

Abstract
Colorectal cancer (CRC) is one of the top three cancer-related causes of death worldwide. FBXW7 is a known tumor-suppressor gene, commonly mutated in CRC and in a variety of other epithelial tumors. Low expression of FBXW7 is also associated with poor prognosis. Loss of FBXW7 sensitizes cancer cells to certain drugs, while making them more resistant to other types of chemotherapies. However, is not fully understood how epithelial cells within normal gut and primary tumors respond to potential cancer therapeutics. We have studied genetically engineered mice in which the fbxw7 gene is conditionally knocked-out in the intestine (fbxw7∆G). To further investigate the mechanism of Fbxw7-action, we grew intestinal crypts from floxed-fbxw7 (fbxw7fl/fl) and fbxw7ΔG mice, in a Matrigel-based organoid (mini-gut) culture. The fbxw7ΔG organoids exhibited rapid budding events in the crypt region. Furthermore, to test organoids for drug response, we exposed day 3 intestinal organoids from fbxw7fl/fl and fbxw7∆G mice, to various concentrations of 5-fluorouracil (5-FU) for 72 hours. 5-FU triggers phenotypic differences in organoids including changing shape, survival, resistance, and death. 5-FU however, rescues the drug-resistance phenotype of fbxw7ΔG through the induction of terminal differentiation. Our results support the hypothesis that a differentiating therapy successfully targets FBXW7-mutated CRC cells.
Introduction
Common anticancer strategies often fail resulting in tumor recurrences and metastasis. Only about 6% of colorectal cancer (CRC) metastatic patients live more than 5 years after diagnosis.1 There is an urgent need to develop molecularly targeted cancer therapies and drug-screening models to stratify patients and achieve maximum benefit from expensive treatments. The recent advent of organoid (mini-gut) culture has allowed development of an in vitro model that mimics many aspects of colon/intestinal homoeostasis and tumors.2–7 Sato and colleagues,7 initially established the ideal conditions for isolated small intestinal crypts or single leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5),8 positive stem cells to develop into organoids: laminin-rich Matrigel mimicking the basal lamina and a finely balanced medium including R-spondin1 (Wnt agonist),9 Noggin (BMP antagonist)10 and epidermal growth factor,11 were found sufficient to maintain the proliferation of the stem cell compartment or single Lgr5-positive cells. Similar to small intestine, the stem cell compartment at the base of the crypt-like domain of the mini-guts maintains epithelial turnover: stem cells give rise to progenitor cells, which differentiate while migrating toward the villus-like domain into enterocytes, enteroendocrine cells, and goblet cells. Paneth cells, instead, are located at the bottom of the crypt. In the organoid model, tumor cells can grow in a more similar manner to that in living organisms, with cell-cell interactions and boundaries. The modeling of cancer mutations in human intestinal/colon organoids using new tools for gene manipulation, allowed both in vitro and in vivo experimental approaches for mutational and functional analysis.3–5 Organoid culture can be used indefinitely. Moreover, changes in the composition of the medium can drive changes in phenotype/structure of organoids that facilitates in vitro testing of drugs, inhibitors, and growth factors. This model may also present a novel method of drug screening.4,12
One of the key cellular mechanisms of regulating activity of transcriptional-activators is through protein degradation using the ubiquitin-proteasome system. Specificity of proteolysis for any particular substrate is determined by its association with a specific E3-receptor subunit. FBXW7 (also called hCDC4, Ago, Sel10, and Fbw7) functions as a receptor subunit for the Skp1/Cullin/F-box (SCF)-E3-ubiquitin-ligase (SCFFBXW7). Recent excellent reviews on FBXW7 confirm that loss of FBXW7 function is likely to result in failed regulation of its downstream proteins targets, which disrupt a variety of critical signaling pathways resulting in acquisition of the hallmarks of cancer.13–20 Underlining the importance of FBXW7 as a tumor suppressor is the fact that loss of function mutations in the FBXW7 gene occur in a variety of human cancers including 10–15% of CRCs.21–23
We and others have previously studied the Fbxw7 conditional knockout in murine intestinal lineages and cancer.24,25 The synergistic contribution of FBXW7 and TP53 to the suppression of gastrointestinal cancer has also been reported.26,27 Interestingly, an intestinal knock-in model of Fbxw7 (R482Q) mutation has recently been described,28 nevertheless both Fbxw7 conditional knockout and knock-in models promote an equivalent intestinal tumorigenesis on an adenomatous polyposis coli (Apc) mutant background.25,28 FBXW7 mutations have been investigated comprehensively in CRC.23,29 Understanding FBXW7 loss-associated mechanism(s) driving drug resistance could therefore be important to successfully target and eradicate CRC cells carrying FBXW7 mutations. Recent literature has reported FBXW7 loss confers resistance to oxaliplatin and cisplatin chemotherapeutic agents,30,31 while CRC cell lines harboring FBXW7 mutations or deletions are sensitive to Rapamycin treatment.32
Here, we employed the murine intestinal organoid system, to identify cellular response mediated by Fbxw7 to various concentrations of 5-fluorouracil (5-FU), a DNA damage agent commonly administered to metastatic CRC patients.1 We show that fbxw7 gut-specific inactivation (fbxw7ΔG) with high proliferative capacity give rise to microadenoma-like structures. 5-FU promotes terminal differentiation and apoptosis in control floxed-fbxw7 (fbxw7fl/fl) organoids. Intestinal organoids from fbxw7∆G are two to three times less sensitive to 5-FU treatment than fbxw7fl/fl intestinal organoids. Moreover, epithelial cells extravasated from the fbxw7∆G organoids were abrogated by the 5-FU treatment, and the induction of cell differentiation.
