

Abstract
The M1 muscarinic acetylcholine receptor (mAChR) subtype has been implicated in the underlying mechanisms of learning and memory and represents an important potential pharmacotherapeutic target for the cognitive impairments observed in neuropsychiatric disorders such as schizophrenia. Patients with schizophrenia show impairments in top-down processing involving conflict between sensory-driven and goal-oriented processes that can be modeled in preclinical studies using touchscreen-based cognition tasks. The present studies used a touchscreen visual pairwise discrimination task in which mice discriminated between a less salient and a more salient stimulus to assess the influence of the M1 mAChR on top-down processing. M1 mAChR knockout (M1 KO) mice showed a slower rate of learning, evidenced by slower increases in accuracy over 12 consecutive days, and required more days to acquire (achieve 80% accuracy) this discrimination task compared to wild-type mice. In addition, the M1 positive allosteric modulator BQCA enhanced the rate of learning this discrimination in wild-type, but not in M1 KO, mice when BQCA was administered daily prior to testing over 12 consecutive days. Importantly, in discriminations between stimuli of equal salience, M1 KO mice did not show impaired acquisition and BQCA did not affect the rate of learning or acquisition in wild-type mice. These studies are the first to demonstrate performance deficits in M1 KO mice using touchscreen cognitive assessments and enhanced rate of learning and acquisition in wild-type mice through M1 mAChR potentiation when the touchscreen discrimination task involves top-down processing. Taken together, these findings provide further support for M1 potentiation as a potential treatment for the cognitive symptoms associated with schizophrenia.
Keywords: Positive allosteric modulators, M1 muscarinic acetylcholine receptors, touchscreen cognition, M1 knockout mice, top-down processing, BQCA
Graphical Abstract
Schizophrenia is a debilitating neuropsychiatric illness affecting approximately 1% of the population worldwide. Characterized by positive (hallucinations, delusions), negative (anhedonia, social withdrawal, apathy), and cognitive (attention, memory, executive function) symptom clusters, current treatments are largely ineffective in treating the negative and cognitive symptoms.1–3 Increasing evidence supports a role between cognitive performance and overall functional outcome in patients with schizophrenia,4–6 substantiating the need to develop pharmacological treatments targeting these cognitive impairments. Disruptions in top-down, goal-oriented, or rule-based processing when competing with bottom-up, sensory-driven processing7,8 are prevalent in schizophrenia and have contributed to impairments in attention, working memory, and social perception.9–12 In clinical studies, top-down processing is assessed by using stimuli of unequal salience such that a less salient, task-relevant stimulus competes with a more salient, task-irrelevant stimulus. For example, patients diagnosed with schizophrenia displayed normal attentional set shifting performance when searching for a highly salient target, but they displayed impaired attentional shifting when the target salience was low.13–15 Thus, incorporating top-down processing into preclinical cognitive assessments may improve our understanding of underlying etiology and enhance development of novel pharmacotherapies for the treatment of schizophrenia.
One key to better preclinical modeling of the complex cognitive impairments in schizophrenia involves the use of touchscreen-based cognitive tasks. These approaches allow a range of parametric manipulations to be employed to alter cognitive demand in similar ways across species from rodents to clinical populations, including assessment of top-down processing.16–19 For example, Dickson and colleagues reported that Fmr1 knockout mice (KO), a murine model of fragile X syndrome, displayed comparable performance to wild-type mice when discriminating between two stimuli of equal salience in a touchscreen task but made increased errors when discriminating between stimuli of unequal salience.20 These findings suggest that utilization of touchscreen-based cognitive tasks need to be further evaluated in rodent models relevant to the top-down processing deficits observed in neuropsychiatric disorders like schizophrenia.
In the current study, we investigated the role of the M1 muscarinic acetylcholine receptor (mAChR) subtype in learning touchscreen-based visual pairwise discrimination tasks under different degrees of cognitive demand using wild-type and M1 mAChR KO mice. By employing a touchscreen task requiring discrimination between a nonpreferred, less salient stimulus from a preferred, more salient stimulus, we are able to model top-down processing functions in mice similar to tasks used clinically that show impairments in top-down processing in patients with schizophrenia (e.g., refs 13–15). Impairments in M1 mAChR function may contribute to cognitive impairments associated with schizophrenia.21–25 A subset of patients with schizophrenia has shown decreased M1 mAChR expression in the PFC, hippocampus, and other forebrain regions,26–28 and previous studies have shown that activation of the M1 mAChR is important for learning and memory.23–25 For example, the prototypical M1 positive allosteric modulator (PAM) BQCA increased spontaneous excitatory postsynaptic potentials in medial PFC (mPFC) pyramidal cells ex vivo and increased cell firing rate in the mPFC of freely moving rats.29 M1 mAChRs are signaling partners with N-methyl-D-aspartate receptor subtype (NMDAR) of glutamate receptors, and NMDAR stimulation is integral for learning and memory.30,31 Furthermore, indirect modulation of NMDAR function may produce therapeutic effects without risk of excitotoxicity associated with direct agonist activity at the NMDAR.32 M1 agonists and PAMs enhanced performance or reversed pharmacologically induced impairments across several tasks assessing learning and memory in rodents and nonhuman primates.33–39 Lastly, in clinical studies, the M1/M4-preferring mAChR agonist xanomeline reduced the psychotic symptoms observed in schizophrenia patients and improved aspects of cognition, although off-target activity impeded further clinical development.40 In the present study, we examined both rate of learning by comparing daily percent accuracy across 12 consecutive testing days and acquisition, as defined by the number of days to achieve 80% accuracy. Using this approach, our studies are the first to demonstrate distinct cognitive impairments on rate of learning and acquisition in M1 KO mice on touchscreen-based tasks. Moreover, daily administration of BQCA, previously characterized by our group and others,29,33,34 also enhanced the rate of learning and acquisition in wild-type mice on this touchscreen task modeling top-down processing, suggesting a potential role for M1 potentiation in the treatment of such cognitive deficits in schizophrenia.
RESULTS
Touchscreen Training
Similar to previous studies,41 there were no differences between wild-type and M1 KO mice in learning stages 1–5 of training to respond on the touchscreen (data not shown).
Experiment 1: Acquisition of a Pairwise Discrimination Task Involving Top-Down Processing
In order to establish a model to assess top-down processing, we first identified a stimulus pair with unequal salience, which engendered a clear response preference in both wild-type and M1 KO mice. Following training to respond on the touchscreen, mice were exposed to a single session of 50 trials during which responding on either stimulus within a pair resulted in delivery of a reward. As shown in Figure 1. A,B, both wild-type and M1 KO mice showed an inherent preference toward stimulus 2, “4 circles”, compared to stimulus 1, “lasers” (wild-type, t = 4.66, df = 9; p < 0.01; M1 KO, t = 3.73, df = 10; p < 0.01).
Figure 1.
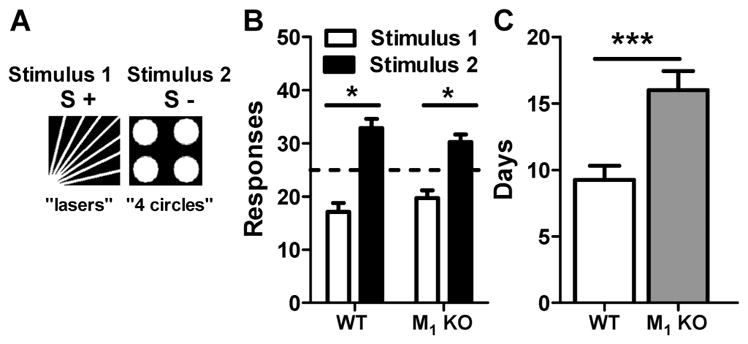
Impaired acquisition of a visual pairwise discrimination task involving top-down processing in M1 KO mice. (A) Stimuli chosen for pairwise discrimination: stimulus 1, “lasers”, and stimulus 2, “4 circles”. (B) Wild-type (WT) and M1 KO mice demonstrate an inherent preference for stimulus 2 when responding on either stimulus was reinforced. (C) M1 KO mice required a greater number of days to acquire the discrimination when discriminating the less salient stimulus (stimulus 1, S+) from the more salient stimulus (stimulus 2, S−). *, p < 0.05; ***, p < 0.001.
To test the hypothesis that M1 mAChRs are involved in top-down processing in mice, we examined, in a new cohort, the number of days required for wild-type mice and M1 KO mice (n = 8/group) to acquire the visual pairwise discrimination task, defined as reaching 80% accuracy when the nonpreferred, less salient stimulus (lasers) was designated as the initial correct stimulus and the preferred stimulus (4 circles) was designated as incorrect. As shown in Figure 1C, the M1 KO mice required a greater number of days to acquire the pairwise discrimination task as compared with the wild-type control mice (t = 3.81, df = 14; p < 0.001).
