

Abstract
Granulins (Grns) are a family of small, cysteine-rich proteins that are generated upon proteolytic cleavage of their precursor, progranulin (Pgrn). All seven Grns (A–G) contain 12 conserved cysteines that form 6 intramolecular disulfide bonds, rendering this family of proteins unique. Grns are known to play multi-functional roles, including wound healing, embryonic growth, and inflammation and are implicated in neurodegenerative diseases. Despite their manifold functions, there exists a dearth of information regarding their structure–function relationship. Here, we sought to establish the role of disulfide bonds in promoting structure by investigating the fully reduced GrnB (rGrnB). We report that monomeric rGrnB is an intrinsically disordered protein (IDP) at low concentrations. rGrnB undergoes dimerization at higher concentrations to form a fuzzy complex without a net gain in the structure—a behavior increasingly identified as a hallmark of some IDPs. Interestingly, we show that rGrnB is also able to activate NF-κB in human neuroblastoma cells in a concentration-dependent manner. This activation correlates with the observed monomer–dimer dynamics. Collectively, the presented data establish that the intrinsic disorder of rGrnB governs conformational dynamics within the reduced form of the protein, and suggest that the overall structure of Grns could be entirely dictated by disulfide bonds.
Keywords: cysteine-rich protein, fuzzy complex, granulin, intrinsically disordered protein, progranulin
Introduction
Granulins (Grns), also known as epithelins, are a family of small (∼6 kDa) proteins involved in a plethora of cellular functions associated with both physiological and pathological states (Bateman and Bennett, 1998). They contain 12 conserved cysteines that form 6 intramolecular disulfide bonds. In mammals, Grns are generated by the proteolytic processing of their precursor protein, progranulin (Pgrn) or granulin/epithilin precursor (GEP). Pgrn is expressed ubiquitously in a variety of cells including epithelial, gastroinstetinal, reproductive and immune cells, as well as cells in the central nervous system (CNS) (Bateman and Bennett, 1998; Daniel et al., 2000, 2003; He et al., 2003). The 68.5 kDa Pgrn protein consists of seven-and-a-half tandem repeats of Grn domains corresponding to Grns A–G. While full-length Pgrn is anti-inflammatory, an inflammatory event triggers the cleavage of Pgrn by proteases to release Grns, of which GrnB is known to be pro-inflammatory (Zhu et al., 2002). Among the proteases involved in Pgrn cleavage are neutrophil elastase, proteinase 3 (a neutrophil protease), MMP-12 (matrix metalloproteinase 12; macrophage elastase), MMP-14, and ADAMTS-7 (a disintegrin and metalloproteinase with thrombospondin motifs) (Zhu et al., 2002; Cenik et al., 2012). Pro-inflammatory Grns play a role in wound healing, tumorigenesis, regulation, embryonic cell development and pro-inflammatory immune responses. Despite their established and emerging roles in biological function, the molecular mechanisms by which Grns exert these functions remain unclear. The knowledge-gap in Grn molecular function is especially evident considering their effect on neuronal cells and CNS pathologies, which has come to bear significance lately (Baker et al., 2006; Cruts et al., 2006; Brouwers et al., 2007; Shankaran et al., 2008). Pgrn and Grns have been implicated in familial frontotemporal lobar degeneration (FTLD), a neurodegenerative disorder characterized by behavioral and personality changes (Rabinovici and Miller, 2010; Sieben et al., 2012). Autosomal dominant mutations in the Pgrn gene that result in null alleles were identified as the primary cause for FTLD (Baker et al., 2006; Cruts et al., 2006). In addition, missense mutations mapped within the GrnB region of the Pgrn gene have been implicated in FTLD (Brouwers et al., 2007; Shankaran et al., 2008). Furthermore, several polymorphisms of the Pgrn gene have also been linked to Alzheimer disease (AD) (Brouwers et al., 2007; Cortini et al., 2008). Despite its pro-inflammatory nature, the potential role of GrnB in immune response frequently associated with neurodegeneration remains poorly understood.
The existing deficit in molecular understanding of Grns has been exacerbated by the difficulty in generating large amounts of pure protein in vitro, primarily due to the complexity imparted by the six intramolecular disulfide bonds within these cysteine-rich proteins. NMR structures of mammalian Grns A, C and F, which were determined at high protein concentrations (∼0.5 to 0.6 mM), indicate a unique fold containing four stacked β-hairpins stabilized by six intramolecular disulfide bonds to form a ladder-like arrangement (Hrabal et al., 1996; Tolkatchev et al., 2000, 2008). Notably, the NMR structure also indicated that only 27 of 58 residues were found to a part of β-hairpins, while the rest of the protein either forms loops or remains disordered (Tolkatchev et al., 2008). This structure closely resembles that of epidermal growth factor (EGF) although EGF contains only three intramolecular disulfide bonds (Kohda and Inagaki, 1991). The presence of highly conserved cysteine residues within the Grn family suggest a key role of disulfide bonds in Grn structure and function, especially considering that the sequence homology of GrnB is only 37–61% with the other six Grns (A, C–F). Based on the NMR structures of Carp-Grn and Grns, A C and F, a putative interlocked fashion of disulfide bonding has been proposed for the Grn family, although precise identification of the S–S bonding pattern has not been established.
In order to understand the significance of disulfide bonds in GrnB's structure and function, in this report, we present the characterization of the fully reduced form of human Y24WGrnB (henceforth denoted as rGrnB). Generation of native GrnB containing correct intramolecular disulfide bonds is challenging and remains to be our future objective. However, we argue that the investigation of the fully reduced form will provide a wealth of information about GrnB structure, especially in identifying whether the conserved cysteine residues (and disulfide bonds) are the sole driving force for the protein's structure and function. Indeed, such an investigation has provided some unique insights into the behavior of rGrnB. Our data suggest that rGrnB exists as an intrinsically disordered protein (IDP) at low concentrations but exists predominantly as a dimer at higher concentrations. Only above 300 µM, does rGrnB display a change toward a more ordered structure. Interestingly, dimerization involves no net gain in overall structure, implicating the formation of a fuzzy complex. Interestingly, we observed that rGrnB is capable of modestly activating a central regulator of inflammation, nuclear factor-κB (NF-κB) (Hayden and Ghosh, 2008), in a concentration-dependent manner. This activity is limited to low rGrnB concentrations that correspond to monomeric protein and correlates well with the observed monomer–dimer dynamics. Although Grns (GrnB in particular) are known to be pro-inflammatory, the ability of rGrnB to activate NF-κB is somewhat surprising, but may implicate a potential role of the reduced form of the protein under conditions of improper folding or cellular stress. This report provides insights into the molecular dynamics of rGrnB, which is observed to play multi-functional roles in its native form. The observed biophysical properties of rGrnB bring forth the role of disulfide bonds in GrnB structure and dynamics, and shed lights on its ability to trigger inflammatory response under reducing environments.
