.g., with respect to the filtration step, the pore size/exclusion size was not communicated). Sterilization—sterile filtration (0.2 µm). *A similar purification process however, devoid of the benzonase and the following diafiltration step was used by Greene et al.56 for the purification of clinical material produced with stable producer cell lines. **The protocol published by Ausubel et al. 43 does not use a final sterile filtration step, thus each batch/sub-batch requires separate testing for sterility before final processing and further use. LV, lentiviral vectors; SEC, size exclusion chromatography; TFF, tangential flow filtration; VSV-g (glycoprotein of the vesicular stomatitis virus).
The usual capture steps, applied to the large-scale purification of LV vectors, consist of clarification using membrane filtration (all protocols shown in Figure 3) followed by a concentration step either based on TFF/ultrafiltration (Beckman Research Institute) or ion-exchange chromatography (Virxsys, Généthon/MolMed, Oxford BioMedica/Henogen, St. Jude Children’s Research Hospital). All protocols, except one using stable cell lines for vector production,56 make use of a benzonase step for reducing the size of residual cellular and, in particular, plasmid-derived DNA contaminants.42,52,54,55,61,105 However, this step is either part of the capture step as in the protocols used by Beckman Research Institute, Généthon/MolMed or Oxford BioMedica/Henogen or is placed after the capture step as in the protocol developed by Virxsys. Both have their advantages and disadvantages: the early use of the benzonase step has the advantage that large DNA pieces are reduced in their size and that there are downstream steps for getting rid of residual benzonase; however, to being efficient, large quantities of benzonase have to be used. On the other side, the late positioning of the benzonase step in a protocol has the advantage that much lower quantities of benzonase have to be applied (thus leading to cost reductions); however, the residual steps of the protocol must have the capacity to remove residual benzonase to nondetectable levels. In addition, the late (downstream) application of the benzonase step has the disadvantage that large size nucleic acid contaminants might lead to the formation of aggregates capturing vector particles thus potentially leading to vector loss during the preceding steps.
The purification protocol developed by Beckman Research Institute is characterized by only one supplementary purification step (intermediate purification = polishing step) which is an ultracentrifugation step. All other protocols make use of a concentration/diafiltration step (as intermediate purification step) and finally of a diafiltration (Virxsys, Oxford BioMedica/Henogen) or size exclusion chromatography (Généthon/MolMed) step for polishing purposes.
For most of the protocols, the final vector preparation is finally sterile filtered with 0.2-µm membranes. This is a standard regulatory requirement to mitigate the risk of microbial contamination of the final product in GMP conditions. Although sterile filtration is strongly recommended, it is possible to skip it provided that the process can be certified as being fully aseptic. This requires the validation of aseptic processing, which is achieved by media process test, and manipulations have to be performed in a clean room with Class 100 (United States) or Grade A (European Union)-controlled environment (less than 3,500 particles of ≥0.5-µm diameter per m3 of air).
Whereas filtration is the most convenient method to guarantee product sterility before filling, the downstream processing protocol developed by Oxford BioMedica has placed this step after the first TFF-based concentration step and before the second (final) TFF step to reduce vector losses due to adsorption to membrane material at very high vector concentrations.113
Finally, there is one large-scale protocol which is devoid of a final sterile filtration step because the production protocol is based on a semiclosed system. In this case, every sub-batch is tested for sterility and pooling is only performed after sterility has been proven.43
The performances of the downstream processing protocols are the following: the concentration factor is ranging between 10 and 80-fold for all protocols, except for that developed by the Beckman Research Institute,61 which published 150 to 200-fold concentration, which is related to the ultracentrifugation step (Table 4). The protocol developed by Oxford BioMedica (Mitrophanous, personal communication) is characterized by a concentration factor of 2,000 due to the use of two succeeding concentration/diafiltration (TFF) steps. The overall process yields range between 20 and 40%. All purification protocols lead to comparable vector concentrations ranging from 1 × 108 to 2 × 109 ip/ml. These large differences are mainly due to the prepurification vector titers, but also to nonstandardized titration methods as well as to the vector construct (promoter, transgene), etc.
Table 4. Results of large-scale downstream processing of VSV-g-pseudotyped LV vectors.
| Company/institution | Concentration factor (x) | Overall yield (%) | Final titer |
|---|---|---|---|
| Beckman Research Institute61 | 150–200 (use of ultracentrifugation) | 40 | 2.6–3.8 × 108 TU/ml |
| Virxsys52 | 20 | 30 | 2.17 × 108 TU/ml |
| Généthon42/MolMed105 | 50/100 | 20 | 1–2 × 109 IG/ml |
| Oxford Biomedica/Henogen (Mitrophanous, personal communication) | 2,000 (use of two succeeding TFF steps) | 30–40 | 0.1–2 × 109 TU/ml |
| St. Jude Children’s Hospital56 | 65–80 | 29–33 | 4.5–7.2 × 108 TU/ml |
LV, lentiviral vectors; VSV-g, glycoprotein of the vesicular stomatitis virus.