Results
FBXW7 loss in human CRC cells promotes resistance to 5-FU
The chemotherapeutic agent 5-FU is used to treat many different types of cancer, including gastrointestinal cancers. In vitro, 5-FU can trigger apoptosis in CRC cells and it appeared to be initiated by caspase-dependent death pathway.33,34 To investigate the role of FBXW7 mutation in drug resistance, we examined the induction of growth inhibition in a dose-dependent manner using FBXW7 knockout HCT116 (HCT116FBXW7(−/−)) and DLD-1 (DLD-1FBXW7(−/−)),23,24 in parallel with FBXW7 wild-type (FBXW7(+/+)) counterpart cell lines (Figure 1a and Supplementary Figure S1a). Seeded cells were synchronized overnight by serum starvation to reduce the FBXW7 deficiency-induced cell division/growth in CRC cells, as reported previously,23,24,30,32,35 prior to 72 hours incubation with 5-FU for cytotoxicity assay. Sulforhodamine B colorimetric assay was carried out to measure the IC50. IC50 of HCT116FBXW7(−/−) cells (14.3 μmol/l) was twofold higher than the IC50 of HCT116FBXW7(+/+) cells (7.1 μmol/l) (Figure 1b,c). In addition, the colony-forming efficiency assay revealed that HCT116FBXW7(−/−) exhibited a significant resistance to 5-FU (Figure 1d,e). In accordance with previous studies using oxaliplatin and cisplatin,30,31 deletion of FBXW7 mediated higher tolerance of 5-FU in HCT116 cells. To test whether re-expression of FBXW7 render CRC cells sensitive to 5-FU, as Fbxw7α was shown to be the preferentially expressed Fbxw7 isoform in intestine/colon,24 we have overexpressed FBXW7α in both HCT116FBXW7(−/−) and DLD-1FBXW7(−/−) cells. CRC cell lines are initially transfected with FLAG-FBXW7α and control pcDNA3 plasmids in 10 cm tissue culture plates. Ten hours after transfection, cells were split and seeded in 96-well plates. Transfection efficiency was determined by western blot (Supplementary Figure S1f). Cells were synchronized after overnight serum starvation, treated with increasing concentrations of the 5-FU for 72 hours and the IC50 was determined using sulforhodamine B assay as outlined above (Figure 1f,g and Supplementary Figure S1d,e). The results showed that Fbxw7α overexpression significantly decreased the 5-FU IC50 and inhibited FBXW7 mutation in mediating 5-FU drug-resistance of CRC cell lines.
Figure 1.

FBXW7 deletion confers 5-FU resistance to human colon cancer HCT116 cells. (a) RT-PCR shows FBXW7 status in HCT116FBXW7(+/+) and HCT116FBXW7(−/−) cell lines. Reverse primer was designed to target the deleted exon 5 of FBXW7. (b,c) Sulforhodamine B (SRB) assay to test the cytotoxicity of 5-FU in both HCT116FBXW7(+/+) and HCT116FBXW7(−/−) cell lines. Vehicle (dimethyl sulfoxide) and 10 doses of 5-FU (from 0.05 to 100 μmol/l) were incubated for 72 hours following cell synchronization. (d,e) Colony-forming efficiency assay of HCT116FBXW7(+/+) and HCT116FBXW7(−/−) cell lines. 0, 2.5, and 7.5 μmol/l 5-FU was replaced after 72 hours incubation with fresh medium. At day 10 of growth, colonies were stained with crystal violet and counted by ImageJ. (f,g) Cytotoxicity of 5-FU was analyzed via SRB assay following transient transfection of HCT116FBXW7(−/−) cells with pcDNA3 (control) or FLAG-FBXW7α plasmids as previously described.30 IC50 values, indicated in the table, were calculated via GraphPad Prism software. Experiments were performed in triplicate and repeated at least in three independent occasions. Data are mean ± standard error of the mean (*P < 0.05, **P < 0.01, ***P < 0.001).
Fbxw7ΔG organoids (mini-guts) possess a peculiar morphology
We have demonstrated intense fbxw7-expression in the crypt base of wild-type mice, including the transit amplifying/progenitor cells, compared with the villus using in-situ hybridization.24,25 Intestinal fbxw7 knockout resulted in impaired cell differentiation, with a reduction in the number of goblet and Paneth cells. However, despite the increase of actively proliferating epithelial cells in the guts of the fbxw7∆G mice, no intestinal tumors were reported up to 11–12 months of age. This could be because—in the guts multiple signaling molecules are emerging from different linages—the underlying mesenchyme, immune and crypt-based epithelial cells may together cooperate tightly to control transformation of intestinal epithelial cells. Recently, Paneth cells were shown to be a critical component of the intestinal epithelial stem cell niche36; though the depletion of Paneth cells failed to alter significantly the crypt phenotype.37 In addition, the crypt culture recapitulates the cellular diversity, resembling proliferative and differentiated compartments of the intestinal epithelium without coculturing with stromal/fibroblast cells. Thus, to explore the function of murine epithelial Fbxw7, we cultured intestinal organoids from fbxw7fl/fl and villin-Cre+:fbxw7fl/fl (fbxw7∆G) mice (Supplementary Figure S2a,b).
Small intestinal crypts isolated from fbxw7fl/fl mice gave rise to enterospheres, spherical organoids formed by a monolayer of epithelial cells, within a few hours following seeding. On day 4 of growth, most of the fbxw7fl/fl enterospheres were fully branched organoids (defined enteroids),38 and characterized by the presence of buds (Figure 2a,b). Morphology of the fbxw7fl/fl organoids represent a normal intestinal organoid culture system which contains crypt and villus domains without a mesenchymal component.7 A low percentage of cystic organoids with an empty lumen, (defined spheres), were also present (17%). Moreover after 7 days of culture, fbxw7fl/fl organoids have mainly manifested by enteroids (Figure 2a). In contrast, Fbxw7 inactivation accelerated organoid growth and altered the phenotype of intestinal organoids (Figure 2a,c). Large fbxw7ΔG organoids were however followed by releasing clusters of cells outside the organoid lumen after 3 days of culture (Supplementary Video S1). Histologically, this peculiar morphology is highly variable in appearance, resembling a microadenoma appearance.39 After 7 days of culture, almost all of the fbxw7ΔG enterospheres had changed to microadenoma-like shape (Figure 2a). Reculturing the released cells from fbxw7ΔG organoids, demonstrated an abnormally high proliferative activity and adapted cancer cells properties (data not shown).