Experimental procedures
Cloning of human GrnB in E. coli
Cloning and modifications of the plasmid were carried out at the molecular cloning facility at Florida State University. The GrnB cDNA containing a Y24W mutation to facilitate fluorescence detection as well as NcoI and XhoI restriction enzyme sites at N- and C-termini, respectively, was synthesized commercially (Genewiz, Inc.). These restriction sites were used to insert GrnB cDNA into a pET32b expression vector (Novagen). The commercially available pET32b plasmid facilitates protein expression as a trxA-GrnB fusion containing both N- and C-terminal hexa-histidine tags. The enterokinase cleavage site separating thioredoxin from the protein leaves an unwanted His-tag at the C-terminus of the expressed protein. In order to circumvent this obstacle, the plasmid was modified to remove the C-terminal His-tag by introducing a stop codon downstream from GrnB cDNA. In addition, the enterokinase cleavage site along with the spacer DNA were deleted so that the GrnB cDNA was present upstream from a preexisting thrombin cleavage site. This modification enabled the expression of GrnB protein with a trxA fusion partner and only the N-terminal His-tag. The modified plasmids containing the GrnB construct were expressed in the E. coli Origami 2(DE3) strain (Novagen). The transformed cells expressing the recombinant trxA-GrnB fusion protein were grown at 37°C in 2 l baffled culture shake flasks containing 1 l LB medium supplemented with 100 μg ml−1 of ampicillin. Expression of the protein was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.9 mM when the optical density of the culture media was between 0.5 and 0.7 AU, as measured by UV-absorbance at 600 nm. After 4 h of induction at 37°C, cells were harvested by centrifuging (14 000 × g, 4°C) and used immediately or stored at −20°C.
Purification of recombinant rGrnB
The overall purification strategy is shown in Supplementary Fig. S1. Cell pellets from 2 l of culture were thawed at room temperature and resuspended in 30–40 ml of equilibration buffer (50 mM Tris, pH 6.5, 300 mM NaCl, 10 mM imidazole, 2 M urea) containing 0.5 mM phenylmethylsulfonyl fluoride (PMSF). The cell suspension was sonicated for 2 min (20 s burst with 1 min rest) on ice and centrifuged at 9200 × g to remove debris. The supernatant was loaded onto Ni2+-NTA beads in a flex column preequilibrated with five bed volumes of the equilibration buffer. The column was then rocked for 1 h at room temperature, and the supernatant was allowed to flow through the beads under gravity and collected as a flow-through fraction. The beads were then sequentially washed with 100 and 120 ml of buffer (50 mM Tris, pH 6.5, 300 mM NaCl) containing 60 and 80 mM imidazole, respectively. The trxA-rGrnB fusion protein was eluted using 20 ml of 300 mM imidazole in 50 mM Tris, pH 6.5 and 300 mM NaCl. The eluate was then reduced with ∼10-fold molar excess of the reducing agent, tris(2-carboxyethyl) phosphine (TCEP), by rocking for 1 h at room temperature and then diluted 10 times with 50 mM Tris, pH 6.5 and 300 mM NaCl. The protein was then dialyzed to remove TCEP and imidazole at room temperature against a buffer of 0.2 mM Tris, pH 6.5 and 0.5 mM NaCl and using a 10 kDa MWCO dialysis membrane (Spectrum Labs). The dialysate was then lyophilized using vacuum evaporation and resuspended in sterile water such that the final buffer concentration was 20 mM Tris, pH 6.5 and 50 mM NaCl. Concentration of the fusion protein was measured using UV–Vis spectroscopy with a molar extinction coefficient of 20 355 M−1cm−1 at 280 nm (Gasteiger et al., 2005). On average, 7–9 mg of fusion protein was obtained per liter of induced cells. The fusion protein was aliquoted in 2 mg ml−1 fractions and used immediately or stored at –20°C. Further purification of rGrnB was accomplished by digesting 2 mg aliquots of the protein via addition of restriction grade thrombin (Novagen) (10 U) to cleave both trxA and the His-tag. The reaction was incubated in a 37°C water bath for 22–24 h with 5 U of enzyme added at the start of the incubation and the remaining 5 U added after 8–10 h of incubation. Completion of the cleavage reaction was confirmed by SDS-PAGE analysis (data not shown). rGrnB protein was then fractionated using a LiChroCART C-18 reverse phase HPLC column (Merck, Inc., Germany) with a gradient of 30–90% acetonitrile containing 0.1% TFA. Freshly purified rGrnB was used for every experiment, and the protein was used within 2 h of purification to avoid potential re-oxidation issues.
Cell culture and treatment
Human neuroblastoma SH-SY5Y cells (Sigma–Aldrich) were maintained in a 1:1 mixture of DMEM and Ham's F12K medium supplemented with FBS (10%), penicillin (100 Units ml−1), and streptomycin (100 μg ml−1). All cultures were maintained at 37°C in a humid atmosphere of 5% CO2 and 95% air. SH-SY5Y cells were seeded onto 22 × 22 mm glass coverslips (Corning) at a density of 4 × 105 cells ml−1 and allowed to stabilize in culture for 24 h. Cells were then treated for 60 min at 37°C with rGrnB diluted into cell culture medium containing 1% FBS to achieve final concentrations of 0.02–2 μM rGrnB. Parallel treatment with an equivalent dilution of buffer (20 mM Tris–HCl, pH 6.5) served as the negative control. Cells were fixed and assessed via immunocytochemistry for normalized cellular NF-κB activation, and results are reported as the mean ± SEM for three to four independent trials. Statistical analysis was performed with a one-way ANOVA using GraphPad Prism 6 software. When the results of the ANOVA analysis demonstrated significance, a Dunnett's test for multiple comparisons identified groups with means significantly different than the control.