Besides the obtained vector titers/vector yield further characterization of vector batches concerns, in particular, the removal of various contaminants. Percent removal of total DNA and total protein contamination ranged from 99.1105 to 99.84%42 and from 99.8542 to 99.9%,52 respectively. Regarding the removal of host cell DNA and host cell protein, removals of 99.8 and of 99.4%, respectively, have been reported.105 Since it is not only the residual DNA which might be a problem but also the size of the DNA and thus the possibility of transferring an entire functional open reading frame, regulatory agencies are more and more demanding on the maximal size of residual contaminating DNA. As an example, in the FDA guidance for “Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications”, it is stated that the residual host cell DNA fragments should not exceed the size of a functional gene, estimated at 200 base pairs. In this context, Ausubel et al.43 reported on a size distribution of <500 bp.
In addition to the quality control requirements, as for all biotech products produced with continuous cell lines, LV vector batches as well as the producer cells at the end of vector production have to be analyzed for the absence of RCLs which might be generated via homologous recombination. Despite the fact that for the current protocol of the LV production, that uses split genome packaging constructs with little or no sequence overlap between the vector components, the unlikely generation of RCLs has never been observed, the absence of RCLs in every batch intended for clinical use has to be indeed proven using very sensitive methods.114,115 In this context, the availability of stable producer cell lines will provide an increased safety level since production can be performed starting from well-defined quality controlled cell banks and end of production cell lines could be tested much easier for absence of RCLs.
Conclusion and Perspectives
As LV gene therapy will be soon a routine treatment not only for rare genetic, but also for acquired diseases, i.e., haematological malignancies and infectious diseases as HIV infection, for which large number of patients are expected to be treated, the implementation of scalable vector production protocols is becoming urgent to satisfy the demand not only of academic and hospital institutions, but also of the industry, which is moving very rapidly towards this type of medicinal products.
Obviously the standard transfection including the suspension culture-based transfection protocols is insufficient for providing the LV vector quantities required for the future routine use of LV vectors, and can only be considered as intermediate solution. The final solution for the production of large vector lots will be the implementation of stable producer cell lines cultured under suspension conditions allowing in principle unlimited scalability.
Several advances have been achieved in the last years in the stable packaging field: (i) the use of integrating vectors rather than plasmids to deliver vector genes; (ii) the codon optimization of the packaging genes to destroy homologous regions to reduce the probability of RCL formation and ψ-gag recombination; it is worth mentioning, however, that RCL formation has never been reported, at the best of our knowledge, even with noncodon optimized genes; (iii) the development of constitutive packaging cells which are simpler and safer than inducible cells both in the upstream and downstream process; (iv) the development of the cSIN-LV to simplify the integration of the TV even though it is only applicable with an inducible system; (v) the application of RMCE approach to the LV system. Major contribution of the RMCE technology is expected though in the integration of the TV to allow the switch of different transgenes in the same selected locus.
Several issues still remain to be addressed: the majority of stable systems were developed using the GFP marker and only a few have used therapeutic genes. It is necessary to collect more data on a larger number of therapeutic genes to understand how the expression of the transgene can influence cell productivity. Furthermore, are there alternatives to HEK293T cells as starting material? Is autotransduction a safety concern during LV manufacturing? Would the removal of the LV envelope receptor by the plasma membrane of the producer cells prevent either autotransduction or vector aggregation without affecting the performance of the producer cells?
Although VSV-g is endowed with the indisputable quality of entering any type of cells either in resting or stimulated conditions,116 its application is highly preferable in ex vivo rather in vivo gene therapy to avoid the risk of possible toxicity in transduced nontarget cells. In fact, even if specific promoters or specific miRNA target sequences117 can be included in the TV to control transgene expression, the risk of possible genotoxicity derived by LV integration in nontarget cells cannot be prevented by these strategies. Furthermore, if directly administrated to the blood stream (intravenous administration) VSV-g pseudotyped vectors are sensible to degradation by human complement; however, this fact does not preclude the administration of VSV-g pseudotyped LV vectors and efficient transduction of target cells in the case of localized delivery (such as treatment of disorders of the central nervous system12 or of the liver, i.e., for the treatment of haemophilia).118
To alleviate most of the drawbacks associated with the use of VSV-g pseudotyped LV vectors, alternative envelopes complement-resistant, such as the RD114-TR and RD114-PR, have already been tested in stable systems,89,92,107,119 whereas other excellent candidates studied only in transient systems are awaiting to be validated also in stable packaging cell lines. Among these, the most promising include the baboon endogenous retrovirus glycoprotein, belonging to the same beta-retroviruses family of RD114. Baboon endogenous retrovirus has recently been shown to transduce at high efficiency resting HSCs.120 Yet, a scFv derived from a specific monoclonal antibody against the CD133 molecule has been developed as LV pseudotyping agent to preferentially transduce a population of human hematopoietic stem cells with high proliferative potential in vitro and multilineage engraftment in vivo.121 Finally, mutants of the measles virus hemagglutinin (H) and fusion (F) glycoproteins H/F have been shown to efficiently transduce quiescent T and B lymphocytes in the presence of high concentrations of measles virus antibody positive human serum.122 Although alternative envelopes have much higher target specificity than VSV-g, it has to be kept in mind that they are not entirely specific because all cells expressing the specific receptor recognized by the env protein can be transduced, signifying the requirement for their careful evaluation for direct in vivo administration.