Figure 2.
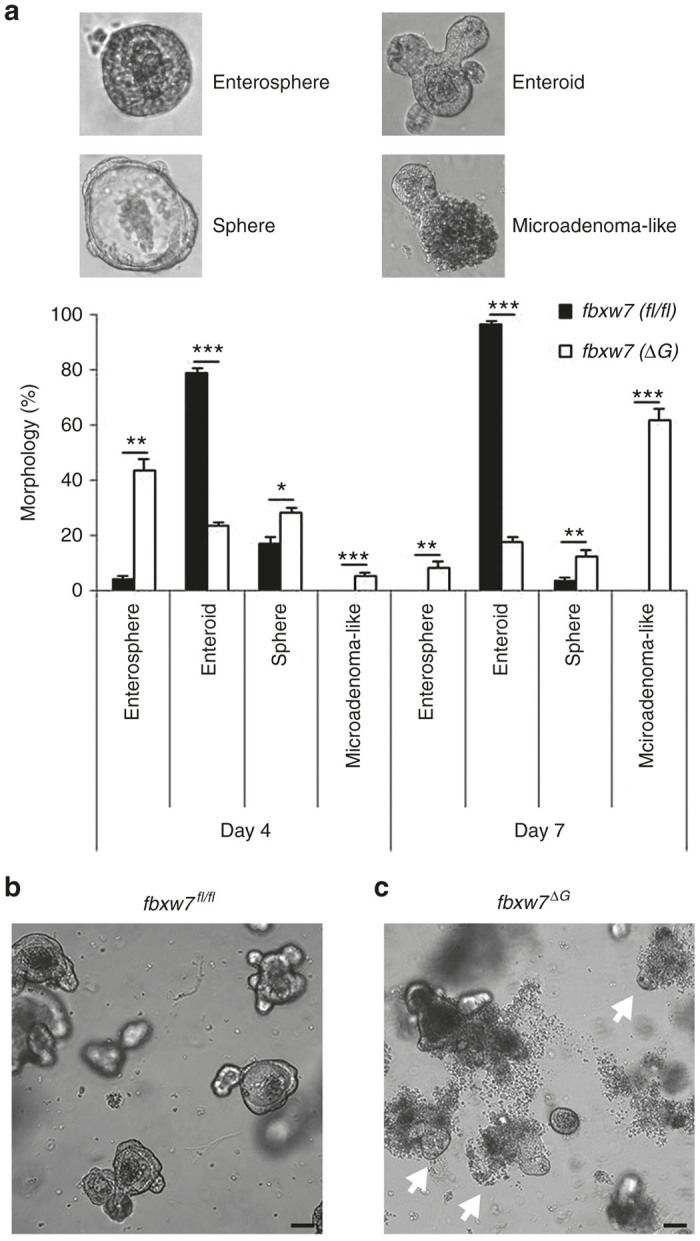
fbxw7-null organoids grow faster and present microadenoma-like morphology. (a) Percentage of enterosphere, enteroid, sphere, and microadenoma-like morphologies at day 4 and day 7 of growth of fbxw7fl/fl and fbxw7ΔG mini-guts. Mini-guts were counted by microscopy (*P < 0.05, **P < 0.01, ***P < 0.001). (b,c) Images are representative of fbxw7fl/fl and fbxw7ΔG mini-gut populations at day 4 of growth. Arrowheads indicate microadenoma-like mini-guts. Scale bars, 75 μm.
Loss of Fbxw7 in intestinal organoid induces resistance to 5-FU
To test the loss of FBXW7 function as a potential mechanism of drug resistance, we next performed 5-FU toxicity assay in fully structured enteroids. In addition, according to a recent report,12 the intestinal organoids are three to four times more sensitive than CRC cell lines. Thus, both fbxw7fl/fl and fbxw7ΔG organoids were exposed to the estimated IC50 of HCT116FBXW7(+/+) cells, 7.5 μmol/l 5-FU (Figure 1c), along with 2.5 and 0.8 μmol/l 5-FU (Figure 3a). We and other groups previously showed that the deficiency of murine intestinal Fbxw7 induced cell division via Ki67 immunohistochemistry and BrdU labelling.24,25 Therefore, to exclude the potential effect of cell proliferation, the 5-FU treatment started after 2 days for fbxw7ΔG organoids, whereas for the fbxw7fl/fl organoids after 3 days (Figure 3a). The toxicity assay showed that mini-guts harboring fbxw7 deletion were approximately two to three times less sensitive to 5-FU treatment than intestinal organoids from fbxw7fl/fl mice (Figure 3b,c), likewise HCT116FBXW7(−/−) and DLD-1FBXW7(−/−) cells (Figure 1b,c and Supplementary Figure S1). These results have confirmed the crucial role of Fbxw7 inactivation in the acquisition of drug resistance. Furthermore, we have compared the morphology of organoids treated with 5-FU by counting the number of viable organoids per each category; enterospheres, enteroids, spheres, and microadenoma-like structure. Quantification revealed that 5-FU promoted large spheres versus enteroids, in a dose-dependent manner, in both wild-type fbxw7fl/fl and fbxw7ΔG mutant organoids after 72 hours (Figure 3d–f). Although the highest concentration of 5-FU (7.5 μmol/l) induced the death of most of the organoids, the remaining viable organoids were spheres only. Notably, the number of microadenoma-like structures from fbxw7ΔG mutant organoids significantly decreased in response to 5-FU (Figure 3d,e and Supplementary Video S2). These findings together suggest that 5-FU-induced DNA damages may cause a reduction in cellular proliferation resulting in morphological changes within the mini-gut structure. In this regard, as the proliferative compartment is predominantly in the crypt-like domains, its impairment may lead to spherical mini-guts. These results support the hypothesis that the organoids can be used as ex vivo tools for investigating drug resistance.