Immunocytochemistry
Following 60 min rGrnB treatment, cells were washed with medium containing 1% FBS and fixed with 4% paraformaldehyde in PBS for 10 min at 25°C. Fixed cells were rinsed three times with DPBS, permeabilized (0.1% Triton X-100, 0.01 mol l−1 glycine in DPBS) and rinsed again with DPBS. To prevent nonspecific binding, cells were blocked first with 5% BSA in DPBS, followed by 5% normal donkey serum, 1% BSA in DPBS. Cells were incubated overnight at 4°C with the primary antibody MAB3026 (Millipore), which is specific for the activated form of the NF-κB p65 subunit, diluted 1:1200 in DPBS. Labeled cells were rinsed with 1% BSA in DPBS prior to blocking with 5% normal donkey serum, 1% BSA in DPBS. Bound primary antibody was detected via incubation for 2 h at 25°C with Alexa Fluor 555 conjugate goat anti-mouse secondary antibody diluted 1:1000 in DPBS. After rinsing three times with 1% BSA in DPBS followed by two times with DPBS, prepared coverslips were mounted onto slides using Fluoroshield with DAPI (Sigma–Aldrich).
Slides were visualized under a Zeiss LSM 510 META Confocal Scanning Laser Microscope (Carl Zeiss) using a plan-neofluar 40×/1.3 oil DIC immersion objective (Carl Zeiss). z-Stacks of the two-channels, corresponding to fluorescently labeled activated NF-κB and DAPI-labeled nuclei, were acquired to encompass the entire cell layer; z-stacks of three different fields were captured for each treatment. ImageJ64 software (Abràmoff et al., 2004; Schneider et al., 2012) was used to create a z-projection of maximum intensity for each of the two channels within every acquired z-stack. The resultant two-channel images were converted to TIFF files for quantitative image analysis using a custom subroutine written in Matlab™ software (MathWorks). Here, images were separated into the two channels, and each channel was converted into 8-bit monochrome images. DAPI images were transformed into 8-bit binary images via comparison of individual pixel values to a common threshold, and noise was removed. Nuclei were then identified by tagging the pixels representative of nuclear boundaries and identifying successive ‘layers’ of pixels toward the interior of this boundary until convergence was reached. Upon convergence, a cell was counted, and a radius of exclusivity was defined within which another cell could not be identified. Next, 8-bit monochrome NF-κB images were used to calculate the fluorescence associated with activated NF-κB, which was divided by the number of cells to yield the activated NF-κB fluorescence per cell. This cellular NF-κB activation was determined using the three images acquired for each treatment. Reported values are normalized to the cellular NF-κB activation observed for the control.
Results
In silico analysis of rGrnB reveals a largely disordered protein
To test whether rGrnB is intrinsically disordered, Predictor of Naturally Disordered Regions (PONDR) was used (www.pondr.com). PONDR utilizes a series of computational predictor tools such as VLXT (Romero et al., 2001), VLS2B (Peng et al., 2006), VL3 (Peng et al., 2005) and a meta-predictor called PONDR-Fit (Xue et al., 2010). All these predictors employ artificial neural networks with primary sequence information as inputs, and individually assign score to each amino acid to predict their disorderness within the protein sequence (Xue et al., 2010). PONDR VLXT analyzes the intermediate and terminal regions using X-ray and NMR-derived protein structures as training datasets. VSL2B and VL3 accurately predict long disordered regions (>30 amino acids) using multiple sequence profiles and secondary structure alignments as training sets. PONDR-Fit is a meta-predictor that uses the aforementioned tools in addition to FoldIndex, IUPred and TopID. Upon analysis, VSL2B and VL3 predicted rGrnB to be completely disordered with a disorder disposition value > 0.6 for entire sequence of the protein (Fig. 1B, blue and red lines). However, VLXT and PONDR-Fit showed the protein to be partially disordered with a disorder disposition value < 0.5 for 30 and 60% of the protein sequence, respectively (Fig. 1B, gray and green lines). This disparity could arise due to several factors: first, the training dataset for VLXT include 15 proteins that contain low amount of cysteines (0.5–2%) both in oxidized and reduced forms (Romero et al., 2001). Second, it is possible that VLS2B and VL3 are more accurate for rGrnB than VLXT as they primarily predict long disordered regions spanning longer than 30 amino acid residues (Xue et al., 2010). To test whether this disparity in prediction arises due to the presence of 12 cysteines in rGrnB, the same predictors were used to analyze Cys to Ala, and Cys to Ser mutations of rGrnB (alaGrnB and serGrnB, respectively) (Fig. 1C and D). The data indicate that VLXT and PONDR-Fit predicted a significant disorder for both alaGrnB and serGrnB (∼90% of the sequence with a disorder disposition > 0.5). The discrepancies surrounding the disorder prediction involving cysteines also bring forth the paucity in attributes for the amino acid (oxidized or reduced) within disorder predicting tools. Nevertheless, the collective analyses of rGrnB, alaGrnB and serGrnB suggest a predominantly disordered structure for rGrnB. We also analyzed rGrnB and the mutants (alaGrnB, and serGrnB) with CIDER (Classification of Intrinsically disordered Ensemble Regions) Holehouse, et al. (2015). This analysis showed that both rGrnB and mutants fall within region 1 (black dot, Fig. 1E) indicating the proteins to be weak polyampholytes or polyelectrolytes. In addition, all three proteins showed κ (indicator of charged amino acid mixing within a sequence) and FCR (fraction of charged residue) values of 0.214 and 0.167, respectively (Fig. 1E, table), further confirming their weak polyampholytic nature. Finally, the disorder promoting scores derived from CIDER analysis also correlate with those obtained from PONDR, corroborating the disordered nature of rGrnB (Fig. 1E, table).
Fig. 1.