Despite these advances there is still an important hurdle to be taken which concerns the development of scalable purification methods of these LV vector pseudotypes before they can be routinely used in preclinical and clinical research. First achievements in view of the purification of GaLV-TR-pseudotyped LV vectors have been reported by Boudeffa et al.,109 recently.
In conclusion, recent successes in the clinical trials using LV for the treatment of rare and acquired diseases as well as advances in vector and manufacturing technologies will bring this promising vector system to routine application for benefit of human mankind.
References
- Salmon, F, Grosios, K and Petry, H (2014). Safety profile of recombinant adeno-associated viral vectors: focus on alipogene tiparvovec (Glybera®). Expert Rev Clin Pharmacol 7: 53–65. [DOI] [PubMed] [Google Scholar]
- Andreadis, ST, Roth, CM, Le Doux, JM, Morgan, JR and Yarmush, ML (1999). Large-scale processing of recombinant retroviruses for gene therapy. Biotechnol Prog 15: 1–11. [DOI] [PubMed] [Google Scholar]
- Delenda, C (2004). Lentiviral vectors: optimization of packaging, transduction and gene expression. J Gene Med 6 (suppl. 1): S125–S138. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Hacein-Bey, S, de Saint Basile, G, Gross, F, Yvon, E, Nusbaum, P et al. (2000). Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288: 669–672. [DOI] [PubMed] [Google Scholar]
- Naldini, L, Blömer, U, Gallay, P, Ory, D, Mulligan, R, Gage, FH et al. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267. [DOI] [PubMed] [Google Scholar]
- Montini, E, Cesana, D, Schmidt, M, Sanvito, F, Bartholomae, CC, Ranzani, M et al. (2009). The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest 119: 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee, S and Thrasher, AJ (2013). Gene therapy for PIDs: progress, pitfalls and prospects. Gene 525: 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Biasco, L, Scaramuzza, S, Ferrua, F, Cicalese, MP, Baricordi, C et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott–Aldrich syndrome. Science 341: 1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier, N, Hacein-Bey-Abina, S, Bartholomae, CC, Veres, G, Schmidt, M, Kutschera, I et al. (2009). Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326: 818–823. [DOI] [PubMed] [Google Scholar]
- Biffi, A, Montini, E, Lorioli, L, Cesani, M, Fumagalli, F, Plati, T et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Payen, E, Negre, O, Wang, G, Hehir, K, Fusil, F et al. (2010). Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 467: 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfi, S, Gurruchaga, JM, Ralph, GS, Lepetit, H, Lavisse, S, Buttery, PC et al. (2014). Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet 383: 1138–1146. [DOI] [PubMed] [Google Scholar]
- Levine, BL (2015). Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther 22: 79–84. [DOI] [PubMed] [Google Scholar]
- Poeschla, E, Gilbert, J, Li, X, Huang, S, Ho, A and Wong-Staal, F (1998). Identification of a human immunodeficiency virus type 2 (HIV-2) encapsidation determinant and transduction of nondividing human cells by HIV-2-based lentivirus vectors. J Virol 72: 6527–6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, SS, Kothari, N, You, XJ, Robinson, WE Jr, Schnell, T, Uberla, K et al. (2001). Generation of replication-defective helper-free vectors based on simian immunodeficiency virus. Virology 282: 154–167. [DOI] [PubMed] [Google Scholar]
- Curran, MA, Kaiser, SM, Achacoso, PL and Nolan, GP (2000). Efficient transduction of nondividing cells by optimized feline immunodeficiency virus vectors. Mol Ther 1: 31–38. [DOI] [PubMed] [Google Scholar]
- Berkowitz, RD, Ilves, H, Plavec, I and Veres, G (2001). Gene transfer systems derived from Visna virus: analysis of virus production and infectivity. Virology 279: 116–129. [DOI] [PubMed] [Google Scholar]
- Mselli-Lakhal, L, Guiguen, F, Greenland, T, Mornex, JF and Chebloune, Y (2006). Gene transfer system derived from the caprine arthritis-encephalitis lentivirus. J Virol Methods 136: 177–184. [DOI] [PubMed] [Google Scholar]
- Mitrophanous, K, Yoon, S, Rohll, J, Patil, D, Wilkes, F, Kim, V et al. (1999). Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther 6: 1808–1818. [DOI] [PubMed] [Google Scholar]
- Ramezani, A and Hawley, RG (2002). Overview of the HIV-1 lentiviral vector system. Curr Protoc Mol Biol (Chapter 16: Unit 16.21). [DOI] [PubMed]