Experiment 2: Role of M1 mAChRs on Rate of Learning and Acquisition of a Pairwise Discrimination Task Involving Top-Down Processing
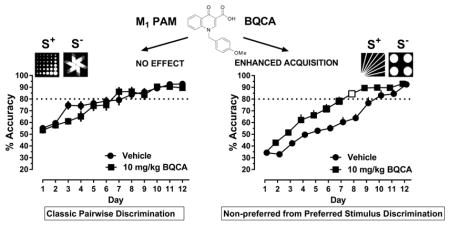
To examine in more detail the role of M1 mAChRs on top-down processing, we examined the rate of learning in wild-type and M1 KO mice when performing the discrimination task established in experiment 1 by comparing daily changes in percent accuracy across 12 consecutive days after pretreatment with vehicle or doses of the selective M1 PAM BQCA. Following training, wild-type (vehicle or 10 or 30 mg/kg BQCA) and M1 KO mice (vehicle, 10 mg/kg BQCA; n = 11–12/group) were administered vehicle or a dose of BQCA 15 min prior to each daily session for 12 consecutive days. As shown in Figure 2A, on day 1, the accuracy for all groups was below 50% (chance), demonstrating increased responding for the more salient, but incorrect, stimulus, and that the same stimulus bias that was seen in experiment 1 was present in this new cohort of mice. Over the 12 day period, there was a significant increase in percent accuracy (F11,599 = 105.40; p < 0.0001), a significant effect of group (F4,599 = 68.56; p < 0.0001), and a significant interaction between day and group (F44,599 = 2.66; p < 0.0001). Posthoc analysis revealed that vehicle-treated M1 KO mice demonstrated a slower rate of learning, as shown by significantly lower percent accuracy across days 5–12, compared to the vehicle-treated wild-type mice (all p < 0.05). However, daily administration of 10 mg/kg BQCA did not improve rate of learning in wild-type mice or M1 KO mice compared to respective vehicle-treated groups (Figure 2A), perhaps due to contingency parameters engendering a ceiling effect on rate of learning that could not be enhanced further (see experiment 3). Moreover, the 30 mg/kg dose of BQCA impaired the rate of learning in wild-type mice, noted by significantly lower percent accuracy compared to vehicle-treated wild-type mice on days 7–10 and 12 (all p < 0.05).
Figure 2.

Impaired rate of learning a visual pairwise discrimination task involving top-down processing in M1 KO mice and enhanced rate of learning in wild-type mice treated with BQCA. (A) Percent accuracy showing rate of learning and (B) survival plots showing acquisition across 12 consecutive days in wild-type (WT, black) and M1 KO (gray) mice treated with vehicle or BQCA prior to discriminating the less salient stimulus from the more salient stimulus when exposed to 100 trials per session. (C) Percent accuracy and (D) survival plots across 12 consecutive days in wild-type (black) and M1 KO (gray) mice treated with vehicle or BQCA when exposed to 60 trials per session. (E) Percent accuracy and (F) survival plots across 12 consecutive days in wild-type (black) and M1 KO (gray) mice exposed to 60 (circles) or 100 trials (triangles); open symbols, p < 0.05 compared to the vehicle-treated wild-type group exposed to 100 trials per session; ^, p < 0.05 compared to wild-type mice completing 60 trials per session.
To examine acquisition between groups, the percent of each group that acquired per test day (>80% accuracy) was plotted as a survival curve. As shown in Figure 2B, a log-rank (Mantel–Cox) test showed a significant effect of group on percent acquisition (χ2 = 22.39; df = 4; p < 0.001). Eighty percent of the vehicle-treated and 10 mg/kg BQCA-treated wild-type mice acquired the discrimination within 7 days, whereas the 30 mg/kg dose of BQCA reduced the total percent that acquired the discrimination to <50% by day 12. In contrast, only 20% of vehicle-treated and 10 mg/kg BQCA-treated M1 KO mice acquired by day 7. By day 12, 40% of vehicle-treated and 70% of M1 KO mice treated with 10 mg/kg BQCA acquired the discrimination. Although fewer vehicle-treated M1 KO mice acquired, effects of BQCA were not significant in the M1 KO mice; 5 of 7 vehicle-treated M1 KO mice that did not acquire the discrimination (e.g., >80% accuracy) achieved >70% accuracy.
We also examined the number of trials completed and overall session length, additional variables that can directly influence rate of learning,16 as well as the response and reinforcer retrieval latencies that provide a measure of motor function or motivation to respond that may indirectly influence learning. As shown in Table 1, there was a significant effect of group on session length on day 1 (F4,50 = 9.6, p < 0.0001) such that vehicle-treated M1 KO mice completed the session significantly faster than vehicle-treated wild-type mice (p < 0.05). There was also a significant effect of group on number of trials completed (F4,47 = 32.19, p < 0.0001), correct response latency (F4,50 = 9.86, p < 0.0001), and reinforcer retrieval latency (F4,50 = 6.39, p < 0.001) on day 1 such that wild-type mice that received the 30 mg/kg dose of BQCA completed fewer trials and had longer response and reinforcer retrieval latencies (all p < 0.001) compared to vehicle-treated wild-type mice.
Table 1.
Effects of BQCA and Trial Number on Performance in Wild-Type and M1 KO Mice on a Pairwise Discrimination Task Involving Top-Down Processinga
| gen | n | trials | Tx | acquisition
|
session length (s)
|
trials completed
|
cor resp lat (s)g
|
SR ret lat (s)g
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| days | trials | day 1 | day 12 | day 1 | day 12 | day 1 | day 12 | day 1 | day 12 | |||||
| exp 2 | WT | 12b | 100 | veh | 6.5 (1.2) | 563.6 (136.2) | 3032.2 (694.2) | 2387.3 (1027.7) | 84.7 (22.7) | 92.2 (14.6) | 5.2 (0.7) | 12.9 (4.1) | 2.1 (0.4) | 2.3 (0.4) |
| WT | 12 | 100 | 10 | 6.3 (1.4) | 513.4 (144.7) | 3391.3 (302.0) | 1939.2 (630.6) | 78 (22.7) | 99.0 (0.3) | 8.7 (1.2) | 6.5 (1.1) | 2.7 (0.2) | 1.6 (0.1) | |
| WT | 11 | 100 | 30 | 8.0 (2.7)d | 655.8 (237.8)d | 3600.0 (0.0) | 2562.5 (599.6) | 28.8 (18.6)h | 96.8 (7.3) | 80.0 (22.0)h | 7.1 (0.9) | 19.5 (6.9)h | 3.9 (1.7) | |
| M1 KO | 11 | 100 | veh | 8.8 (2.4)f | 801.5 (158.3)f | 2274.3 (717.0)h | 2940.7 (748.4) | 91.4 (28.6) | 92.1 (12.8) | 13.1 (9.8) | 7.3 (1.8) | 3.3 (1.6) | 5.0 (2.1) | |
| M1 KO | 11b | 100 | 10 | 8.3 (2.0)c | 787.0 (201.0)c | 2653.8 (795.9) | 3024.6 (817.3) | 99.3 (2.2) | 93.8 (8.0) | 4.9 (0.6) | 9.1 (2.7) | 1.8 (0.2) | 3.2 (0.6) | |
| exp 3 | WT | 10 | 60 | veh | 10.0 (1.8) | 597.2 (110.4) | 2087.0 (664.3) | 1168.1 (638.0) | 57.9 (6.6) | 60.0 (0.0) | 5.4 (1.1) | 8.2 (2.9) | 2.1 (0.3) | 1.8 (0.2) |
| WT | 10 | 60 | 1 | 7.6 (2.5) | 434.8 (134.8) | 2600.4 (1117.5) | 953.6 (152.1) | 54 (13.8) | 60.0 (0.0) | 6.2 (1.4) | 4.7 (0.5) | 1.8 (0.1) | 1.4 (0.1) | |
| WT | 10 | 60 | 10 | 6.8 (1.8) | 401 (105.8) | 2066.6 (759.1) | 866.2 (219.9) | 55.6 (13.9) | 60.0 (0.0) | 4.4 (0.6) | 5.0 (1.0) | 1.7 (0.1) | 1.3 (0.1) | |
| M1 KO | 11 | 60 | veh | 9.7 (2.0)c | 569.7 (124.8)c | 1597.3 (759.6) | 1547.6 (732.2) | 58.4 (5.4) | 59.2 (2.7) | 9.5 (2.4) | 6.0 (1.3) | 2.1 (0.2) | 1.7 (0.2) | |
| M1 KO | 10 | 60 | 10 | 8.6 (2.2)e | 516.0 (131.5)e | 1414.3 (382.3) | 987.2 (247.3) | 60.0 (0.0) | 60.0 (0.0) | 4.6 (0.9) | 3.7 (0.7) | 1.6 (0.1) | 1.4 (0.2) | |
Data are expressed as mean (standard deviation) except where noted. Abbreviations: wild-type (WT), genotype (gen), treatment (Tx), correct response latency (cor resp lat), reinforcer retrieval latency (SR ret lat), seconds (s). As some mice did not acquire the discrimination, statistical analyses were not conducted on days or trials to acquisition and are shown for qualitative comparison only.