In silico analysis of intrinsic disorder of rGrnB, alaGrnB and serGrnB. (A) Primary sequence of the fully reduced rGrnB. The * at position 28 (position 24 in the sequence of native GrnB) indicates the Y to W substitution made within the construct. (B–D) predictor of naturally disordered regions (PONDR) analysis of rGrnB, alaGrnB and serGrnB, respectively, using VLXT (light gray), VSL2B (black), VL3 (dark gray) and PONDR-Fit (medium gray) predictors. (E) CIDER analysis of rGrnB, alaGrnB and serGrnB shows that all three of them (black dot) fall in region 1 (lightest gray) of the diagram of states (plot of positively charged residues vs. fraction of negatively charged residues. The table, obtained from the CIDER analysis, shows a low kappa (κ) value of 0.214, low FCR (fraction of charged residues) value of 0.167 and disorder promoting value of 0.6 for rGrnB and 0.8 for alaGrnB and serGrnB.
rGrnB displays potential to dimerize via noncovalent interactions
To facilitate experimental characterization of rGrnB by intrinsic fluorescence, the point mutation, Y24W (position 28 in our construct, Fig. 1A) was introduced and the clone was recombinantly expressed in E. coli (see Experimental procedures section). This clone represents a conservative substitution from native GrnB, as all Grns except GrnB and GrnG contain a conserved W at residue 24. Protein concentration was estimated using the calculated molar extinction coefficient of 6250 M−1 cm−1 at 280 nm (Gasteiger et al., 2005). HPLC fractionation of reduced thioredoxin fusion rGrnB (trxA-rGrnB) cleaved by thrombin resulted in elution of rGrnB in fractions 10 and 11 (Fig. 2A, arrow). Examination of rGrnB by SDS-PAGE performed under reducing and nonreducing conditions revealed that the protein remains monomeric in both conditions confirming the absence of any intermolecular disulfide bond (Fig. 2B, Lanes 1 and 2). Purified rGrnB was confirmed by MALDI-ToF mass spectrometric analysis, which indicated a major peak at an m/z value of 6367.73 Da corresponding to a monoisotopic (or average) mass of 6367.39 Da for fully reduced rGrnB. However, without heat treatment, a diffuse band near ∼13 kDa in addition to monomeric rGrnB was observed in SDS-PAGE gel, which correlates with dimeric rGrnB (Fig. 2B, Lane 3). As intermolecular disulfide bonds are ruled out, the dimerization must be predominantly due to noncovalent interactions. Indeed, a peak at m/z 12735.3 Da, corresponding to dimeric rGrnB was also observed in MALDI-Tof analysis (Fig. 2C). Ellman's assay performed on freshly purified rGrnB samples indicated >95% of the cysteines were in the reduced form. In order to unambiguously confirm that all the 12 cysteines are indeed present as free thiols, aliquot of the purified sample was alkylated with iodoacetamide and analyzed by MALDI-Tof mass spectrometry. As shown in Fig 2D, the treatment yielded a single m/z value of 6993.7 corresponding to 10.7 (∼11) cysteines, which correlated with the ∼5% of oxidized protein by Ellman's assay, confirming that over 90% of the rGrnB was present in a reduced form. In order to see the extent of rGrnB re-oxidation, free sulfhydryl content was assessed using Ellman's assay for up to 6 h at room temperature, a condition within which all the experiments were performed (Supplementary Fig. S2A). The results indicate that 95% of cysteines remained reduced within this time. Furthermore, incubation of the protein up to 7 days at 4°C also showed no change in oxidation content (data not shown). Only after 20 days at 4°C, 4 out of 12 cysteines (33%) of rGrnB re-oxidized based on iodoacetamide alkylation (Supplementary Fig. S2B).
Fig. 2.

Characterization of recombinant rGrnB. (A), HPLC profile of rGrnB purification after thrombin cleavage. The peak marked with an arrow indicates the fraction corresponding to the native form of rGrnB. The other peak corresponds to thioredoxin. (B) SDS-PAGE analysis of the rGrnB fraction marked with an arrow on the HPLC profile. Lane 1 contains the sample reduced using 2-mercaptoethanol (β-ME) and boiled; Lane 2 contains nonreduced and boiled sample; Lane 3 contains nonreduced and not boiled sample. (C) MALDI-ToF analysis of rGrnB indicating a molecular weight of 6367.39 Da, corresponding to monomeric mass (theoretical MW 6367.4 Da). In addition, the spectrum shows the presence of rGrnB dimer (12735.3 Da). (D) Alkylation of rGrnB with iodoacetamide showing a signal corresponding to 6993.7 Da indicating presence of 10.7 (∼11) free thiols (reduced cysteines). The results are representative of three or more consistent repeats.
rGrnB is intrinsically disordered at low concentrations
The secondary structure of rGrnB as a function of protein concentration was analyzed using far-UV circular dichroism (CD) spectroscopy. Between concentrations of 10 and 100 μM, rGrnB displayed a predominantly disordered structure, reflected by a signature random coil CD spectrum exhibiting a minimum at λ = 200 nm (Fig. 3A). Concentrations below 10 µM also exhibited random coil structure (Supplementary Fig. S3A). In contrast, as the rGrnB concentration increased above 100 μM, the degree of disorder diminished, as indicated by a decrease in ellipticity at 200 nm and a concomitant increase in negative ellipticity at 222 nm, indicative of α-helical structure (Fig. 3A). This change became more pronounced for rGrnB concentrations exceeding 300 µM. At high concentrations, a pronounced net negative ellipticity at 225 nm (Fig. 3A, inset) suggests a shift toward α-helical structure (222 nm) with contributions from turns and loops (230 nm). While CD spectra above 800 μM were partly obscured by fluctuations associated with high voltage at low wavelengths, these spectra did indicate a sharp negative minimum at 230 nm, suggesting a conformation dominated by loops and turns (Supplementary Fig. S3B). Zonal fractionation of rGrnB by size exclusion chromatography (SEC) resulted in elution of the protein at fractions corresponding to three times its own molecular weight, based on globular protein standards (BioRad) (Fig. 3B). This result is not surprising considering that IDPs typically exhibit a 2- to 3-fold increase in molecular weight during SEC fractionation due to the increased hydration sphere around the unfolded structure (Csizmók et al., 2006).
Fig. 3.

rGrnB is an IDP. (A) Concentration-dependent conformational changes in rGrnB observed by far-UV CD. (Inset) Spectra as a difference in the molar ellipticities at 222 nm (helix) and 200 nm (random coil). (B) Fractionation of rGrnB using a Superdex™ 75 size exclusion column (solid line) and compared with the fractionation profile of gel filtration standards (dotted line)—1-bovine thyroglobulin (670 kDa), 2-bovine γ-globulin (158 kDa), 3-chicken ovalbumin (44 kDa), 4-horse myoglobin (17 kDa) and 5-vitamin B12 (1.35 kDa).