- Dull, T, Zufferey, R, Kelly, M, Mandel, RJ, Nguyen, M, Trono, D et al. (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72: 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, VN, Mitrophanous, K, Kingsman, SM and Kingsman, AJ (1998). Minimal requirement for a lentivirus vector based on human immunodeficiency virus type 1. J Virol 72: 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi, H, Blömer, U, Takahashi, M, Gage, FH and Verma, IM (1998). Development of a self-inactivating lentivirus vector. J Virol 72: 8150–8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey, R, Dull, T, Mandel, RJ, Bukovsky, A, Quiroz, D, Naldini, L et al. (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol 72: 9873–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schambach, A, Swaney, WP and van der Loo, JC (2009). Design and production of retro- and lentiviral vectors for gene expression in hematopoietic cells. Methods Mol Biol 506: 191–205. [DOI] [PubMed] [Google Scholar]
- Throm, RE, Ouma, AA, Zhou, S, Chandrasekaran, A, Lockey, T, Greene, M et al. (2009). Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood 113: 5104–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frecha, C, Szécsi, J, Cosset, FL and Verhoeyen, E (2008). Strategies for targeting lentiviral vectors. Curr Gene Ther 8: 449–460. [DOI] [PubMed] [Google Scholar]
- Swanstrom, R and Wills, JW (1997). Synthesis, assembly, and processing of viral proteins. In: Coffin, J, Hughes, SH and Varmus, H (eds.). Retroviruses. Cold Spring Harbor Laboratory Press: Cold Spring Harbor/NY, pp. 263–334. [PubMed] [Google Scholar]
- Kotsopoulou, E, Kim, VN, Kingsman, AJ, Kingsman, SM and Mitrophanous, KA (2000). A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J Virol 74: 4839–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, R, Graf, M, Bieler, K, Wolf, H, Grunwald, T, Foley, P et al. (2000). Rev-independent expression of synthetic gag-pol genes of human immunodeficiency virus type 1 and simian immunodeficiency virus: implications for the safety of lentiviral vectors. Hum Gene Ther 11: 2403–2413. [DOI] [PubMed] [Google Scholar]
- Sastry, L, Xu, Y, Johnson, T, Desai, K, Rissing, D, Marsh, J et al. (2003). Certification assays for HIV-1-based vectors: frequent passage of gag sequences without evidence of replication-competent viruses. Mol Ther 8: 830–839. [DOI] [PubMed] [Google Scholar]
- Tareen, SU, Nicolai, CJ, Campbell, DJ, Flynn, PA, Slough, MM, Vin, CD et al. (2013). A Rev-independent gag/pol eliminates detectable psi-gag recombination in lentiviral vectors. Biores Open Access 2: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes, J, Jayaraman, B and Frankel, A (2012). The HIV-1 Rev response element: an RNA scaffold that directs the cooperative assembly of a homo-oligomeric ribonucleoprotein complex. RNA Biol 9: 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin, AS, Valentin, A, Pavlakis, GN and Felber, BK (1994). Continuous propagation of RRE(−) and Rev(−)RRE(−) human immunodeficiency virus type 1 molecular clones containing a cis-acting element of simian retrovirus type 1 in human peripheral blood lymphocytes. J Virol 68: 7944–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasmi, M, Glynn, J, Jin, MJ, Jolly, DJ, Yee, JK and Chen, ST (1999). Requirements for efficient production and transduction of human immunodeficiency virus type 1-based vectors. J Virol 73: 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nappi, F, Schneider, R, Zolotukhin, A, Smulevitch, S, Michalowski, D, Bear, J et al. (2001). Identification of a novel posttranscriptional regulatory element by using a rev- and RRE-mutated human immunodeficiency virus type 1 DNA proviral clone as a molecular trap. J Virol 75: 4558–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohll, JB, Mitrophanous, KA, Martin-Rendon, E, Ellard, FM, Radcliffe, PA, Mazarakis, ND et al. (2002). Design, production, safety, evaluation, and clinical applications of nonprimate lentiviral vectors. Methods Enzymol 346: 466–500. [DOI] [PubMed] [Google Scholar]
- Levine, BL, Humeau, LM, Boyer, J, MacGregor, RR, Rebello, T, Lu, X et al. (2006). Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA 103: 17372–17377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, X, Humeau, L, Slepushkin, V, Binder, G, Yu, Q, Slepushkina, T et al. (2004). Safe two-plasmid production for the first clinical lentivirus vector that achieves >99% transduction in primary cells using a one-step protocol. J Gene Med 6: 963–973. [DOI] [PubMed] [Google Scholar]
- Schonely, K, Afable, C, Slepushkin, V, Lu, X, Andre, K, Boehmer, J, et al. (2003). QC release testing of an HIV-1 based lentiviral vector lot and transduced cellular product. Bioproc J 2: 39–47. [Google Scholar]