Excluded 1 mouse that completed <40 trials in 8 or more days.
Two mice were excluded from analysis that did not acquire within 12 days.
Six mice were excluded from analysis that did not acquire within 12 days.
Five mice were excluded from analysis that did not acquire within 12 days.
Seven mice were excluded from analysis that did not acquire within 12 days.
Mean (SEM).
p < 0.05 compared to vehicle-treated wild-type mice within each respective experiment
Experiment 3: Influence of Trial Number on Rate of Learning and Acquisition of a Pairwise Discrimination Task Involving Top-Down Processing
Previous studies in rats have shown that the number of trials per session can influence rate of learning touchscreen-based pairwise discrimination tasks.16 Here, we reduced the number of trials from 100 to 60 trials per session to test the hypothesis that exposure to fewer trials per session would decrease the baseline rate of learning and that M1 potentiation might improve learning this pairwise discrimination involving top-down processing. Following training, wild-type (vehicle or 1 or 10 mg/kg BQCA) and M1 KO mice (vehicle, 10 mg/kg BQCA; n = 10–11/group) were administered vehicle or BQCA 15 min prior to daily cognition sessions for 12 consecutive days using 60 trials per session. As shown in Figure 2C, similar to experiment 2, percent accuracy for all groups was below 50% on day 1. As in experiment 2 over the 12 day period, there was a significant improvement in percent accuracy (F11,551 = 101.7; p < 0.0001), a significant effect of group (F4,551 = 31.21; p < 0.0001), and a significant interaction (F44,551 = 1.41; p < 0.05). In addition, the overall percent accuracy in wild-type mice treated with 1 and 10 mg/kg BQCA was higher than the vehicle-treated wild-type mice, significant on days 7 and 8, respectively (all p < 0.05). Percent accuracy in vehicle-treated M1 KO mice was not significantly different from vehicle-treated wild-type mice. As shown in Figure 2D, a log-rank (Mantel–Cox) test showed a significant effect of group on percent acquisition (χ2 = 19.26; df = 4; p < 0.001). Eighty percent of wild-type mice treated with 10 mg/kg BQCA acquired the discrimination in 8 days, compared to 10 and 12 days for 1 mg/kg BQCA and vehicle-treated wild-type mice, respectively. M1 KO mice treated with vehicle or 10 mg/kg BQCA showed similar rates of learning as those of vehicle-treated wild-type mice across the first 11 days, although 80% or fewer acquired the discrimination when exposed to 60 trials.
There was a significant effect of treatment on session length on day 1 (F4,50 = 3.68, p < 0.05; Table 1) and day 12 (F4,50 = 3.52, p < 0.05), but none of the groups were different from vehicle-treated wild-type mice. There was not an effect of treatment on number of trials completed, correct response latency, or reinforcer retrieval latency on day 1 or 12 compared to vehicle-treated wild-type mice (Table 1).
As shown in Figure 2E, data from vehicle-treated groups from experiments 2 and 3 were replotted to directly compare influence of trial number on rate of learning between wild-type and M1 KO mice. There was a significant effect of day (F11,480 = 77.13; p < 0.0001) and trial number (F3,480 = 34.46; p < 0.0001) and a significant interaction (F33,480 = 2.0; p < 0.05). Percent accuracy in wild-type and M1 KO mice exposed to 60 trials was not different from each other at any time point. Percent accuracy in wild-type mice completing 100 trails per session was higher than wild-type mice completing 60 trails per session and was significantly higher on days 7 and 8. Percent accuracy was not different in M1 KO mice exposed to 60 and 100 trials, demonstrating that increasing exposure does not enhance rate of learning in M1 KO mice. Compared to wild-type mice exposed to 100 trials per session, percent accuracy in M1 KO mice exposed to 60 or 100 trials was lower on days 7–9 and on days 7–10, respectively (all p < 0.05). Compared to wild-type mice exposed to 60 trials per session, percent accuracy in M1 KO mice exposed to 100 trials was lower on day 12 (p < 0.05). As shown in Figure 2F, a log-rank (Mantel–Cox) test showed a significant effect of group on percent acquisition (χ2 = 27.35; df = 3; p < 0.0001). While 80% of the wild-type mice exposed to 100 and 60 trials acquired the task within 7 and 12 days, respectively, only 80% and <50% of M1 KO mice exposed to 60 and 100 trials acquired the discrimination by day 12.
To examine effects of repeated dosing of BQCA on plasma and brain concentrations, mice from experiment 3 were dosed with 1 or 10 mg/kg BQCA (wild-type) and 10 mg/kg BQCA (M1 KO mice) for one additional day (day 13) after the last cognition session. Plasma and brain were collected 30 min following administration of vehicle or BQCA. For comparison, an additional set of wild-type and M1 KO mice were administered a single dose of 1 or 10 mg/kg BQCA to examine plasma and brain concentrations following acute administration. As shown in Table 2, there was a modest but statistically significant elevation in plasma and brain concentrations of BQCA following repeated dosing with the 10 mg/kg dose but not following repeated dosing with the 1 mg/kg dose in wild-type mice compared to acute dosing. There was a main effect of dose (F1,28 = 124.5, p < 0.001), treatment duration (F1,28 = 6.00; p < 0.05), and an interaction (F1,28 = 8.95; p < 0.001) on observed plasma concentrations in wild-type mice, and, similarly, there was a main effect of dose (F1,28 = 158.9, p < 0.001), treatment duration (F1,28 = 27.18; p < 0.001), and an interaction (F1,28 = 24.17; p < 0.001) on observed brain concentrations in wild-type mice. There were no differences in plasma or brain concentrations of BQCA following repeated dosing with the 10 mg/kg dose in M1 KO mice compared to acute dosing. There was not an effect of genotype (F1,32 = 0.90; p > 0.05) or treatment duration (F1,32 = 3.87; p > 0.05), but there was a significant interaction (F1,32 = 6.40; p,0.05) on absolute plasma concentrations following 10 mg/kg BQCA. Brain concentrations of BQCA were higher in wild-type mice compared to M1 KO mice following repeated dosing with 10 mg/kg BQCA. There was a significant effect of treatment duration (F1,32 = 21.99; p < 0.001), no effect of genotype (F1,32 = 3.4; p > 0.05), but a significant interaction between genotype and duration (F1,32 = 14.63; p < 0.001) on absolute brain concentrations; see Table 2 for posthoc analyses. Interestingly, calculated free (unbound) brain concentrations after repeated dosing with 1 or 10 mg/kg BQCA, which both produced similar magnitudes of enhancements in rate of learning (experiment 3), were below (68 ± 17 nM) and slightly above (693 ± 176 nM), respectively, previously reported in vitro estimates of the EC50 inflection points for BQCA when determined in the presence of an EC20 concentration of ACh (~300 nM29,33,34).
Table 2.
In Vivo Drug Exposure Analysis of BQCAa
| 1 mg/kg
|
10 mg/kg
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| acute (1 day)
|
repeated (13 days)
|
acute (1 day)
|
repeated (13 days)
|
||||||
| obs | unb | obs | unb | obs | unb | obs | unb | ||
| WT | plasma [nM] | 2606 (628) | 123 (29) | 1864 (248) | 88 (12) | 13786b (2756) | 648 (130) | 21236c (7198) | 998 (338) |
| brain [nM] | 462 (199) | 58 (25) | 546 (135) | 68 (17) | 2635b (752) | 332 (95) | 5499d (1400) | 693 (176) | |
| Kp,uu | 0.48 | 0.79 | 0.51 | 0.69 | |||||
| M1 KO | plasma [nM] | 19550 (4480) | 919 (211) | 18612 (4192) | 874 (197) | ||||
| brain [nM] | 3301 (998) | 422 (131) | 3592e (706) | 453 (89) | |||||
| Kp,uu | 0.46 | 0.52 | |||||||
Mean (standard deviation) of total (observed, obs) and calculated unbound (unb) plasma and brain concentrations of BQCA in wild-type (WT) and M1 KO mice 30 min after intraperitoneal administration of 1 or 10 mg/kg BQCA once (acute, 1 day) or once daily for 13 days (repeated, 13 days); n = 6–7/group for acute and 9–11/group for chronic dosing studies. Kp,uu, unbound brain/unbound plasma ratio.
p < 0.001 compared to observed values following 1 mg/kg acute (1 day) dosing in WT mice.
p < 0.01 compared to observed values following 10 mg/kg acute (1 day) dosing in WT mice.
p < 0.001 compared to observed values following 10 mg/kg acute (1 day) dosing in WT mice.
p < 0.01 compared to observed values following 10 mg/kg repeated (13 days) dosing in WT mice mouse plasma free fraction, 0.047 rat brain free fraction, 0.126.