Figure 3.

fbxw7ΔG organoids tolerate higher dose of 5-FU. (a) Layout of the drug response experiments and following analysis considering faster growth of fbxw7ΔG mini-guts. (b,c) Toxicity assay of 5-FU in fbxw7fl/fl and fbxw7ΔG mini-guts and IC50 values. (d,e) Morphological analysis following 72 hours incubation with 0.8, 2.5, and 7.5 μmol/l 5-FU. Percentage of enterosphere, enteroid, sphere, and microadenoma-like morphologies was assessed by microscopy in fbxw7fl/fl and fbxw7ΔG mini-gut populations. (f) Images represent phenotypical changes of both fbxw7fl/fl and fbxw7ΔG mini-guts in response to 5-FU. Scale bars, 50 μm. Experiments were performed in triplicate and repeated at least in two independent occasions (*P < 0.05, **P < 0.01, ***P < 0.001).
Fbxw7 loss counteracts organoids differentiation induced by 5-FU
To examine the effects of 5-FU on proliferation and differentiation, surviving organoids were fixed, embedded in paraffin, and processed for immunohistochemistry. As outlined above, a treatment with 7.5 μmol/l 5-FU induced cell death in almost but not all wild-type fbxw7fl/fl organoids, and very few remaining live organoids formed spheres (Figure 3e), therefore we used the surviving organoids treated with 2.5 and 0.8 μmol/l 5-FU. Previous studies reported that Fbxw7 loss in murine intestinal epithelium resulted a reduction in secretory cells together with an increase in actively cycling cells within the crypts.24,25 Consistent with this, by comparing untreated samples, fbxw7ΔG mini-guts exhibited a significantly lower percentage of secretory cells such as enteroendocrine cells positively stained for Chromogranin A (chg-A) (Figure 4a,b), goblet cells stained for mucin 2 (muc2) (Figure 4a,c) and Paneth cells stained for lysozyme (Figure 4a,d). Moreover, we examined whether there was correlation between Ki-67 proliferative activity and apoptosis in treated organoids. In line with previous studies of mouse models,24–26 we observed that the ratio between proliferative cells (Ki-67 positive) and apoptotic cells (active caspase 3 positive) was higher in mutant mini-guts (Figure 4a,e). Interestingly, 5-FU treatment induced goblet, Paneth, and enteroendocrine cells differentiation in a concentration-dependent manner (Figure 4a–d), indicating that 5-FU improves organoid peculiar structure by rescuing the cellular differentiation driven by loss of fbxw7. In addition, consistent with fbxw7ΔG mice,24,25 expression of secretory progenitor markers math-1 (mouse atonal homologue-1) and ngn-3 (neurogenin-3) were also significantly repressed in organoids lacking of fbxw7 (Figure 5a). Interestingly, following 5-FU treatment, math1 and ngn3 expression was significantly increased about fourfolds in fbxw7ΔG mini-guts (Figure 5b), in contrast with a slight increase in fbxw7fl/fl mini-guts (Figure 5c). These data indicate that 5-FU promotes terminal differentiation and apoptosis in normal intestinal epithelium, potentially by triggering the DNA damage/repair pathway. Also, CRC harboring FBXW7 mutations may acquire drug resistance via induction of proliferative progenitor cells and impairment of their terminal differentiation in colon/intestine.
Figure 4.

Fbxw7 loss impairs 5-FU-induced differentiation and, thus, apoptosis. (a) H&E staining and immunohistochemistry (IHC) analysis of enteroendocrine cells positive for chromogranine A (chgA), goblet cells positive for mucine (muc2), Paneth cells positive for lysozyme (lys), cycling cells (Ki67), and apoptotic cells positive for active caspase 3 (active cas 3) on fbxw7fl/fl and fbxw7ΔG mini-guts following incubation with 0.8 μmol/l 5-FU. Black arrowheads show positively stained cells. Scale bars, 50 μm. (b–d) Histograms show percentage of terminally differentiated cells and ratio between cycling cells and apoptotic cells (e) following IHC analyses. The relative number of positively stained cells in 50-untreated organoids was counted for each genotype (fbxw7fl/fl versus fbxw7ΔG mini-guts) and in 30-treated organoids for each dose of 5-FU and presented as % of positively stained cells to the total number of cells (*P < 0.05, **P < 0.01, ***P < 0.001).
Figure 5.

5-FU treatment promotes secretory progenitor cells in absence of Fbxw7. (a) fbxw7, math1, and ngn3 expression was analyzed by quantitative real-time polymerase chain reaction (qRT-PCR) in fbxw7ΔG mini-guts at day 7 of growth. Expression level was first normalized to β-actin and then to fbxw7fl/fl mini-guts. (b,c) math1 and ngn3 expression was analyzed by qRT-PCR in fbxw7fl/fl and fbxw7ΔG mini-guts after 72 hours treatment with 0.8 μmol/l 5-FU. Expression was first normalized to β-actin and then to the corresponding untreated mini-guts (*P < 0.05, **P < 0.01, ***P < 0.001). (d) Schematic shows 5-FU induction of terminal differentiation and apoptosis in fbxw7ΔG mini-guts.