To further the experimental and computational assessment of rGrnB's intrinsic disorder, the ability of osmolytes trifluorethanol (TFE) and trimethylamine N-oxide (TMAO) to induce structural changes within the protein was probed. TFE is a known helix inducer, while TMAO aids the formation of folded structures (Uversky et al., 2001). As expected, TFE was able to induce α-helical conformation within rGrnB at concentrations as low as 20–30% TFE (Fig. 4A). The low mid-point of this transition (equivalent to the melting point during protein unfolding), was ∼16 ± 5% TFE (Fig. 4A, inset), which provides further evidence that rGrnB is intrinsically disordered. Similarly, addition of TMAO resulted in significant conformational changes in rGrnB (Fig. 4B). The loss of random coil structure (decrease in negative minimum at 198 nm) with increasing concentrations of TMAO was profound (Fig. 4B), indicating that this osmolyte was able to induce folding. At higher TMAO concentrations, the CD spectrum exhibits a negative minimum moving toward 216 nm and a positive maximum at 198 nm, reflecting a β-sheet conformation. This transition was markedly observed at 198 nm, where the negative minimum observed in the absence of TMAO became a positive maximum at high TMAO concentrations (Fig. 4B). Collectively, these observed data suggest that rGrnB exists as an IDP as predicted by in silico analyses.
Fig. 4.

Effect of osmolytes on rGrnB conformation. (A) Far UV CD spectra of rGrnB measured in the presence of increasing concentrations of TFE. (Inset) The difference in the ellipticities between 208 and 222 nm (helix) for each TFE concentration plotted against the concentration of TFE. The data were fitted with Boltzman's sigmoidal equation to obtain a melting concentration of 16 ± 5% TFE. (B) Far UV CD spectra of rGrnB in the presence of increasing concentrations of trimethylamine N-oxide (TMAO).
rGrnB dimerizes as a fuzzy complex
As described earlier, SDS-PAGE results suggest that rGrnB forms noncovalent dimers (Fig. 2B). rGrnB dimerization was further investigated using electrophoretic, spectroscopic and sedimentation analyses. Upon subjecting rGrnB (pI = 5.33) to electrophoresis under nondenaturing conditions using 25 mM Tris, pH 8.8, containing 192 mM glycine as the running buffer, a diffuse band encompassing dimeric (D; 12.6 kDa) as well as monomeric (M; 6.3 kDa) rGrnB was observed (Fig. 5A, inset). Under similar electrophoretic conditions, the positive control, Aβ (pI = 5.31), yielded three distinct bands corresponding to monomer (M; 4.5 kDa), dimer (D; 9 kDa), and trimer (T; 13.5 kDa) (Fig. 5A, inset). Sedimentation velocity analysis of 100 µM rGrnB by analytical ultracentrifugation (AUC) showed a major peak at 1.5 s, which corresponds to a calculated molecular weight of 13.1 kDa and thus correlates to dimeric rGrnB (Fig. 5A). The peak at 0.4 s, which was deconvoluted to 4.5 kDa, could indicate the presence of monomeric rGrnB (Fig. 5A). Dimerization was also probed using ANS, which binds specifically to solvent-exposed hydrophobic surfaces on proteins. Here, the rGrnB concentration was varied from 1 to 25 µM while keeping the ANS concentration constant at 500 µM (Fig. 5B, inset). Upon titration, the normalized ANS fluorescence emission, monitored at 512 nm, showed an exponential decrease with increasing rGrnB concentration (Fig. 5B, closed circle). This exponential decrease suggests that lower concentrations of rGrnB contain more solvent-exposed hydrophobic binding surfaces. As the protein concentration increases, the available hydrophobic surfaces for ANS binding decrease, which is likely due to the involvement of hydrophobic surfaces in dimerization, rendering those unavailable for ANS to bind. The data were fitted to a monomer–dimer model (Eq. (1)) (see Experimental procedures section in Supplementary data) to yield an apparent dissociation constant, Kdapp of 0.24 ± 0.02 μM. In contrast, BSA, which is known to undergo weak dimerization, showed an increase in normalized ANS fluorescence intensity with an increase in protein concentration (Fig. 5B, open circle). In parallel, the intrinsic fluorescence emission arising from solvent exposure of the single tryptophan residue (Y24W) within rGrnB was monitored upon diluting the protein with 20 mM Tris, pH 6.5, containing 0.01% NaN3. At low rGrnB concentrations, a tryptophan emission at 355 nm, typical for a solvent exposed residue in an IDP, was observed. An increase in rGrnB concentration yielded only a slight blue shift (to 352 nm) (Fig. 5C), indicating a small change in the hydrophobic environment around the tryptophan. The normalized florescence intensity, however, indicated a notable increase with increasing protein concentration (Fig. 5D, closed circle), suggesting a distinct conformational change around the tryptophan residue, similar to that observed previously for human interferon γ (Boteva et al., 1996). Fitting the data with the same monomer-dimer model (Eq. 1) yielded a Kdapp value of 0.9 ± 0.17 μM, comparable to the value obtained from ANS binding data. In contrast, BSA showed relatively weak transitions in normalized fluorescence intensity, reflecting its diminished ability to undergo dimerization (Fig. 5D, open circle). To obtain further insight into such a mechanism, solvent accessibility of tryptophan at low and high rGrnB concentrations was investigated by Stern–Volmer analysis (Lakowicz, 2007), in which tryptophan fluorescence in the presence of a quencher (acrylamide) was analyzed. At both low concentration (10 μM, Fig. 5E, closed circle) and high concentration (100 μM, Fig. 5E, open circle), stark deviations from linearity were observed, with an upward curvature indicative of the presence of both static and dynamic quenching processes due to solvent exposed tryptophan. Moreover, normalized fluorescence intensities for the two concentrations indicate that the deviation is more pronounced at a lower rGrnB concentration, suggesting that tryptophan is more solvent exposed in the monomeric state than in the dimer. Unfortunately, quenching experiments at concentrations below 10 µM could not be performed as intensities dropped below detection limits. However, since 10 and 100 µM rGrnB would contain significant monomer and dimer populations, respectively, the data provides a reliable insight into solvent exposure surrounding the tryptophan residue.
Fig. 5.