In addition, the potential effects of repeated dosing of BQCA on M1 mAChR mRNA levels in the hippocampus, prefrontal cortex (PFC) and striatum were assessed from tissue dissected from each brain region 30 min after BQCA administration on day 13 (1 day after the last cognition test session) from the wild-type mice treated with vehicle or 1 or 10 mg/kg BQCA in experiment 3. As shown in Figure 3, a one-way ANOVA per brain region showed that there was no effect of repeated BQCA administration on M1 mRNA expression levels in hippocampus (F2,27 = 3.22; p > 0.05), PFC (F2,24 = 2.03; p > 0.05), or striatum (F2,26 = 0.18; p > 0.05). Data are expressed as percent of M1 mRNA levels in vehicle-treated mice.
Figure 3.
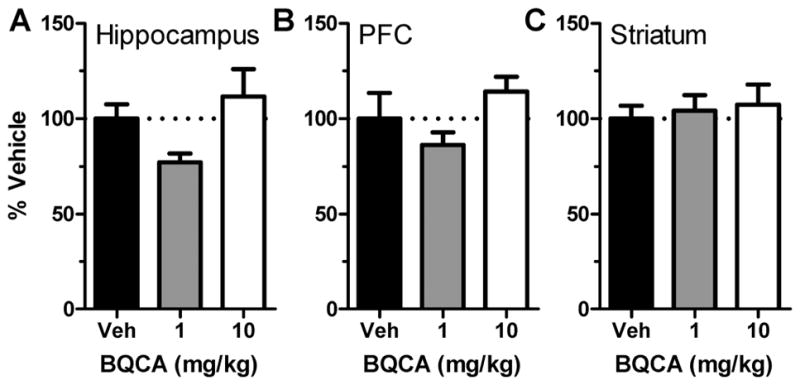
Repeated BCQA administration does not alter M1 mAChR mRNA expression. M1 mAChR mRNA levels expressed as a percent of mRNA levels in vehicle-treated wild-type mice in the (A) hippocampus, (B) prefrontal cortex (PFC), and (C) striatum following 13 days of repeated BQCA administration.
Experiment 4: Role of M1 mAChRs on Rate of Learning and Acquisition of a Pairwise Discrimination Task Not Involving Top-Down Processing
For comparison with the experiments 1–3, we tested the hypothesis that M1 KO mice would not show impaired learning of a pairwise discrimination task between relatively equal salient stimuli that do not involve top-down processing. First, relative salience was determined in wild-type and M1 KO mice between two stimuli used in prior touchscreen studies without purported stimulus bias (e.g., refs 16, 19, and 41). As shown in Figure 4. A,B, wild-type mice showed no inherent preference for either stimulus 1, “marbles”, or stimulus 2, “fan” (t = 2.055, df = 9; p > 0.05); M1 KO mice showed a slight but significant preference for stimulus 1 (marbles; t = 2.43, df = 10; p < 0.05). Rate of learning this pairwise discrimination across 12 days was examined in separate groups of vehicle-treated wild-type and M1 KO mice exposed to either 60 or 100 trials when stimulus 1 (marbles) was designated as the correct stimulus. As shown in Figure 4C, there was a significant effect of day (F11,491 = 57.51; p < 0.0001) and trial number (F3,491 = 60.0; p < 0.0001) but no interaction (F33,491 = 1.25; p > 0.05) on percent accuracy. There were no significant differences in percent accuracy between wild-type mice exposed to 60 or 100 trials or between groups of M1 KO mice completing 60 or 100 trials (all p > 0.05). However, percent accuracy in M1 KO mice completing 60 trials was significantly lower than wild-type mice completing 60 trials on days 9 and 10 (p < 0.05); percent accuracy in M1 KO mice was also significantly lower on days 2–4 and 9 compared to wild-type mice when each group was exposed to 100 trials (all p < 0.05). Compared to wild-type mice exposed to 100 trials per session, percent accuracy in M1 KO mice exposed to 60 trials was significantly lower on days 3–11 (all p < 0.05). Lastly, percent accuracy in M1 KO mice exposed to 100 trials was not different from wild-type mice exposed to 60 trials. As shown in Figure 4D, a log-rank (Mantel–Cox) test showed a significant effect of group on percent acquisition (χ2 = 12.33; df = 3; p < 0.01). Eighty percent of the wild-type mice exposed to 100 and 60 trials acquired the task in 5 and 10 days, respectively, whereas 80% of the M1 KO mice acquired in 11 days regardless of trial number.
Figure 4.

Increasing trial numbers enhanced rate of learning a visual pairwise discrimination task not involving top-down processing in wild-type (WT) and M1 KO mice. (A) Common stimuli used for pairwise discrimination: Stimulus 1, “marbles”, and Stimulus 2, “fan”. (B) Wild-type mice do not show an inherent preference for either stimulus; M1 KO mice show a slight preference for Stimulus 1 when responding on either stimulus was reinforced. (C) Percent accuracy showing rate of learning and (D) survival plots showing acquisition across 12 consecutive days in wild-type (black) and M1 KO (gray) mice when discriminating between relatively equal salient stimuli (Stimulus 1, S+; Stimulus 2, S−) when exposed to 60 (circles) or 100 trials (triangles). (E) Percent accuracy and (F) survival plots across 12 consecutive days following vehicle or BQCA administration in wild-type mice completing 60 trials per session. open symbols, p < 0.05 compared to vehicle-treated wild-type group exposed to 100 trials per session; *, p < 0.05 compared to vehicle-treated wild-type group exposed to 60 trials per session.
As expected, there was a significant difference between trials completed on both day 1 (F3,44 = 16.09, p < 0.0001; Table 3) and day 12 (F3,44 = 7851, p < 0.0001) such that, regardless of genotype, mice exposed to 100 trials completed more trials than mice exposed to 60 trials (all p < 0.05). There was also a significant effect of genotype on session length on day 1 (F3,44 = 8.55, p < 0.001) and day 12 (F3,44 = 31.59, p < 0.0001). On day 1, session length for the M1 KO group exposed to 60 trials was significantly shorter than the session lengths for both wild-type and M1 KO mice exposed to 100 trials (p < 0.05). On day 12, regardless of genotype, the session lengths for mice exposed to 60 trials were significantly shorter than the session lengths for both groups of mice exposed to 100 trials (all p < 0.05). There was a significant effect of trial number (60 or 100) on correct response latency and reinforcer retrieval latency on day 12 (F3,44 = 3.07, p < 0.05) and (F3,44 = 3.54, p < 0.05), respectively, but not day 1. On day 12, reinforcer retrieval latencies were faster in wild-type mice exposed to 60 versus 100 trials (p < 0.05).
Table 3.
Effects of BQCA and Trial Number on Performance in Wild-Type and M1 KO Mice on a Pairwise Discrimination Task Not Involving Top-Down Processinga
| gen | n | trials | Tx | acquisition
|
session length (s)
|
trials completed
|
cor resp lat (s)e
|
SR ret lat (s)e
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| days | trials | day 1 | day 12 | day 1 | day 12 | day 1 | day 12 | day 1 | day 12 | |||||
| exp 4 | WT | 12 | 60 | veh | 6.3 (2.1)c | 379.1 (122.4)c | 2533.8 (788.6) | 1311.8 (528.3)f,g | 57.4 (8.1)f,g | 60.0 (0.0)f,g | 7.7 (1.4) | 3.9 (1.0) | 2.7 (0.8) | 1.5 (0.2)f |
| WT | 12 | 100 | veh | 4.9 (1.2) | 425.9 (73.1) | 3303.2 (381.1) | 2750.9 (434.0) | 85.9 (17.0) | 99.8 (0.9) | 6.0 (0.7) | 7.9 (1.6) | 2.4 (0.4) | 2.4 (0.4) | |
| M1 KO | 11 | 60 | veh | 9.2 (2.1)d | 532.1 (128.8)d | 1828.9 (887.6)f,g | 1139.3 (275.5)f,g | 57.8 (4.9)f,g | 60.0 (0.0)f,g | 8.1 (2.5) | 4.2 (0.4) | 1.7 (0.2) | 1.5 (0.1) | |
| M1 KO | 11b | 100 | veh | 6.9 (2.8)d | 610.6 (219.5)d | 2861.2 (722.2) | 2314.4 (590.7) | 89.6 (22.5) | 99.5 (1.6) | 10.1 (3.7) | 6.0 (0.9) | 1.7 (0.1) | 1.7 (0.2) | |
| WT | 10 | 60 | veh | 5 (2.8) | 287.1 (146.9) | 1777.2 (944.1) | 1166.6 (251.6) | 55.2 (15.2) | 60.0 (0.0) | 6.2 (2.2) | 5.1 (0.9) | 2.0 (0.3) | 2.1 (0.5) | |
| WT | 10 | 60 | 1 | 4.7 (1.4) | 282.0 (85.1) | 1409.6 (189.2) | 1070.5 (183.6) | 60.0 (0.0) | 60.0 (0.0) | 3.9 (0.6) | 4.5 (0.7) | 1.6 (0.1) | 1.5 (0.2) | |
| WT | 10 | 60 | 10 | 5.7 (1.8) | 342.0 (109.7) | 1682.3 (690.0) | 1235.5 (754.7) | 60.0 (0.0) | 60.0 (0.0) | 7.0 (1.4) | 8.1 (2.8) | 2.0 (0.2) | 2.3 (1.0) | |
Data are expressed as mean (standard deviation) except as noted. Abbreviations: wild-type (WT), genotype (gen), treatment (Tx), correct response latency (cor resp lat), reinforcer retrieval latency (SR ret lat), seconds (s). As some mice did not acquire the discrimination, statistical analyses were not conducted on days or trials to acquisition and are shown for qualitative comparison only.