Discussion
Murine intestinal fbxw7-deletion blocks differentiation and drives hyperproliferation in the intestine.24,25,28 In vitro studies also reported that loss of FBXW7 function leads to the acquisition of drug resistance in CRC cell lines, through the induction of an epithelial-mesenchymal transition (EMT) pathway,32 and/or alteration of MCL1,40 mTOR,41 and TP53 status.30 In this study, to demonstrate that intestinal organoids are a useful and physiologically relevant model system to study the role of Fbxw7 in cancer biology and drug resistance, we have cultured organoids in the presence and absence of 5-FU. Due to their comparable structure and behavior,12,38 treated and untreated organoids (seeded initially with the same number of crypts) are analyzed across several key parameters over time; integrity, morphology, survival and apoptosis, and stained for various intestinal cell lineages. Here, we describe that fbxw7ΔG organoids exhibited higher proliferative activity, thus driving faster maturation of the enterospheres into enteroids and, ultimately microadenoma-like structures, which disseminated cells outside the mini-gut lumen in a burst (Figure 2c and Supplementary Video S1).
Consistent with CRC cell lines, we further confirmed that Fbxw7 deficiency confers higher tolerance to 5-FU, a DNA damaging agent; the IC50 of fbxw7 mutant mini-guts was significantly higher than controls (5.7 versus 2.6 μmol/l 5-FU, respectively) (Figure 3c). Analyzing the complex phenotypical changes in organoid culture, 5-FU treatment induced the transformation of the common crypt-villus morphology (enteroids) into cystic structures (spheres), in a dose-dependent manner (Figure 3d–f). Likewise the loss of Fbxw7-induced cellular transformation, the transformation of enteroids into spheres was also found in Apc-inactivated intestinal organoids,42 a key component of the Wnt/β-catenin signaling pathway in regulating intestinal epithelial homeostasis and the early stages of colorectal carcinogenesis.43 As predicted, the number of microadenoma-like mini-guts significantly decreased after 5-FU treatment (Figure 3e,f). Fbxw7-depleted mini-guts, showed an active proliferative compartment (Figure 4e) along with reduction of secretory progenitor cells (Figure 5a). According to these, loss of Fbxw7 leads to impaired intestinal terminal differentiation and therefore fbxw7ΔG organoids appears smaller than fbxw7fl/fl organoids. On the other hand, due to an early increased rate of proliferation, most of fbxw7ΔG organoids quickly enlarged and exploded. Consequently, fbxw7ΔG organoids were not homogeneous populations to quantify and compare their size with fbxw7fl/fl organoids.
Our experiments do not point to molecular mechanism underneath FBXW7-rendered resistance to 5-FU. The role of FBXW7 in drug resistance is just beginning to emerge. Based on our own and recent published studies, there is a strong link between FBXW7 and cell cycle regulation as well as the EMT-mediated drug resistance in cancer cells.30,32,40,44,45 However, beyond the scope of this current study and the importance of given data here and our published and unpublished data on links between EMT and FBXW7, more recently, we have initiated a study using different approaches to explore molecular mechanism(s) underneath FBXW7-rendered resistance to DNA-damage agents including 5-FU in CRC, organoid, and animal models. Our current analysis however highlights significant downregulation of the cyclin-dependent kinase inhibitor p21 in fbxw7ΔG organoids while the p53 related transcription factor p63 expression appeared to be not significantly changed. 5-FU led to the induction of p21 and p63 in wild-type fbxw7fl/fl organoids but in fbxw7ΔG organoids induced p63 and not p21 (Supplementary Figure S3a–c). The differential expression of p53 related genes further support the hypothesis of an existing relationship between mutated-Fbxw7 and p53 in response to drug treatment.30 Previously published studies showed that p53 upregulates Fbxw727,46 whereas it represses ZEB family transcriptional regulators (ZEB1 and ZEB2), implicated in EMT.47
These findings clearly indicated that the percentage of enteroendocrine cells, goblet cells and Paneth cells were increased along with a reduction of cycling versus apoptotic cell ratio following 5-FU-mediated DNA damage. Fbxw7 depletion was found to influence cell commitment and proliferation in murine intestinal epithelium via activation of c-Jun and Notch signaling pathways.24,25,28 However, since the amount of proteins available for extraction from 5-FU-treated organoids, it is difficult to analyze how 5-FU modulates math1 and ngn3 in organoids. Previously published reports indicated that intestinal Notch activation upregulates Hes-1 and consequently, represses the transcription of math1 and ngn3.48,49 Therefore, protein extracts were isolated from untreated and 5-FU treated HCT116 and DLD-1 cell lines with FBXW7(−/−) and FBXW7(+/+) alleles and Western blotted for active NICD1 antibody, as a FBXW7 target for degradation.50 These observation further support that expression level of active NICD1 is increased in FBXW7(−/−) cells while decreased in both FBXW7(+/+) and FBXW7(−/−) cells treated with 5-FU (Supplementary Figure S4a). We have also studied the relative mRNA expression of MATH1 and NGN3 genes by quantitative real-time polymerase chain reaction in CRC cell lines. Unfortunately with both Cyber green and TaqMan assays, Ct values of MATH1 and NGN3 were >32 (representing even higher Ct value than FBXW7 gene in K/O cells). We have additionally increased the amount of cDNA which had also no effect on Ct value (Supplementary Figure S4c). These suggest that MATH1 and NGN3 are very low expressed genes in these cells and may not be reliable to calculate their relative gene expression for the purpose of the experiment. In addition, terminal differentiation of the enteroendocrine producing cells has been shown by coordinating transcription of the secretin gene with cell cycle arrest.51,52 Hence, the modulation of Math1 and ngn3 by 5-FU could also be linked to the changes of cyclin-dependent kinase inhibitor p21 level as outlined above (Supplementary Figure S3b,c). However, further studies should be carried out in future, to further precisely explore these possibilities via loss and gain of functional analysis. Progenitor cells, abundant in fbxw7ΔG mini-guts, may drive tumor recurrences and/or metastasis post-treatment. However, contrary to the organoid model, the fbxw7ΔG mice alone did not promote intestinal tumorigenesis up to 11–12 months of age,24,25 whereas we predict that this genotype causes sufficient functional rearrangement to promote/recapitulate human tumorigenesis. Nevertheless, the organoid culture method allows a specific analysis of epithelial components of the intestine/colon and therefore these changes in morphology suggest an alteration between proliferative and differentiating compartments, induced independently of intestinal microenvironment.