- Gama-Norton, L, Botezatu, L, Herrmann, S, Schweizer, M, Alves, PM, Hauser, H et al. (2011). Lentivirus production is influenced by SV40 large T-antigen and chromosomal integration of the vector in HEK293 cells. Hum Gene Ther 22: 1269–1279. [DOI] [PubMed] [Google Scholar]
- Merten, OW, Charrier, S, Laroudie, N, Fauchille, S, Dugué, C, Jenny, C et al. (2011). Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum Gene Ther 22: 343–356. [DOI] [PubMed] [Google Scholar]
- Ausubel, LJ, Hall, C, Sharma, A, Shakeley, R, Lopez, P, Quezada, V et al. (2012). Production of CGMP-grade lentiviral vectors. Bioprocess Int 10: 32–43. [PMC free article] [PubMed] [Google Scholar]
- Smith, SL and Shioda, T (2009). Advantages of COS-1 monkey kidney epithelial cells as packaging host for small-volume production of high-quality recombinant lentiviruses. J Virol Methods 157: 47–54. [DOI] [PubMed] [Google Scholar]
- Marino, MP, Luce, MJ and Reiser, J (2003). Small- to large-scale production of lentivirus vectors. Methods Mol Biol 229: 43–55. [DOI] [PubMed] [Google Scholar]
- Graham, FL and van der Eb, AJ (1973). A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52: 456–467. [DOI] [PubMed] [Google Scholar]
- Boussif, O, Lezoualc’h, F, Zanta, MA, Mergny, MD, Scherman, D, Demeneix, B et al. (1995). A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA 92: 7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda, H, Kutner, RH, Bazan, NG and Reiser, J (2009). Simplified lentivirus vector production in protein-free media using polyethylenimine-mediated transfection. J Virol Methods 157: 113–121. [DOI] [PubMed] [Google Scholar]
- Pham, PL, Kamen, A and Durocher, Y (2006). Large-scale transfection of mammalian cells for the fast production of recombinant protein. Mol Biotechnol 34: 225–237. [DOI] [PubMed] [Google Scholar]
- Kutner, RH, Zhang, XY and Reiser, J (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4: 495–505. [DOI] [PubMed] [Google Scholar]
- Patel, M, Wilcox, DA, Giddings, AM, McKay, TR, Olsen, JC (2004). Modification of HEk 293 cell integrin expression profile allows convenient large-scale roller bottle production of lentiviral vectors. Mol Ther 9:S33. [Google Scholar]
- Slepushkin, V, Chang, N, Cohen, R, Gan, Y, Jiang, B, Deausen, E, et al. (2003). Large-scale purification of a lentiviral vector by size exclusion chromatography or Mustang Q ion exchange chromatography. Bioproc J 2:89–95. [Google Scholar]
- Karolewski, BA, Watson, DJ, Parente, MK and Wolfe, JH (2003). Comparison of transfection conditions for a lentivirus vector produced in large volumes. Hum Gene Ther 14: 1287–1296. [DOI] [PubMed] [Google Scholar]
- Negré, O, Denaro, M, Gillet-Legrand, B, Fusil, F, Hehir, K, Dorazio, R, et al. (2008). Long-term correction of murine beta-thalassemia following busulfan conditioning and transplant of bone marrow transduced with clinical-grade lentiviral vector (LentiGlobin™). Mol Ther 16:S85. [Google Scholar]
- Dupont, F (2008). Large Scale Manufacturing of a Lentiviral Vector (ProSavin®) for Phase I/II Clinical Trial. Presented at the CONSERT Labcourse, Evry/F, 29 June–1 July 2008.
- Greene, MR, Lockey, T, Mehta, PK, Kim, YS, Eldridge, PW, Gray, JT et al. (2012). Transduction of human CD34+ repopulating cells with a self-inactivating lentiviral vector for SCID-X1 produced at clinical scale by a stable cell line. Hum Gene Ther Methods 23: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, LJ and Zaiss, AK (2002). Lentiviral vectors. Preparation and use. Methods Mol Med 69: 303–318. [DOI] [PubMed] [Google Scholar]
- Follenzi, A and Naldini, L (2002). HIV-based vectors. Preparation and use. Methods Mol Med 69: 259–274. [PubMed] [Google Scholar]
- Reiser, J (2000). Production and concentration of pseudotyped HIV-1-based gene transfer vectors. Gene Ther 7: 910–913. [DOI] [PubMed] [Google Scholar]
- Ballas, C, Johnson, TT and Cornetta, K (2005). Harvest Timing and Media Composition Effects on Lentiviral Vector Production. Presented at the 8th Annual Meeting of the ASGT, 1 June–5 June 2005, St. Louis, MO.
- Couture, LA (2008). Vector Production in Support of Early Clinical Trials. Presented at the ASGT Meeting, 28 May–1 June 2008, Boston, MA.
- Sheu, J, Beltzer, J, Fury, B, Wilczek, K, Tobin, S, Falconer, D et al. (2015). Large-scale production of lentiviral vector in a closed system hollow fiber bioreactor. Mol Ther Methods Clin Dev 2: 15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau, N and Gasmi, M, Scalable lentiviral vector production system compatible with industrial pharmaceutical applications, WO 2013076309 A1 (2013).