Excluded 1 mouse that completed <40 trials in 8 or more days.
Two mice were excluded from analysis that did not acquire within 12 days
One mouse was excluded from analysis that did not acquire within 12 days
Mean (SEM).
p < 0.05 compared to wild-type group exposed to 100 trials.
p < 0.05 compared to M1 KO group exposed to 100 trials
Finally, to understand if M1 potentiation enhances discrimination learning in general or is specific to discrimination learning involving top-down processing, separate groups of wild-type mice were administered vehicle or BQCA (1, 10 mg/kg) prior to each session when exposed to 60 trials for 12 consecutive days when discriminating between stimuli of equal salience. As shown in Figure 4E, there was a significant effect of day (F11,321 = 43.66; p < 0.0001) and dose (F2,321 = 7.17; p < 0.001) but no significant interaction (F22,321 = 1.20; p > 0.05) on percent accuracy; no specific time points were different from the vehicle-treated group. As shown in Figure 4F, a log-rank (Mantel–Cox) test showed a significant effect of group on percent acquisition (χ2 = 11.46; df = 3; p < 0.01). Eighty percent of the wild-type mice treated with vehicle or 1 or 10 mg/kg BQCA acquired in 6, 5, and 7 days, respectively. There were no differences in session length, correct response latencies, or reinforcer retrieval latencies between groups on day 1 or 12 (Table 3).
Experiment 5: Relative Reinforcing Strength of the Liquid Reinforcer Using a Progressive Ratio Schedule of Reinforcement
To confirm that reinforcing strength of the liquid reward was not different between genotypes, a potential confound that could influence motivation to perform cognitive tasks, separate groups of wild-type and M1 KO mice were trained to nose poke under a progressive ratio schedule of reinforcement to obtain different concentrations of liquid Ensure. As shown in Figure 5, both genotypes showed a concentration-dependent increase in the number of reinforcers earned, demonstrated by a significant main effect of concentration (F3,91 = 73.24; p < 0.0001), but there was no difference between genotypes (F1,91 = 1.73; p > 0.05) or dose by genotype interaction (F3,91 = 0.02; p > 0.05).
Figure 5.
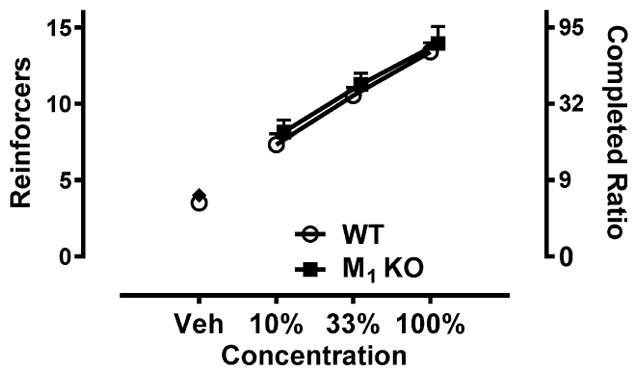
Similar breakpoints in wild-type (WT) and M1 KO mice responding via a nose poke under a progressive ratio schedule of reinforcement. Responding maintained by different concentrations of a liquid reinforcer in wild-type (open circles) and M1 KO mice (closed squares) under a progressive ratio schedule of reinforcement. Increasing concentrations of liquid Ensure are shown on the x-axis. The 3 day mean (±SEM) number of reinforcers achieved (left y-axis) and corresponding number of nose pokes emitted to complete each ratio (right y-axis) are shown (e.g., the 5th reinforcer required 9 nose pokes).
DISCUSSION
Selective activation of M1 mAChRs has been proposed as a novel mechanism for enhancement of cognitive deficits associated with schizophrenia. However, previous reports examining cognitive performance in M1 KO mice have shown equivocal results, questioning the role of M1 mAChR involvement in specific cognitive domains. By implementing a discrimination task assessing top-down processing, we revealed cognitive impairments using touchscreen assessments in the M1 KO mice for the first time. Moreover, we also showed that selective activation of M1 mAChRs by the M1 PAM BQCA in wild-type mice can enhance cognitive performance during top-down processing tasks. Importantly, our studies also demonstrated that altering trial number affected rate of learning, stressing the need to understand parametric influences on baseline cognitive performance when assessing genetic models or conducting pharmacological challenge studies.
By employing a touchscreen task requiring discrimination between a less salient from a more salient stimulus, we were able to model top-down processing functions in mice similar to tasks used clinically that show impairments in top-down processing in patients with schizophrenia (e.g., refs 13–15). Bias toward one stimulus in a discrimination set can affect discrimination learning; thus, pairs of stimuli with similar salience are suggested to avoid this confound (e.g., ref 16). However, selecting stimuli with different salience, and designating the less salient stimulus as the correct stimulus, introduces conflict between sensory-based, (bottom-up) and rule-based (top-down) processing (see refs 7, 8, and 20). Using this approach, we demonstrated that M1 KO mice have a slower rate of learning and required more days to acquire pairwise discriminations involving top-down processing functions.
Our findings with BQCA support a critical role for the modulation of M1 mAChRs in learning and memory under conditions of more complex cognitive processing. Supporting these findings, CDD-0102A, an M1 mAChR partial agonist, did not affect acquisition of a place or visual discrimination, yet it enhanced shifting between the place and visual cues.42 Similarly, in Tg2576 mice, a genetic model of Alzheimer’s disease, acute administration of BQCA reduced errors on a tactile and olfactory-based compound discrimination task when irrelevant stimuli were present, but it did not affect the simple discrimination component in the absence of irrelevant stimuli.29 Although additional studies are warranted to understand the circuitry mediating this preclinical assay modeling top-down processing, the present data support a specific role of M1 mAChR function in top-down processing.
Interestingly, the performance deficits of the M1 KO mice in top-down processing were observed only when completing 100 trials per session. Historically, murine studies implement 20–30 “test trials” and an unlimited number of “correction trials” that are not incorporated in overall accuracy or trial number (e.g., refs 16, 20, 41, and 43). These correction trials provide response feedback following incorrect responding, effectively providing supplemental training that may influence number of total trials completed within a session (test + correction trials), session duration, and overall learning. To avoid this possible confound on learning and importantly to align murine tasks with the parameters implemented in nonhuman primate and human studies (e.g., refs 44 and 45), we eliminated correction trials and increased total trial number to 60 and 100 trials per session. Increasing the trial number per session influenced rate of learning in wild-type mice only, such that rate of learning and acquisition was faster when completing 100 trials as compared to 60 trials. Importantly, exposure to 100 trials per session did not appear to have a detrimental effect on performance in either genotype, as demonstrated by similar within-session percent accuracies across 20-trial bins (Supporting Information Figures S1 and S2). M1 potentiation enhanced rate of learning and acquisition in wild-type mice only when 60 trials were completed, and the baseline rate of learning was slower. Interestingly, a pattern emerged regarding performance of the discrimination task not involving top-down processing such that increasing trial number appeared to have a beneficial effect in both genotypes, suggesting that additional exposure could enhance learning in wild-type and M1 KO mice alike on more simple discrimination tasks not involving top-down processing. BQCA did not significantly enhance rate of learning in wild-type mice when discriminating between equal salient stimuli, supporting a more selective role of M1 mAChR function on learning and memory when top-down processing is involved. Lastly, these data stress the importance of understanding baseline cognitive performance and demonstrate that different baseline levels, such as rate of learning, are necessary for assessing potential cognitive deficiencies in genetic models or pharmacological challenge studies examining potential cognitive enhancement.