We conclude that Fbxw7-mutated cancer cells are highly proliferative and resistant to the terminal differentiation and subsequent cell death that is normally triggered by chemotherapeutic DNA damage (Figure 5d). Furthermore, DNA damaging agents could cause mutations in cycling cells promoting the acquisition of prosurvival properties in drug-resistant cancer cells.53,54 Our findings suggest that a differentiation therapy may be one strategy to validate current chemotherapeutic drugs against the FBXW7-mutated CRCs. These results support the hypothesis that human mini-guts derived from cancer patients,4 and/or the modeling of cancer mutations in human intestinal/colon organoids using new tools for gene manipulation,3,5 may allow both ex vivo and in vivo experimental approaches investigating drug resistance, drug screening, and preclinical toxicological studies in molecularly targeted systems that mimic endogenous cancer driver mutations and eventually design efficacious cancer-specific drugs.
Materials and Methods
Mouse lines
Both fbxw7fl/fl and fbxw7∆G were described previously.24 Only same sex littermates of 4–6-week mice were used. Mice were housed and bred in a pathogen-free transgenic animal facility of the Biomedical Service Unit and all procedures complied with regulations and guidelines of the Biomedical Service Unit, University of Nottingham.
Small intestine crypt isolation and organoid culture
Small intestine was resected from fbxw7fl/fl and fbxw7ΔG mice.7,24 Intestine was repeatedly washed with cold phosphate-buffered saline, then longitudinally opened and sectioned in small fragments. Crypts were released from the epithelium by incubation with 3 mmol/l ethylenediaminetetraacetic acid phosphate-buffered saline for 35 minutes at 4 °C. Following counting and isolation by centrifuge, crypts were resuspended with Matrigel (BD Biosciences, Oxford, UK), seeded into a 48-well plate and fed with advanced Dulbecco’s modified eagle medium/F-12 medium containing; N2 supplement (Invitrogen, Warrington, UK), B27 supplement (Invitrogen), 1 mmol/l N-acetylcysteine (Sigma-Aldrich, Dorset, UK), 50 ng/ml murine recombinant epidermal growth factor (Invitrogen), 100 ng/ml Noggin (Peprotech, London, UK), and 1 μg/ml R-Spondin1.7 0.8, 2.5, and 7.5 μmol/l 5-FU (Tocris Bioscience, Bristol, UK) was administered at day 3 or day 4 of growth respectively to fbxw7ΔG and fbxw7fl/fl mini-guts, for viability and morphological analyses in response to drug treatment. Organoid were also monitored with integrated optics for phase-contrast imaging and machine vision technology in a humidified, 5% CO2 atmosphere at 37 °C for 5 days (Cell-IQ, CM Technologies Oy; www.c-mtechnologies.com). Media replaced every 48 hours and images captured automatically per well at half an hour intervals. Cell-IQ analyzer software was used according to manufacturer’s instructions.
Cell culture, transfection, and western blotting analysis
Human colon cancer HCT116 and DLD-1 cells were fed with RPMI-1640 medium supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 2 mmol/l L-glutamine (Life Technologies, Warrington, UK). Transfection of CRC cell lines with DNA plasmids was performed via Lipofectamine 3000 (Invitrogen) transfection reagent following the manufacturer’s instructions. RIPA lysis buffer supplemented with protease inhibitor cocktail was used to extract the protein content from both live and dead cells. Equal amount of total proteins was denaturized, loaded into a 10% sodium dodecyl sulfate-polyacrylamide gradient gel for separation and then transferred overnight to polyvinylidene fluoride membranes. Membranes were blocked with 3% bovine serum albumin in Tris-buffered saline and probed with different antibodies as indicated as previously described.30
Cytotoxicity assay and colony-forming efficiency assay
Human colon cancer HCT116 and DLD-1 cells were fed with RPMI-1640 medium supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 2 mmol/l L-glutamine (Life Technologies). 3,000 cells/well were seeded for cytotoxicity assay into 96-well plate in triplicate, synchronized by serum-starvation, and incubated with increasing concentrations of 5-FU for 72 hours. Cells were fixed with 6% trichloroacetic acid for 1 hour at 4 °C and then washed with water. 0Air dried plates were stained with 0.4% (w/v) sulforhodamine B in acetic acid for 20 minutes at room temperature and washed five times with 1% acetic acid. 10 mmol/l Trizma base was added and the absorbance read at 540 nm.30
To perform colony-forming efficiency assay, 500 cells (HCT116) were seeded into a six-well plate in triplicate. Then, 0, 2.5, and/or 7.5 μmol/l 5-FU was administered prior to cell-doubling, to reduce survival advantages associated to different proliferative capabilities. 72 hours after, 5-FU containing medium was replaced with a complete culture medium. Colonies were fixed with 4% paraformaldehyde for 20 minutes at room temperature and stained with 0.1% crystal violet for the following procedures to count number of colonies using Image J software.30
Immunohistochemistry
Organoids were fixed in 4% paraformaldehyde overnight at 4 °C and resuspended in 2% low-melting agarose prior to paraffin embedding. 4-µm sections were cut and processed for immunohistochemistry analysis or Hematoxylin and Eosin (H&E) staining as previously reported.24 The following primary antibodies were used: active caspase 3 (R&D systems, Abingdon, UK), Chromogranin A and Ki-67 (Abcam, Cambridge, UK), Mucin 2 (Santa Cruz Biotechnology, Heidelberg, Germany), and Lysozyme (Dako, Cambridge, UK).