- Witting, SR, Li, LH, Jasti, A, Allen, C, Cornetta, K, Brady, J et al. (2012). Efficient large volume lentiviral vector production using flow electroporation. Hum Gene Ther 23: 243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansorge, S, Lanthier, S, Transfiguracion, J, Durocher, Y, Henry, O and Kamen, A (2009). Development of a scalable process for high-yield lentiviral vector production by transient transfection of HEK293 suspension cultures. J Gene Med 11: 868–876. [DOI] [PubMed] [Google Scholar]
- Durocher, Y, Perret, S and Kamen, A (2002). High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res 30: E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom, R, Bisson, L and Durocher, Y (2008). Transfection of HEK293-EBNA1 cells in suspension with linear PEI for production of recombinant proteins. CSH Protoc 2008: pdb.prot4977. [DOI] [PubMed] [Google Scholar]
- Jaalouk, DE, Crosato, M, Brodt, P and Galipeau, J (2006). Inhibition of histone deacetylation in 293GPG packaging cell line improves the production of self-inactivating MLV-derived retroviral vectors. Virol J 3: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena-Esteves, M, Tebbets, JC, Steffens, S, Crombleholme, T and Flake, AW (2004). Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods 122: 131–139. [DOI] [PubMed] [Google Scholar]
- Gossen, M and Bujard, H (1992). Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA 89: 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H, Rabson, AB, Kaul, M, Ron, Y and Dougherty, JP (1996). Inducible human immunodeficiency virus type 1 packaging cell lines. J Virol 70: 4530–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul, M, Yu, H, Ron, Y and Dougherty, JP (1998). Regulated lentiviral packaging cell line devoid of most viral cis-acting sequences. Virology 249: 167–174. [DOI] [PubMed] [Google Scholar]
- Xu, K, Ma, H, McCown, TJ, Verma, IM and Kafri, T (2001). Generation of a stable cell line producing high-titer self-inactivating lentiviral vectors. Mol Ther 3: 97–104. [DOI] [PubMed] [Google Scholar]
- Cockrell, AS, Ma, H, Fu, K, McCown, TJ and Kafri, T (2006). A trans-lentiviral packaging cell line for high-titer conditional self-inactivating HIV-1 vectors. Mol Ther 14: 276–284. [DOI] [PubMed] [Google Scholar]
- Ni, Y, Sun, S, Oparaocha, I, Humeau, L, Davis, B, Cohen, R et al. (2005). Generation of a packaging cell line for prolonged large-scale production of high-titer HIV-1-based lentiviral vector. J Gene Med 7: 818–834. [DOI] [PubMed] [Google Scholar]
- Ikeda, Y, Takeuchi, Y, Martin, F, Cosset, FL, Mitrophanous, K and Collins, M (2003). Continuous high-titer HIV-1 vector production. Nat Biotechnol 21: 569–572. [DOI] [PubMed] [Google Scholar]
- Wielgosz, MM, Kim, YS, Carney, GG, Zhan, J, Reddivari, M, Coop, T et al. (2015). Generation of a lentiviral vector producer cell clone for human Wiskott–Aldrich syndrome gene therapy. Mol Ther Methods Clin Dev 2: 14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner, M, Ma, Z, Zhou, S, Ren, A, Chandrasekaran, A, Gray, JT, et al. (2015). Development of a Second Generation Stable Lentiviral Packaging Cell Line In Support Of Clinical Gene Therapy Protocols. http://wwwabstracts2viewcom/asgct/indexphp. In: 18th ASGCT Annual Meeting, New Orleans.
- Hu, P, Li, Y, Sands, MS, McCown, T and Kafri, T (2015). Generation of a stable packaging cell line producing high-titer PPT-deleted integration-deficient lentiviral vectors. Mol Ther Methods Clin Dev 2: 15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, HJ, Leroux-Carlucci, MA, Sion, CJ, Mitrophanous, KA and Radcliffe, PA (2009). Development of inducible EIAV-based lentiviral vector packaging and producer cell lines. Gene Ther 16: 805–814. [DOI] [PubMed] [Google Scholar]
- Stewart, HJ, Fong-Wong, L, Strickland, I, Chipchase, D, Kelleher, M, Stevenson, L et al. (2011). A stable producer cell line for the manufacture of a lentiviral vector for gene therapy of Parkinson’s disease. Hum Gene Ther 22: 357–369. [DOI] [PubMed] [Google Scholar]
- Broussau, S, Jabbour, N, Lachapelle, G, Durocher, Y, Tom, R, Transfiguracion, J et al. (2008). Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol Ther 16: 500–507. [DOI] [PubMed] [Google Scholar]
- Pacchia, AL, Adelson, ME, Kaul, M, Ron, Y and Dougherty, JP (2001). An inducible packaging cell system for safe, efficient lentiviral vector production in the absence of HIV-1 accessory proteins. Virology 282: 77–86. [DOI] [PubMed] [Google Scholar]
- Sparacio, S, Pfeiffer, T, Schaal, H and Bosch, V (2001). Generation of a flexible cell line with regulatable, high-level expression of HIV Gag/Pol particles capable of packaging HIV-derived vectors. Mol Ther 3: 602–612. [DOI] [PubMed] [Google Scholar]
- Sanber, KS, Knight, SB, Stephen, SL, Bailey, R, Escors, D, Minshull, J et al. (2015). Construction of stable packaging cell lines for clinical lentiviral vector production. Sci Rep 5: 9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coroadinha, AS, Schucht, R, Gama-Norton, L, Wirth, D, Hauser, H and Carrondo, MJ (2006). The use of recombinase mediated cassette exchange in retroviral vector producer cell lines: predictability and efficiency by transgene exchange. J Biotechnol 124: 457–468. [DOI] [PubMed] [Google Scholar]
- Schucht, R, Coroadinha, AS, Zanta-Boussif, MA, Verhoeyen, E, Carrondo, MJ, Hauser, H et al. (2006). A new generation of retroviral producer cells: predictable and stable virus production by Flp-mediated site-specific integration of retroviral vectors. Mol Ther 14: 285–292. [DOI] [PubMed] [Google Scholar]