Dimerization of rGrnB. (A) Molecular size distribution of rGrnB at 100 μM obtained by sedimentation velocity indicates a predominant dimer (MW 13.1 kDa). (Inset) Native PAGE analysis of rGrnB at 100 μM run on a 14% gel indicates a diffuse dimeric band corresponding to ∼13 kDa, based on the Aβ sample electrophoresed in parallel for which monomer (M: 4.5 kDa), dimer (D: 9.0 kDa) and trimer (T: 13.5 kDa) are observed. (B, D) rGrnB (closed circle) concentration-dependent ANS binding and intrinsic tryptophan fluorescence, respectively. The normalized fluorescence was plotted against protein concentration, and data were fitted (solid line) to a monomer–dimer model as described in Supplementary data. Bovine serum albumin (BSA) (open circle) was used as a negative control. (Inset) (B) Representative normalized ANS fluorescence scans of 25 and 1 μM rGrnB. (C) Representative normalized tryptophan fluorescence scans of 25 and 1 μM rGrnB. E, Normalized Stern-Volmer plots for 10 μM (closed circle) and 100 μM (open circle) rGrnB using acrylamide as the quenching agent.
In combination with findings that rGrnB exists in a predominantly disordered state within the concentration range of 10–100 µM (Fig. 3A), these data suggest that the intrinsically disordered rGrnB undergoes dimerization in this concentration range without a net gain in the overall structure. As such, rGrnB forms a fuzzy complex containing intrinsically disordered, homogenous dimers, a property that an increasing number of IDPs have been reported to possess (Tompa and Fuxreiter, 2008; Libich et al., 2010; Brzovic et al., 2011; Fuxreiter, 2012; Fuxreiter and Tompa, 2012). Collectively, the data indicate that rGrnB displays concentration-dependent changes in the following regimes: (i) <1 μM—intrinsically disordered, monomeric; (ii) 50–100 μM—intrinsically disordered, dimeric and (iii) >300 μM—partially α-helical/turns.
Low concentrations of rGrnB activate NF-κB in SH-SY5Y human neuroblastoma cells
Since Grns, especially GrnB, are known to be pro-inflammatory (Zhu et al., 2002), we wondered whether rGrnB, although likely to be present in rare circumstances such as cellular stress and/or during the breakdown of protein folding machinery, could elicit inflammatory response. Therefore, the ability of rGrnB to activate NF-κB was assessed in SY-SH5Y human neuroblastoma cells. Here, immunocytochemistry was coupled with an antibody specific for an epitope that is accessible on the p65, or RelA, subunit of NF-κB only when NF-κB is in an unbound state. When NF-κB is bound to inhibitor of κB (IκB), it remains inactive in the cytoplasm. Introduction of an inflammatory stimulus provokes the phosphorylation, ubiquitination and degradation of IκB and subsequent release of NF-κB leading to its activation and nuclear translocation (Verma et al., 1995; Baldwin, 1996; Hayden and Ghosh, 2008). Thus, the primary antibody employed in these immunocytochemistry studies is specific for the activated form of NF-κB.
SH-SY5Y cells that underwent negative control treatment displayed low NF-κB activation (Fig. 6A–C). When cells were exposed for 60 min to medium containing 0.02 μM rGrnB, a slight increase in NF-κB activation was observed (Fig. 6D–F). However, image analysis employed to quantify activated NF-κB staining per cell revealed that this increase did not reach significance (Fig. 6P). In contrast, SH-SY5Y cells treated with medium containing 0.2 or 0.5 μM rGrnB demonstrated a significant increase in NF-κB activation when compared with the control (Fig. 6G–L, P). Thus, at rGrnB concentrations of 0.5 μM or lower, a dose-dependent increase in rGrnB activation of NF-κB was observed. Treatment of SH-SY5Y cells with higher rGrnB concentrations of 0.8, 1 and 2 μM (Fig. 6M–O, P); however, failed to elicit any increase in NF-κB activation. This latter result indicates that the dose-dependent increase in rGrnB activation of NF-κB does not extend to concentrations 0.8 μM and above.
Fig. 6.
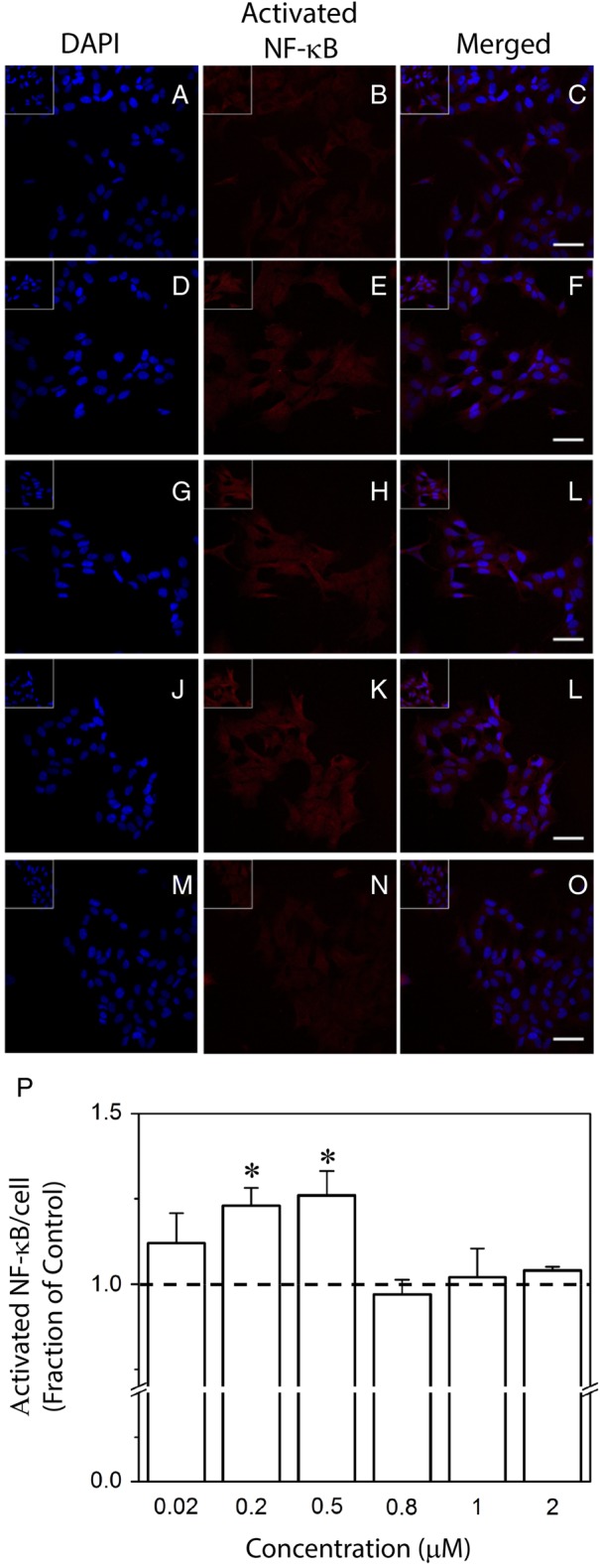
Effect of rGrnB on NF-κB activation in SH-SY5Y cells. SH-SY5Y cells were incubated alone (control, A–C) or in the presence of 0.02 (D–F), 0.2 (G–I), 0.5 (J–L), 0.8 (not shown), 1 (not shown), or 2 μM (M–O) rGrnB. Immunofluorescence staining was performed for the activated form of NF-κB in conjunction with nuclear DAPI staining. Scale bars = 50 μm. Inserts located in top left corners represent ×1.5 magnification. (P) Activated NF-κB staining intensity per cell was analyzed using a custom MATLAB routine as described in the Experimental procedures. Results are reported relative to the control. Error bars indicate SEM, n = 3–4. *P < 0.05, relative to the control.