The present studies are the first to demonstrate cognitive impairments in M1 KO mice using touchscreen assays. Previous studies involving M1 KO mice have demonstrated deficits in some nonmatching-to-sample tasks requiring hippocampal–cortical interactions but not matching-to-sample tasks touted as hippocampal-dependent.41,46,47 Previous evaluation of M1 KO mice in touchscreen-based assessments of attention, learning, and behavioral flexibility, including a pairwise discrimination task using identical stimulus as in experiment 4, in which the slightly preferred stimulus (marbles) was the correct stimulus, reported largely intact cognitive performance.41 While these classic touchscreen tasks examined specific cognitive domains, they do not model the more complex cognitive processes such as top-down processing that require integration of multiple circuits including fronto-parietal and corticolimbic circuits. 22,48,49 Cognitive assessments using touchscreen-based assays in preclinical species are touted to have high translatability to human touchscreen assessments. The present studies support this claim and reiterate the need to constantly evaluate and understand the role of various task parameters to improve translatability of preclinical assays.
The present studies are also the first to demonstrate that M1 potentiation enhances the rate of learning and acquisition of a touchscreen discrimination task. Moreover, doses of BQCA that enhanced learning achieved brain levels that were either below or slightly above the reported in vitro EC50,29,33,34 and repeated dosing with BQCA did not alter M1 mRNA expression. Importantly, the modest increases in plasma and brain concentrations following repeated dosing with 10 mg/kg BQCA in wild-type mice were not observed following repeated dosing with 1 mg/kg BQCA, suggesting that the effects on rate of learning were not due to a pharmacokinetic confound. BQCA has a low affinity for the M1 mAChR receptor but a very high degree of positive cooperativity.50 This high degree of cooperativity associated with BQCA may account for the in vivo effects at doses lower than the in vitro EC50. Future studies examining the relationship between in vitro potencies and in vivo effects are necessary. Of note, 30 mg/kg BQCA impaired acquisition in the first cohort of mice. Due to the high degree of selectivity, we do not hypothesize this effect to be attributed to off-target activity. However, pharmacological effects on cognition commonly produce an “inverted-U-shaped” effect such that optimal doses enhance performance, yet over-stimulation, regardless of the mechanism of action, may be disruptive.51,52 Additionally, disruptions at this high dose of BQCA may be attributed to potential allosteric agonist activity recently reported at high concentrations in specific in vitro assays.50 While comparisons of the effects of BQCA in this task with M1 mAChR orthosteric agonists are needed to further understand potential differences in performance relative to therapeutic index, our current data demonstrate evidence that positive allosteric modulation of the M1 mAChR may provide a larger therapeutic window for enhancement of cognition without development of tolerance and/or downregulation of M1 mAChRs as compared to ligands acting at the orthosteric binding site.53–55
Finally, while the underlying neural circuitry mediating the observed effects of the M1 PAM BQCA on top-down processing remains unknown, one possible mechanism may involve activation of the N-methyl-D-aspartate receptor subtype (NMDARs) of glutamate receptors. Previous studies have shown that M1 is a closely associated signaling partner with NMDAR and may be important in regulating NMDAR function in forebrain regions implicated in the pathophysiology of schizophrenia.29–31 For example, BQCA increased spontaneous excitatory postsynaptic potentials in medial PFC (mPFC) pyramidal cells ex vivo and increased cell firing rate in the mPFC of freely moving rats.29 Multiple studies have also demonstrated that activation of NMDARs modulates mechanisms of synaptic plasticity, including long-term potentiation in the hippocampus and frontal cortex, integral for various learning and memory tasks (e.g., refs 56–58). In addition, genetic knockdown of the NMDAR subunits NR1 or NR2A or pharmacological blockade of NMDARs results in cognitive disturbances, including acquisition and maintenance of pairwise discrimination learning.59–62 Similarly, NMDAR antagonists exacerbate cognitive impairments in patients with schizophrenia and impair cognition in healthy humans (for reviews, see refs 23, 63, and 64). Future studies will examine this hypothesis by assessing the ability of M1 PAMs to improve top-down processing in rodent models of NMDAR hypofunction modeling the cognitive impairments associated with schizophrenia such as chronic NMDAR antagonism or the NR1 NMDAR transgenic knockdown mouse model.
METHODS
Subjects
All behavioral studies were conducted with adult male M1 KO mice (n = 84) and wild-type mice (n = 137) with the same genetic background (C57BL/6NTac). Mice were group-housed 2–5 mice per cage in a temperature- and humidity-controlled environment under a 12/12 h light–dark cycle with water available ad libitum. For all studies, 8–12 week old mice of each genotype were gradually food restricted and maintained at ~85% free-feeding weight. All experiments were approved by the Vanderbilt University Animal Care and Use Committee, and experimental procedures conformed to guidelines established by the National Research Council Guide for the Care and Use of Laboratory Animals.
Touchscreen Training
Mice were trained in operant chambers (Lafayette Instruments, Lafayette, IN) to respond to stimuli presented on a computer screen by breaking an infrared beam in close proximity to the stimuli (e.g., a nose poke) according to convention (e.g., refs 19 and 41). Throughout training and testing, a mask was placed over the touchscreen such that responses could be made only in one of two (2 × 2 in.) windows on the screen. In stage 1, mice were habituated for 1 day to the operant chamber and trained to collect a liquid reward (33% diluted Ensure; 30 μL delivered via a peristaltic pump) from a receptacle located on the opposite wall from the touchscreen. In stage 2, mice were required to collect a liquid reward following a 3 s presentation and removal of a stimulus on one of the two touchscreen windows. In stage 3, mice were required to make a nose poke on either touchscreen window (breaking the infrared beam in front of the touchscreen) to receive a reward, followed by a 5 s intertrial interval (ITI). In stage 4, mice were trained to initiate each trial by registering a nose poke in the reward receptacle. Trial availability was signaled by illumination of a light within the receptacle. For stages 1–4, sessions lasted 30 min or until 30 trials were completed. The criterion for advancement to the next training stage was the completion of 30 trials within each session. In stage 5, mice were trained to track and respond via a nose poke to a stimulus appearing in the response window. A response to a blank window was considered to be an incorrect response, terminating the trial and extinguishing the houselight for 5 s. The duration of stage 5 was 60 min or 50 trials, and mice had to complete 50 trials with >80% accuracy for two consecutive sessions before initiation of the pairwise discrimination task. Prior to initiating discrimination tasks involving manipulation of trial number or pharmacological challenge (experiments 2–5), mice were distributed into counterbalanced groups such that weight, percent accuracy, total session length, correct response latencies (duration of time from trial initiation to a registered nosepoke on the stimulus), and reinforcer retrieval latencies (duration of time to make a head entry into the reward receptacle following a correct response) on the last day of training were not different.
Experiment 1: Acquisition of a Pairwise Discrimination Task Involving Top-Down Processing
To assess relative stimulus salience between pairs of visual stimuli, mice (WT, n = 10; M1 KO, n = 11) were exposed to a single session of 50 trials during which responding on either stimulus within a pair when psueudorandomly distributed across touchscreen windows resulted in delivery of a reward. This was repeated twice on different days separated by 24 h: once when stimuli were “lasers and 4 circles” and once when the stimuli were the classic “fan and marbles”. A two-tailed paired t-test was used to determine significant differences between number of responses for each stimulus within each pair; p < 0.05 was considered to be significant.
To examine the role of M1 mAChR function on top-down learning and memory processing, we examined acquisition of a pairwise discrimination when the initial correct stimulus was the less salient, nonpreferred stimulus. Two stimuli were presented on the screen, pseudorandomly across trials. Responding on the less-preferred stimulus (S+, lasers) resulted in reward delivery, followed by a 5 s intertrial interval (ITI), whereas responding on the more preferred stimulus (S−, 4 circles) terminated the trial, extinguished the house light, and initiated the 5 s ITI before the house light illuminated again to signal the next trial. In order to align murine discrimination tasks with the parameters implemented in nonhuman primates and human studies (e.g., refs 44 and 45), correction trials were not implemented following incorrect responses. Sessions lasted for a total of 100 trials or 60 min; 100 total trials were chosen to account for lack of correction trials. Daily sessions continued for each mouse until each mouse acquired the discrimination defined as >80% accuracy. A two-tailed, unpaired t-test was used to examine significant differences in days to acquire the discrimination between wild-type and M1 KO mice; p < 0.05 was considered to be significant.