- Carrondo, M, Panet, A, Wirth, D, Coroadinha, AS, Cruz, P, Falk, H et al. (2011). Integrated strategy for the production of therapeutic retroviral vectors. Hum Gene Ther 22: 370–379. [DOI] [PubMed] [Google Scholar]
- Stornaiuolo, A, Piovani, BM, Bossi, S, Zucchelli, E, Corna, S, Salvatori, F et al. (2013). RD2-MolPack-Chim3, a packaging cell line for stable production of lentiviral vectors for anti-HIV gene therapy. Hum Gene Ther Methods 24: 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa, H, Persons, DA and Nienhuis, AW (2005). Mobilization and mechanism of transcription of integrated self-inactivating lentiviral vectors. J Virol 79: 8410–8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W, Russ, JL and Eiden, MV (2012). Evaluation of residual promoter activity in γ-retroviral self-inactivating (SIN) vectors. Mol Ther 20: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin, V, Stornaiuolo, A, Piovan, C, Corna, S, Bossi, S, Pema, M, et al. (2016). RD-MolPack technology for the constitutive production of self-inactivating lentiviral vectors pseudo-typed with the non-toxic RD114-TR envelope. Mol Ther Methods Clin Dev (in press). [DOI] [PMC free article] [PubMed]
- Porcellini, S, Alberici, L, Gubinelli, F, Lupo, R, Olgiati, C, Rizzardi, GP et al. (2009). The F12-Vif derivative Chim3 inhibits HIV-1 replication in CD4+ T lymphocytes and CD34+-derived macrophages by blocking HIV-1 DNA integration. Blood 113: 3443–3452. [DOI] [PubMed] [Google Scholar]
- Porcellini, S, Gubinelli, F, Alberici, L, Piovani, BM, Rizzardi, GP and Bovolenta, C (2010). Chim3 confers survival advantage to CD4+ T cells upon HIV-1 infection by preventing HIV-1 DNA integration and HIV-1-induced G2 cell-cycle delay. Blood 115: 4021–4029. [DOI] [PubMed] [Google Scholar]
- Di Nunzio, F, Piovani, B, Cosset, FL, Mavilio, F and Stornaiuolo, A (2007). Transduction of human hematopoietic stem cells by lentiviral vectors pseudotyped with the RD114-TR chimeric envelope glycoprotein. Hum Gene Ther 18: 811–820. [DOI] [PubMed] [Google Scholar]
- Kim, YS, Wielgosz, MM, Hargrove, P, Kepes, S, Gray, J, Persons, DA et al. (2010). Transduction of human primitive repopulating hematopoietic cells with lentiviral vectors pseudotyped with various envelope proteins. Mol Ther 18: 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge, GD, Wu, RA, Hansen, M, Ironside, C, Watts, KL, Olsen, P et al. (2010). Cocal-pseudotyped lentiviral vectors resist inactivation by human serum and efficiently transduce primate hematopoietic repopulating cells. Mol Ther 18: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisti, Y (2007). Strategies in downstream processing. In: Subramanian, G (ed.). Bioseparation and Bioprocessing: A Handbook, vol. 1, 2nd edn. Wiley-VCH: New York, pp. 29–62. [Google Scholar]
- Tiscornia, G, Singer, O and Verma, IM (2006). Production and purification of lentiviral vectors. Nat Protoc 1: 241–245. [DOI] [PubMed] [Google Scholar]
- Scherr, M, Battmer, K, Eder, M, Schüle, S, Hohenberg, H, Ganser, A et al. (2002). Efficient gene transfer into the CNS by lentiviral vectors purified by anion exchange chromatography. Gene Ther 9: 1708–1714. [DOI] [PubMed] [Google Scholar]
- Baekelandt, V, Eggermont, K, Michiels, M, Nuttin, B and Debyser, Z (2003). Optimized lentiviral vector production and purification procedure prevents immune response after transduction of mouse brain. Gene Ther 10: 1933–1940. [DOI] [PubMed] [Google Scholar]
- Yamada, K, McCarty, DM, Madden, VJ and Walsh, CE (2003). Lentivirus vector purification using anion exchange HPLC leads to improved gene transfer. Biotechniques 34: 1074–1078, 1080. [DOI] [PubMed] [Google Scholar]
- Segura, MM, Kamen, A and Garnier, A (2006). Downstream processing of oncoretroviral and lentiviral gene therapy vectors. Biotechnol Adv 24: 321–337. [DOI] [PubMed] [Google Scholar]
- Rodrigues, T, Carrondo, MJ, Alves, PM and Cruz, PE (2007). Purification of retroviral vectors for clinical application: biological implications and technological challenges. J Biotechnol 127: 520–541. [DOI] [PubMed] [Google Scholar]
- Bellintani, F, Piacenza, L, Sciarretta, R, Martelli, L, Massa, S, Vallanti, G, et al. (2008). Large scale process for the production and purification of lentiviral vectors for clinical applications. Hum Gene Ther 19:1089. [Google Scholar]
- Segura, MM, Garnier, A, Durocher, Y, Coelho, H and Kamen, A (2007). Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol Bioeng 98: 789–799. [DOI] [PubMed] [Google Scholar]
- Koldej, R, Cmielewski, P, Stocker, A, Parsons, DW and Anson, DS (2005). Optimisation of a multipartite human immunodeficiency virus based vector system; control of virus infectivity and large-scale production. J Gene Med 7: 1390–1399. [DOI] [PubMed] [Google Scholar]
- Geraerts, M, Michiels, M, Baekelandt, V, Debyser, Z and Gijsbers, R (2005). Upscaling of lentiviral vector production by tangential flow filtration. J Gene Med 7: 1299–1310. [DOI] [PubMed] [Google Scholar]
- Boudeffa, D, Fenard, D, Mormin, M, Galy, A, Merten, O-W (2015). Development of innovative scalable protocols for the purification of lentiviral vectors pseudotyped with GaLV-TR or mutated measles virus glycoproteins. Presented at the 18th Annual Meeting of the ASGCT, 13 May–16 May 2015, New Orleans, LA/USA.