Quantitative real-time polymerase chain reaction analysis
To dissociate the Matrigel, organoids were incubated with cell recovery solution (Corning, Corning, NY) for 3 hours in cold room. Total RNA was separated by using TRIZOL reagent (Sigma-Aldrich) and precipitated following manufacturer’s instructions. cDNA synthesis was obtained by using PrimeScript Reverse Transcriptase (TAKARA, Saint-Germain-en-Laye, France) according to the manufacturer’s instructions. Quantitative real-time polymerase chain reaction was performed based on the incorporation of SYBR green (Life Technologies). Primers24 used are listed in Supplementary Table S1.
Statistics
All statistical analyses were evaluated with Student’s t-tests and the Mann-Whitney U-test, as appropriate. A P value of < 0.05 was considered statistically significant, where experiments were repeated at least two and three times in triplicates, respectively.
Acknowledgments
We thank B. Vogelstein for providing HCT116 knockout cells, and N. Li and P. Clarke for technical advice, M. Ahmed for reading the manuscript, and the fantastic fundraising efforts of Alison Sims and her family in memory of Daz Sims to support the work in our laboratory. This study was supported by Cancer Research UK, research projects A9275 and A10247, and the University of Nottingham Vice Chancellor’s Scholarship for Research Excellence (European Union).
The authors declare no conflict of interest.
References
- Stintzing, S (2014). Management of colorectal cancer. F1000Prime Rep 6: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, N, Ridgway, RA, van Es, JH, van de Wetering, M, Begthel, H, van den Born, M et al. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457: 608–611. [DOI] [PubMed] [Google Scholar]
- Matano, M, Date, S, Shimokawa, M, Takano, A, Fujii, M, Ohta, Y et al. (2015). Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med 21: 256–262. [DOI] [PubMed] [Google Scholar]
- van de Wetering, M, Francies, HE, Francis, JM, Bounova, G, Iorio, F, Pronk, A et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161: 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost, J, van Jaarsveld, RH, Ponsioen, B, Zimberlin, C, van Boxtel, R, Buijs, A et al. (2015). Sequential cancer mutations in cultured human intestinal stem cells. Nature 521: 43–47. [DOI] [PubMed] [Google Scholar]
- Sato, T and Clevers, H (2013). Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340: 1190–1194. [DOI] [PubMed] [Google Scholar]
- Sato, T, Vries, RG, Snippert, HJ, van de Wetering, M, Barker, N, Stange, DE et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265. [DOI] [PubMed] [Google Scholar]
- Barker, N, van Es, JH, Kuipers, J, Kujala, P, van den Born, M, Cozijnsen, M et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007. [DOI] [PubMed] [Google Scholar]
- de Lau, W, Peng, WC, Gros, P and Clevers, H (2014). The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev 28: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon, JA, Takada, S, Zimmerman, LB, Fan, CM, Harland, RM and McMahon, AP (1998). Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev 12: 1438–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normanno, N, De Luca, A, Bianco, C, Strizzi, L, Mancino, M, Maiello, MR et al. (2006). Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366: 2–16. [DOI] [PubMed] [Google Scholar]
- Grabinger, T, Luks, L, Kostadinova, F, Zimberlin, C, Medema, JP, Leist, M et al. (2014). Ex vivo culture of intestinal crypt organoids as a model system for assessing cell death induction in intestinal epithelial cells and enteropathy. Cell Death Dis 5: e1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z, Inuzuka, H, Fukushima, H, Wan, L, Gao, D, Shaik, S et al. (2012). Emerging roles of the FBW7 tumour suppressor in stem cell differentiation. EMBO Rep 13: 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker, M and Clurman, BE (2008). FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8: 83–93. [DOI] [PubMed] [Google Scholar]
- Wang, Z, Inuzuka, H, Zhong, J, Wan, L, Fukushima, H, Sarkar, FH et al. (2012). Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett 586: 1409–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa, K, Kotake, Y and Kitagawa, M (2009). Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Sci 100: 1374–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Y and Li, G (2012). Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev 31: 75–87. [DOI] [PubMed] [Google Scholar]
- Tan, Y, Sangfelt, O and Spruck, C (2008). The Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett 271: 1–12. [DOI] [PubMed] [Google Scholar]
- Davis, RJ, Welcker, M and Clurman, BE (2014). Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 26: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona, CA, Sancho, R, Diefenbacher, ME and Behrens, A (2016). Fbw7 and its counteracting forces in stem cells and cancer: Oncoproteins in the balance. Semin Cancer Biol 36: 52–61. [DOI] [PubMed] [Google Scholar]
- Sjöblom, T, Jones, S, Wood, LD, Parsons, DW, Lin, J, Barber, TD et al. (2006). The consensus coding sequences of human breast and colorectal cancers. Science 314: 268–274. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan, H, Jallepalli, PV, Rago, C, Velculescu, VE, Kinzler, KW, Vogelstein, B et al. (2004). Inactivation of hCDC4 can cause chromosomal instability. Nature 428: 77–81. [DOI] [PubMed] [Google Scholar]
- Babaei-Jadidi, R, Li, N, Saadeddin, A, Spencer-Dene, B, Jandke, A, Muhammad, B et al. (2011). FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for degradation. J Exp Med 208: 295–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho, R, Jandke, A, Davis, H, Diefenbacher, ME, Tomlinson, I and Behrens, A (2010). F-box and WD repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology 139: 929–941. [DOI] [PubMed] [Google Scholar]