Experiment 2: Role of M1 mAChRs on Rate of Learning and Acquisition of a Pairwise Discrimination Task Involving Top-Down Processing
Following training (above), wild-type and M1 KO mice were administered vehicle or the M1 positive allosteric modulator (PAM) BQCA (synthesized within the Vanderbilt Center for Neuroscience Drug Discovery), intraperitoneally (i.p.) 15 min prior to the start of each discrimination session for 12 consecutive days starting on day 1 of the pairwise discrimination task. Separate groups of wild-type mice were administered vehicle (5% beta-cyclodextran in sterile H2O), or 10 or 30 mg/kg BQCA; initial doses were based on previous studies showing cognitive enhancing effects in transgenic models of Alzheimer’s disease.29 Separate groups of M1 KO mice were administered vehicle to assess potential differences in rate of learning from wild-type mice or 10 mg/kg BQCA to confirm M1 selectivity prior to each of 12 consecutive cognition test days. Total sessions lasted 100 trials or a maximum of 1 h.
Experiment 3: Influence of Trial Number on Rate of Learning and Acquisition of a Pairwise Discrimination Task Involving Top-Down Processing
Following training, separate groups of wild-type and M1 KO mice were administered vehicle or BQCA (1, 10 mg/kg, i.p.) for 12 consecutive days when the total number of trials per session was decreased to 60 trials. Lower doses were chosen based on the disruptive effects of 30 mg/kg BQCA in experiment 2. Primary dependent variables included percent accuracy per day to examine rate of learning across the 12 day period and percent of total mice that acquired the discrimination per day of testing. A two-way, nonrepeated measures analysis of variance (ANOVA) was conducted using group (dose/genotype) and day as factors. Significant main effects were followed by Bonferonni posthoc tests. Log-rank (Mantel–Cox) tests were conducted to compare survival plots for each dose/genotype. The number of days required for 80% of each group to acquire the discrimination is presented. In addition, one-way nonrepeated measures ANOVAs were conducted to examine influence of dose/genotype on days to acquisition, trials to acquisition, total trials completed, session length, correct response latency, and reward retrieval latency on day 1 of testing. Significant main effects were followed by Bonferonni posthoc tests. In all cases, p < 0.05 was considered to be significant. Data from sessions in which less than 20 responses were completed were omitted from analyses.
To assess effects of repeated dosing on mRNA expression, mice that completed the pairwise discrimination with 12 consecutive days of vehicle or 1 or 10 mg/kg BQCA were dosed with BQCA 1 day after the last cognition session. Thirty minutes following administration, mice were lightly anesthetized with isoflurane and decapitated, and the striatum, hippocampus, and PFC were dissected and flash frozen on dry ice and stored at −80 °C until analysis via quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR). Aqueous Micro kits (Ambion by Life Technologies, USA) were used for RNA extraction followed by DNase I treatment. The quantity of purified RNA was assessed by NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc. USA). Total RNA (0.5 μg) was reverse-transcribed into complementary DNA (cDNA) at 42 °C for 0.5 h using QuantiTect reverse transcription kit (QIAGEN, Germany). qRT-PCR reactions were performed in a CFX96 real-time PCR detection system (Bio-Rad, USA) using primers from TaqMan gene expression assays (ABI-Life Technologies, USA) for rat Chrm1/M1 (Rn00589936-s1) and TaqMan fast universal PCR master mix (ABI-Life Technologies, USA). The thermocycle reaction conditions were as follows: one cycle at 50 °C for 2 min, one cycle at 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control; data are presented using the comparative cycle threshold (CT) method normalized to vehicle-treated mice. One-way NR measure ANOVAs were conducted within each brain region to examine differences in M1 mRNA expression compared to respective vehicle groups.
To assess effects of repeated dosing of BQCA on plasma and brain concentrations, mice were dosed with 1 or 10 mg/kg BQCA (wild-type) and 10 mg/kg BQCA (M1 KO mice) 1 day after the last cognition session (day 13 dosing). An additional set of age-matched and food-deprived wild-type and M1 KO mice was administered a single dose of BQCA (wild-type 1, 10 mg/kg; M1 KO, 10 mg/kg). Thirty minutes following administration, mice were lightly anesthetized with isoflurane and decapitated, and trunk blood was collected and stored on ice in EDTA-coated blood collection tubes until centrifuged (10 min, 3000 rpm, 4 °C). Brains were extracted and flash frozen on dry ice. Plasma was collected, and plasma and whole brain were stored at −80 °C until analysis. Total plasma and brain concentrations of BQCA were determined using LC-MS/MS methods as previously described.29 A two-way, nonrepeated measures ANOVA was conducted, comparing dose (1 or 10 mg/kg BQCA) and treatment (acute, 1 day; repeated, 13 days) as factors to compare observed plasma and brain concentrations in wild-type mice. A separate two-way ANOVA compared observed plasma and brain concentrations between genotype (wild-type or M1 KO) and treatment duration (acute, 1 day; repeated, 13 days) in mice dosed with 10 mg/kg BQCA. Significant main effects were followed by Bonferonni posthoc tests. Calculated unbound plasma and brain concentrations were determined based on plasma free fraction (0.047) and brain free fraction (0.126) determined from historical rat brain homogenate data since brain nonspecific binding is species-independent. 65 Data are presented as mean ± standard deviation (n = 6–11/dose).
Experiment 4: Role of M1 mAChRs on Rate of Learning and Acquisition of a Pairwise Discrimination Task Not Involving Top-Down Processing
Following training, mice were exposed to a pairwise discrimination test in which both stimuli were of relatively equal salience (S+, “marbles”; S−, “fan”); salience was determined as above. Separate groups of wild-type and M1 KO mice were administered vehicle 15 min prior to each session for 12 days and were exposed to 60 or 100 trials per session. Additional groups of wild-type mice were administered vehicle or BQCA (1, 10 mg/kg, i.p.) 15 min prior to each session when exposed to 60 trials for 12 consecutive days. Dependent variables and analyses were same as above.
Experiment 5: Relative Reinforcing Strength of the Liquid Reinforcer Using a Progressive Ratio Schedule of Reinforcement
To assess the reinforcing strength of the liquid reinforcer between wild-type and M1 KO mice, we trained a separate cohort of wild-type and M1 KO mice to respond via a nose poke on a progressive ratio schedule. Mice (age 9–12 weeks at training; n = 12–13/genotype) were maintained at 85% of their free-feeding weight and first trained to respond via a nose poke in operant chambers (Med Associates) with 3 nose poke holes on one wall and a reward receptacle on the opposite wall to allow reinforcement delivery from a dipper. Mice were initially trained such that a single response in the middle nose poke hole when a light was illuminated would be reinforced via delivery of 0.2 mL of 33% diluted Ensure (fixed ratio 1 schedule of reinforcement). The dipper would remain elevated until the mouse entered the reward receptacle and for 5 s thereafter. Sessions lasted 1 h or until completion of 100 trails. The fixed ratio (FR) was increased to 10 responses over the course of subsequent sessions. When mice completed greater than 50 trials under a FR10 schedule for a minimum of 3 days, the reinforcement schedule was switched to a progressive ratio. The number of responses necessary for reinforcement delivery increased following each completed ratio based on the equation described by Richardson and Roberts;66 ratio = [5injection number × 0.2] − 5. The first 15 ratios in the series were 1, 2, 4, 6, 9, 12, 15, 20, 25, 32, 40, 50, 62, 77, and 95. Following each completed ratio, the stimulus light was extinguished for 5 s (ITI). Sessions lasted for 2 h or until a 20 min period elapsed during which a ratio was not completed; the last ratio completed was termed the break point and served as the dependent measure to assess reinforcing strength between genotypes. Responding was initially maintained by 33% diluted Ensure, and then a concentration–response curve was determined (water and 10, 33, and 100% Ensure) in random order. Each concentration was available for a minimum of 5 days, and a 3 day stable average was determined such that the number of reinforcers delivered did not deviate from the mean by more than 2. If stability was not achieved within 10 sessions, a 5 day average was calculated. Following each determination, the reinforcer concentration was returned to 33% for 2–3 days to ensure that baseline responding was similar prior to a new concentration determination. The primary dependent variable was the number of ratios completed at each dose. A two-way nonrepeated measures ANOVA examined effects of concentration and genotype followed by Bonferonni posthoc comparisons, p < 0.05.
Supplementary Material
Acknowledgments
Funding
Grant nos. MH086601 (C.K.J.), MH087965 (P.J.C.), MH093366 (P.J.C.), MH082867 (C.W.L.), and NS065867 (Z.X.), a PhRMA Foundation postdoctoral fellowship grant in Pharmacology and Toxicology (R.W.G.), the Psychiatric Center Copenhagen Research Foundation (D.D.), and the Carlsberg Foundation (D.D.).
We thank Weimin Peng and Josh Luffman for technical assistance and Drs. Randy Barrett, John Allison, and Greg Stanwood for helpful advice in optimizing touchscreen assays. Studies were performed in part through the use of the Murine Neurobehavior Core lab at the Vanderbilt University Medical Center. A portion of these studies was presented at the 2014 Experimental Biology meeting in San Diego, CA, and the 2015 Behavioral Pharmacology Society meeting in Boston, MA.