Discussion
The objective of this report is to identify the role of six intramolecular disulfide bonds in the structure and dynamics of GrnB by investigating the fully reduced form of protein (rGrnB) that abrogates the disulfide bond formation. This approach provides the convenience of avoiding the traditional, cumbersome approach in generating cysteine to alanine mutations, and any impact such mutations may have upon structure and conformation of GrnB. The challenge, however, is to ensure that GrnB is maintained in the reduced form during the course of experiments. Surprisingly, as described in the results section, rGrnB showed resistance to re-oxidation in vitro within our experimental timeframe that facilitated our investigations (Supplementary Fig. S2). This report establishes that rGrnB, as predicted by the computational tools, PONDR and CIDER, exists predominantly as an IDP at low concentrations. This result implicates that intramolecular disulfide bonds within the native, oxidized GrnB are the main forces dictating the structure. Furthermore, the intrinsically disordered rGrnB exhibited some interesting dynamics such as concentration-dependent changes and self-association. It is clear from our data that rGrnB exists as a dimer at high concentrations and that dimerization does not result in a disorder-to-order transition, but rather leads to a fuzzy dimer complex lacking significant structural organization—a behavior that is increasingly identified as a hallmark among IDPs (Tompa and Fuxreiter, 2008; Fuxreiter, 2012; Fuxreiter and Tompa, 2012).
We now know that the structure of oxidized GrnB is primarily due to intramolecular disulfide bonds, and the closest members that fall into this category are the kringle domains, which contain six conserved cysteines that form three intramolecular disulfide bonds (van Zonneveld et al., 1986; Cao et al., 1996; Castellino and McCance, 1997). These proteins include the family of growth factors comprised of EGF and transforming growth factor-α (TGF-α). NMR structures of these two growth factors are identical, exhibiting a single antiparallel β-sheet and other regions with a high degree of disorder (Montelione et al., 1989; Kohda and Inagaki, 1991). The reported NMR structures for Carp-Grn and Grns A, C, and F show significant structural homology with EGF (Kohda and Inagaki, 1991). The data presented here collectively suggest that the overall structure of oxidized, native GrnB is predominantly driven by the intramolecular disulfide bonds rather than the inherent propensity of the amino acids to form secondary and tertiary structures.
From a functional perspective, GrnB has been implicated as pro-inflammatory but rGrnB may not hold much relevance in homeostatic conditions. However, under the conditions of cellular stress or the breakdown of folding machinery, GrnB may be present in the reduced form. Interestingly, IDPs are evolutionarily adapted to be involved in multiple biological functions (Uversky, 2013) and hence, rGrnB could be involved in physiological activity within the cells. Our data provides evidence that rGrnB is able to directly activate NF-κB, the central regulator of inflammation (Hayden and Ghosh), in human neuroblastoma (SH-SY5Y) cells in a dose-dependent manner. Importantly, activation of NF-κB was attenuated at higher concentrations of rGrnB, indicating that dimeric rGrnB may be less pro-inflammatory than the monomeric form. The concentration range of rGrnB at which the attenuation of NF-κB activation occurs (0.5–0.8 µM) is also in close agreement with the observed apparent dissociation constant (Kdapp) for rGrnB dimerization (0.2–0.9 µM).
Conclusions
The report presented here demonstrates that rGrnB exits as an IDP at low concentrations and suggests that the structure of native GrnB is predominantly driven by disulfide bonds similar to the kringle domains. Furthermore, rGrnB by itself showed interesting conformational dynamics. rGrnB dimerizes at elevated concentrations and forms a fuzzy complex. This report also implicates a pro-inflammatory function for rGrnB to the CNS, indicated by the activation of NF-κB in neuroblastoma cells. The ongoing investigations on native GrnB in our laboratory will provide further insights into its functions.
Authors’ contributions
V.R. conceptualized the research, while V.R. and G.G. designed biophysical experiments. M.A.M. was also involved in research design and interpretation. G.G. and R.G.R. performed and analyzed the biophysical experiments. C.M. performed the AUC experiments. L.M.W. performed and analyzed cell culture experiments to probe the ability of rGrnB to activate NF-κB. N.P.V. developed the Matlab™ code for determination of cellular NF-κB activation and drafted the associated section of the manuscript. V.R., M.A.M. and G.G. prepared and edited the manuscript while L.M.W. drafted the manuscript associated with cell culture sections, and assisted with manuscript editing.
Supplementary data
Funding
This work was partly funded by National Center for Research Resources (5P20RR016476-11) and the National Institute of General Medical Sciences (8 P20 GM103476-11) from the National Institutes of Health for funding through INBRE (to V.R.) as well as the National Institute of General Medical Sciences (P20GM103641) from the National Institutes of Health for funding through COBRE (to M.A.M.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors wish to thank Dr Sabine Heinhorst for sharing valuable resources and Dr Andrew Bateman (McGill University, Canada) for generously gifting GrnA plasmids.