- Grim, JE, Knoblaugh, SE, Guthrie, KA, Hagar, A, Swanger, J, Hespelt, J et al. (2012). Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Mol Cell Biol 32: 2160–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokobori, T, Mimori, K, Iwatsuki, M, Ishii, H, Onoyama, I, Fukagawa, T et al. (2009). p53-Altered FBXW7 expression determines poor prognosis in gastric cancer cases. Cancer Res 69: 3788–3794. [DOI] [PubMed] [Google Scholar]
- Davis, H, Lewis, A, Behrens, A and Tomlinson, I (2014). Investigation of the atypical FBXW7 mutation spectrum in human tumours by conditional expression of a heterozygous propellor tip missense allele in the mouse intestines. Gut 63: 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp, Z, Rowan, A, Chambers, W, Wortham, N, Halford, S, Sieber, O et al. (2005). CDC4 mutations occur in a subset of colorectal cancers but are not predicted to cause loss of function and are not associated with chromosomal instability. Cancer Res 65: 11361–11366. [DOI] [PubMed] [Google Scholar]
- Li, N, Lorenzi, F, Kalakouti, E, Normatova, M, Babaei-Jadidi, R, Tomlinson, I et al. (2015). FBXW7-mutated colorectal cancer cells exhibit aberrant expression of phosphorylated-p53 at serine-15. Oncotarget 6: 9240–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y, Zhou, X, Bai, W and Ma, X (2015). FBW7 increases drug sensitivity to cisplatin in human nasopharyngeal carcinoma by downregulating the expression of multidrug resistance-associated protein. Tumour Biol 36: 4197–4202. [DOI] [PubMed] [Google Scholar]
- Wang, Y, Liu, Y, Lu, J, Zhang, P, Wang, Y, Xu, Y et al. (2013). Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem Biophys Res Commun 434: 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz, F, Hwang, PM, Torrance, C, Waldman, T, Zhang, Y, Dillehay, L et al. (1999). Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 104: 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhaidat, NM, Bouklihacene, M and Thorne, RF (2014). 5-Fluorouracil-induced apoptosis in colorectal cancer cells is caspase-9-dependent and mediated by activation of protein kinase C-δ. Oncol Lett 8: 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mao, JH, Perez-Losada, J, Wu, D, Delrosario, R, Tsunematsu, R, Nakayama, KI et al. (2004). Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 432: 775–779. [DOI] [PubMed] [Google Scholar]
- Sato, T, van Es, JH, Snippert, HJ, Stange, DE, Vries, RG, van den Born, M et al. (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, A, Donahue, B, Peignon, G, Letourneur, F, Cagnard, N, Slomianny, C et al. (2012). Functional intestinal stem cells after Paneth cell ablation induced by the loss of transcription factor Math1 (Atoh1). Proc Natl Acad Sci USA 109: 8965–8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzner, M, Helmrath, M, Dunn, JC, Henning, SJ, Houchen, CW, Kuo, C et al. NIH Intestinal Stem Cell Consortium. (2012). A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointest Liver Physiol 302: G1359–G1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney, RP, Ballantyne, GH and Modlin, IM (1990). The adenoma to carcinoma sequence. Surg Gynecol Obstet 171: 81–94. [PubMed] [Google Scholar]
- Wertz, IE, Kusam, S, Lam, C, Okamoto, T, Sandoval, W, Anderson, DJ et al. (2011). Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471: 110–114. [DOI] [PubMed] [Google Scholar]
- Mao, JH, Kim, IJ, Wu, D, Climent, J, Kang, HC, DelRosario, R et al. (2008). FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321: 1499–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuma, K, Ochiai, M, Orihashi, K, Takahashi, M, Imai, T, Nakagama, H et al. (2013). Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc Natl Acad Sci USA 110: 11127–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde, R and Brabletz, T (2007). Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol 19: 150–158. [DOI] [PubMed] [Google Scholar]
- Inuzuka, H, Shaik, S, Onoyama, I, Gao, D, Tseng, A, Maser, RS et al. (2011). SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471: 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X, Jin, W, Jia, H, Yan, J and Zhang, G (2015). MiR-223 promotes the cisplatin resistance of human gastric cancer cells via regulating cell cycle by targeting FBXW7. J Exp Clin Cancer Res 34: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, A, Onoyama, I and Nakayama, KI (2006). Expression of mouse Fbxw7 isoforms is regulated in a cell cycle- or p53-dependent manner. Biochem Biophys Res Commun 350: 114–119. [DOI] [PubMed] [Google Scholar]
- Kim, T, Veronese, A, Pichiorri, F, Lee, TJ, Jeon, YJ, Volinia, S et al. (2011). p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 208: 875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fre, S, Huyghe, M, Mourikis, P, Robine, S, Louvard, D and Artavanis-Tsakonas, S (2005). Notch signals control the fate of immature progenitor cells in the intestine. Nature 435: 964–968. [DOI] [PubMed] [Google Scholar]
- Jensen, J, Pedersen, EE, Galante, P, Hald, J, Heller, RS, Ishibashi, M et al. (2000). Control of endodermal endocrine development by Hes-1. Nat Genet 24: 36–44. [DOI] [PubMed] [Google Scholar]
- O’Neil, J, Grim, J, Strack, P, Rao, S, Tibbitts, D, Winter, C et al. (2007). FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med 204: 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh, H, Naya, FJ, Tsai, MJ and Leiter, AB (1998). The basic helix-loop-helix protein BETA2 interacts with p300 to coordinate differentiation of secretin-expressing enteroendocrine cells. Genes Dev 12: 820–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratineau, C, Petry, MW, Mutoh, H and Leiter, AB (2002). Cyclin D1 represses the basic helix-loop-helix transcription factor, BETA2/NeuroD. J Biol Chem 277: 8847–8853. [DOI] [PubMed] [Google Scholar]
- Blanpain, C, Mohrin, M, Sotiropoulou, PA and Passegué, E (2011). DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 8: 16–29. [DOI] [PubMed] [Google Scholar]
- Metcalfe, C, Kljavin, NM, Ybarra, R and de Sauvage, FJ (2014). Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 14: 149–159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.