- Segura, MM, Garnier, A and Kamen, A (2006). Purification and characterization of retrovirus vector particles by rate zonal ultracentrifugation. J Virol Methods 133: 82–91. [DOI] [PubMed] [Google Scholar]
- Charrier, S, Laroudie, N, Viornery, A, Seye, K, Bucher-Laurent, S, Audit, M, et al. (2008). Reproducible transduction of CD34+ cells with a highly-purified lentiviral vector developed for Wiskott–Aldrich Syndrome gene therapy. Mol Ther 16:S167. [Google Scholar]
- Merten, O-W (2008). Retro- and lentiviral production platforms. Hum Gene Ther 19:1067. [Google Scholar]
- Truran, R, Buckley, R, Radcliffe, P, Miskin, J, Mitrophanous, K, Virus purification, WO 2009153563 A1 (2009).
- Escarpe, P, Zayek, N, Chin, P, Borellini, F, Zufferey, R, Veres, G et al. (2003). Development of a sensitive assay for detection of replication-competent recombinant lentivirus in large-scale HIV-based vector preparations. Mol Ther 8: 332–341. [DOI] [PubMed] [Google Scholar]
- Miskin, J, Chipchase, D, Rohll, J, Beard, G, Wardell, T, Angell, D et al. (2006). A replication competent lentivirus (RCL) assay for equine infectious anaemia virus (EIAV)-based lentiviral vectors. Gene Ther 13: 196–205. [DOI] [PubMed] [Google Scholar]
- Amirache, F, Lévy, C, Costa, C, Mangeot, PE, Torbett, BE, Wang, CX et al. (2014). Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 123: 1422–1424. [DOI] [PubMed] [Google Scholar]
- Brown, BD, Venneri, MA, Zingale, A, Sergi Sergi, L and Naldini, L (2006). Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med 12: 585–591. [DOI] [PubMed] [Google Scholar]
- Cantore, A, Ranzani, M, Bartholomae, CC, Volpin, M, Valle, PD, Sanvito, F et al. (2015). Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med 7: 277ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, Y, Collins, MK, Radcliffe, PA, Mitrophanous, KA and Takeuchi, Y (2002). Gene transduction efficiency in cells of different species by HIV and EIAV vectors. Gene Ther 9: 932–938. [DOI] [PubMed] [Google Scholar]
- Girard-Gagnepain, A, Amirache, F, Costa, C, Lévy, C, Frecha, C, Fusil, F et al. (2014). Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 124: 1221–1231. [DOI] [PubMed] [Google Scholar]
- Brendel, C, Goebel, B, Daniela, A, Brugman, M, Kneissl, S, Schwäble, J et al. (2015). CD133-targeted gene transfer into long-term repopulating hematopoietic stem cells. Mol Ther 23: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy, C, Amirache, F, Costa, C, Frecha, C, Muller, CP, Kweder, H et al. (2012). Lentiviral vectors displaying modified measles virus gp overcome pre-existing immunity in in vivo-like transduction of human T and B cells. Mol Ther 20: 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebben, M (2015). Pre-industrial Manufacturing of Lentiviral Vectors by Transient Transfection in Single Use Systems. Presented at the Spring Meeting of ISBiotech, 9 March–11 March 2015, Washington/DC.
- Kafri, T, van Praag, H, Ouyang, L, Gage, FH and Verma, IM (1999). A packaging cell line for lentivirus vectors. J Virol 73: 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klages, N, Zufferey, R and Trono, D (2000). A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol Ther 2: 170–176. [DOI] [PubMed] [Google Scholar]
- Farson, D, Witt, R, McGuinness, R, Dull, T, Kelly, M, Song, J et al. (2001). A new-generation stable inducible packaging cell line for lentiviral vectors. Hum Gene Ther 12: 981–997. [DOI] [PubMed] [Google Scholar]