Conflict of interest
None declared.
Supplementary Material
References
- Abràmoff M.D., Magalhães P.J., Ram S.J. (2004) Biophotonics Int., 11, 36–43. [Google Scholar]
- Baker M., Mackenzie I.R., Pickering-Brown S.M. et al. (2006) Nature, 442, 916–919. [DOI] [PubMed] [Google Scholar]
- Baldwin A.S. (1996) Annu. Rev. Immunol., 14, 649–681. [DOI] [PubMed] [Google Scholar]
- Bateman A., Bennett H.P. (1998) J. Endocrinol., 158, 145–151. [DOI] [PubMed] [Google Scholar]
- Boteva R., Zlateva T., Dorovska-Taran V., Visser A.J., Tsanev R., Salvato B. (1996) Biochemistry, 35, 14825–14830. [DOI] [PubMed] [Google Scholar]
- Brouwers N., Nuytemans K., van der Zee J. et al. (2007) Arch. Neurol., 64, 1436–1446. [DOI] [PubMed] [Google Scholar]
- Brzovic P.S., Heikaus C.C., Kisselev L. et al. (2011) Mol. Cell, 44, 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Ji R.W., Davidson D. et al. (1996) J. Biol. Chem., 271, 29461–29467. [DOI] [PubMed] [Google Scholar]
- Castellino F.J., McCance S.G. (1997), (2007) Ciba Foundation Symposium, Plasminogen-Related Growth Factors, Vol. 212, pp. 46–65. John Wiley & Sons, Ltd. [DOI] [PubMed] [Google Scholar]
- Cenik B., Cenik B.K., Herz J., Yu G. (2012) J. Biol. Chem., 393, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortini F., Fenoglio C., Guidi I. et al. (2008) Eur. J. Neurol., 15, 1111–1117. [DOI] [PubMed] [Google Scholar]
- Cruts M., Gijselinck I., van der Zee J. et al. (2006) Nature, 442, 920–924. [DOI] [PubMed] [Google Scholar]
- Csizmók V., Szollosi E., Friedrich P., Tompa P. (2006) Mol. Cell. Proteomics, 5, 265–273. [DOI] [PubMed] [Google Scholar]
- Daniel R., He Z., Carmichael K.P., Halper J., Bateman A. (2000) J. Histochem. Cytochem., 48, 999–1009. [DOI] [PubMed] [Google Scholar]
- Daniel R., Daniels E., He Z., Bateman A. (2003) Dev. Dyn., 227, 593–599. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M. (2012) Mol. Biosyst., 8, 168–177. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M., Tompa P (2012) In Fuxreiter M., Tompa P (eds.), Fuzziness, Advances in Experimental Medicine and Biology, vol. 725, U.S.A: Springer, pp. 1–14. [Google Scholar]
- Gasteiger E, Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. (2005) In Walker J.M. (ed.), The Proteomics Protocols Handbook. Humana Press, pp. 571–607. [Google Scholar]
- Hayden M.S., Ghosh S. (2008) Cell, 132, 344–362. [DOI] [PubMed] [Google Scholar]
- He Z., Ong C.H., Halper J. et al. (2003) Nat. Med., 9, 225–229. [DOI] [PubMed] [Google Scholar]
- Holehouse A.S., et al. (2015) Biophys. J., 108 (Suppl. 1), 228a.25606670 [Google Scholar]
- Hrabal R., Chen Z., James S., Bennett H.P., Ni F. (1996) Nat. Struct. Mol. Biol., 3, 747–752. [DOI] [PubMed] [Google Scholar]
- Kohda D., Inagaki F. (1991) Anal. Sci., 7(Suppl.), 853–856. [Google Scholar]
- Lakowicz J.R. (2013), Principles of Fluorescence Spectroscopy. Springer Science & Business Media. [Google Scholar]
- Libich D.S., Ahmed M.A.M., Zhong L., Bamm V.V., Ladizhansky V., Harauz G. (2010) Biochem. Cell Biol., 88, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montelione G.T., Winkler M.E., Burton L.E., Rinderknecht E., Sporn M.B., Wagner G. (1989) Proc. Natl. Acad. Sci., 86, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K., Vucetic S., Radivojac P., Brown C.J., Dunker A.K., Obradovic Z. (2005) J. Bioinform. Comput. Biol., 03, 35–60. [DOI] [PubMed] [Google Scholar]
- Peng K., et al. (2006) BMC Bioinformatics, 7, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici G.D., Miller B.L. (2010) CNS Drugs, 24, 375–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P., Obradovic Z., Li X., Garner E.C., Brown C.J., Dunker A.K. (2001) Proteins, 42, 38–48. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. (2012) Nat. Meth., 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran S.S., Capell A., Hruscha A.T., Fellerer K., Neumann M., Schmid B., Haass C. (2008) J. Biol. Chem., 283, 1744–1753. [DOI] [PubMed] [Google Scholar]
- Sieben A., Van Langenhove T., Engelvorghs S. et al. (2012) Acta Neuropathol., 124, 353–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkatchev D., Ng A., Vranken W., Ni F. (2000) Biochemistry, 39, 2878–2886. [DOI] [PubMed] [Google Scholar]
- Tolkatchev D., Malik S., Vinogradova A., Wang P., Chen Z., Xu P., Bennett H.P.J., Bateman A., Ni F. (2008) Protein Sci., 17, 711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P., Fuxreiter M. (2008) Trends Biochem. Sci., 33, 2–8. [DOI] [PubMed] [Google Scholar]
- Uversky V.N. (2013) Protein Sci., 22, 693–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V.N., Li J., Fink A.L. (2001) FEBS Lett., 509, 31–35. [DOI] [PubMed] [Google Scholar]
- van Zonneveld A.J., Veerman H., Pannekoek H. (1986) Proc. Natl. Acad. Sci., 83, 4670–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma I.M., Stevenson J.K., Schwarz E.M., Van Antwerp D., Miyamoto S. (1995) Genes Dev., 9, 2723–2735. [DOI] [PubMed] [Google Scholar]
- Xue B., Dunbrack R.L., Williams R.W., Dunker A.K., Uversky V.N. (2010) Biochim. Biophys. Acta., 1804, 996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Nathan C., Jin W. et al. (2002) Cell, 111, 867–878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.