Footnotes
Author Contributions
R.W.G., D.D., and C.K.J. designed the experiments. R.W.G., D.D., M.G., M.B., X.Z., and C.L. performed the experiments. J.W., Z.X, C.W.L, P.J.C., and C.K.J. contributed reagents and other resources. R.W.G., D.D., X.Z., C.L., and C.K.J. performed data analyses. R.W.G. and C.K.J. wrote the manuscript.
ASSOCIATED CONTENT
Effects of BQCA and trial number on daily percent accuracy across 20-trial bins. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.5b00123.
Notes
The authors declare the following competing financial interest(s): Over the past year, C.W.L. consulted for Abbott. M.B., C.W.L., P.J.C., and C.K.J. received research/salary support from AstraZeneca and/or Bristol Myers Squibb. C.W.L. and P.J.C. are inventors on multiple composition of matter patents protecting allosteric modulators of GPCRs.
References
- 1.Nuechterlein KH, Barch DM, Gold JM, Goldberg TE, Green MF, Heaton RK. Identification of separable cognitive factors in schizophrenia. Schizophr Res. 2004;72:29–39. doi: 10.1016/j.schres.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Publishing; Washington, DC: 2000. [Google Scholar]
- 3.Barch DM, Ceaser A. Cognition in schizophrenia: core psychological and neural mechanisms. Trends Cognit Sci. 2012;16:27–34. doi: 10.1016/j.tics.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bobes J, Garcia-Portilla MP, Bascaran MT, Saiz PA, Bousono M. Quality of life in schizophrenic patients. Dialogues Clin Neurosci. 2007;9:215–226. doi: 10.31887/DCNS.2007.9.2/jbobes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green MF, Kern RS, Heaton RK. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res. 2004;72:41–51. doi: 10.1016/j.schres.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- 7.Sarter M, Givens B, Bruno JP. The cognitive neuroscience of sustained attention: where top-down meets bottom-up. Brain Res Rev. 2001;35:146–160. doi: 10.1016/s0165-0173(01)00044-3. [DOI] [PubMed] [Google Scholar]
- 8.Kastner S, Ungerleider LG. Mechanisms of visual attention in the human cortex. Annu Rev Neurosci. 2000;23:315–341. doi: 10.1146/annurev.neuro.23.1.315. [DOI] [PubMed] [Google Scholar]
- 9.Hahn B, Robinson BM, Kaiser ST, Harvey AN, Beck VM, Leonard CJ, Kappenman ES, Luck SJ, Gold JM. Failure of schizophrenia patients to overcome salient distractors during working memory encoding. Biol Psychiatry. 2010;68:603–609. doi: 10.1016/j.biopsych.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neuhaus AH, Karl C, Hahn E, Trempler NR, Opgen-Rhein C, Urbanek C, Hahn C, Ta TM, Dettling M. Dissection of early bottom-up and top-down deficits during visual attention in schizophrenia. Clin Neurophysiol. 2011;122:90–98. doi: 10.1016/j.clinph.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 11.Barbalat G, Chambon V, Domenech PJ, Ody C, Koechlin E, Franck N, Farrer C. Impaired hierarchical control within the lateral prefrontal cortex in schizophrenia. Biol Psychiatry. 2011;70:73–80. doi: 10.1016/j.biopsych.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Cook J, Barbalat G, Blakemore SJ. Top-down modulation of the perception of other people in schizophrenia and autism. Front Hum Neurosci. 2012;6:175. doi: 10.3389/fnhum.2012.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luck SJ, Fuller RL, Braun EL, Robinson B, Summerfelt A, Gold JM. The speed of visual attention in schizophrenia: electrophysiological and behavioral evidence. Schizophr Res. 2006;85:174–195. doi: 10.1016/j.schres.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 14.Fuller RL, Luck SJ, Braun EL, Robinson BM, McMahon RP, Gold JM. Impaired control of visual attention in schizophrenia. J Abnorm Psychol. 2006;115:266–275. doi: 10.1037/0021-843X.115.2.266. [DOI] [PubMed] [Google Scholar]
- 15.Gold JM, Fuller RL, Robinson BM, Braun EL, Luck SJ. Impaired top-down control of visual search in schizophrenia. Schizophr Res. 2007;94:148–155. doi: 10.1016/j.schres.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bussey TJ, Padain TL, Skillings EA, Winters BD, Morton AJ, Saksida LM. The touchscreen cognitive testing method for rodents: how to get the best out of your rat. Learn Mem. 2008;15:516–523. doi: 10.1101/lm.987808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mar AC, Horner AE, Nilsson SR, Alsio J, Kent BA, Kim CH, Holmes A, Saksida LM, Bussey TJ. The touchscreen operant platform for assessing executive function in rats and mice. Nat Protoc. 2013;8:1985–2005. doi: 10.1038/nprot.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oomen CA, Hvoslef-Eide M, Heath CJ, Mar AC, Horner AE, Bussey TJ, Saksida LM. The touchscreen operant platform for testing working memory and pattern separation in rats and mice. Nat Protoc. 2013;8:2006–2021. doi: 10.1038/nprot.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horner AE, Heath CJ, Hvoslef-Eide M, Kent BA, Kim CH, Nilsson SR, Alsio J, Oomen CA, Holmes A, Saksida LM, Bussey TJ. The touchscreen operant platform for testing learning and memory in rats and mice. Nat Protoc. 2013;8:1961–1984. doi: 10.1038/nprot.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dickson PE, Corkill B, McKimm E, Miller MM, Calton MA, Goldowitz D, Blaha CD, Mittleman G. Effects of stimulus salience on touchscreen serial reversal learning in a mouse model of fragile X syndrome. Behav Brain Res. 2013;252:126–135. doi: 10.1016/j.bbr.2013.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terry AV, Jr, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. 2003;306:821–827. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- 22.Hasselmo ME, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2011;36:52–73. doi: 10.1038/npp.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones CK, Byun N, Bubser M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology. 2012;37:16–42. doi: 10.1038/npp.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bubser M, Byun N, Wood MR, Jones CK. Muscarinic receptor pharmacology and circuitry for the modulation of cognition. Handb Exp Pharmacol. 2012:121–166. doi: 10.1007/978-3-642-23274-9_7. [DOI] [PubMed] [Google Scholar]
- 25.Carruthers SP, Gurvich CT, Rossell SL. The muscarinic system, cognition and schizophrenia. Neurosci Biobehav Rev. 2015;55:393–402. doi: 10.1016/j.neubiorev.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Dean B, McLeod M, Keriakous D, McKenzie J, Scarr E. Decreased muscarinic1 receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2002;7:1083–1091. doi: 10.1038/sj.mp.4001199. [DOI] [PubMed] [Google Scholar]
- 27.Gibbons AS, Scarr E, Boer S, Money T, Jeon WJ, Felder C, Dean B. Widespread decreases in cortical muscarinic receptors in a subset of people with schizophrenia. Int J Neuropsychopharmacol. 2013;16:37–46. doi: 10.1017/S1461145712000028. [DOI] [PubMed] [Google Scholar]
- 28.Scarr E, Craig JM, Cairns MJ, Seo MS, Galati JC, Beveridge NJ, Gibbons A, Juzva S, Weinrich B, Parkinson-Bates M, Carroll AP, Saffery R, Dean B. Decreased cortical muscarinic M1 receptors in schizophrenia are associated with changes in gene promoter methylation, mRNA and gene targeting microRNA. Transl Psychiatry. 2013;3:e230. doi: 10.1038/tp.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, Jadhav SB, Menon UN, Xiang Z, Watson ML, Christian EP, Doherty JJ, Quirk MC, Snyder DH, Lah JJ, Levey AI, Nicolle MM, Lindsley CW, Conn PJ. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marino MJ, Conn PJ. Direct and indirect modulation of the N-methyl D-aspartate receptor. Curr Drug Targets: CNS Neurol Disord. 2002;1:1–16. doi: 10.2174/1568007023339544. [DOI] [PubMed] [Google Scholar]
- 31.Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1998;95:11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lynch DR, Guttmann RP. Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. J Pharmacol Exp Ther. 2002;300:717–723. doi: 10.1124/jpet.300.3.717. [DOI] [PubMed] [Google Scholar]
- 33.Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Sur C, Kinney G, Seabrook GR, Ray WJ. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chambon C, Jatzke C, Wegener N, Gravius A, Danysz W. Using cholinergic M1 receptor positive allosteric modulators to improve memory via enhancement of brain cholinergic communication. Eur J Pharmacol. 2012;697:73–80. doi: 10.1016/j.ejphar.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Chambon C, Wegener N, Gravius A, Danysz W. A new automated method to assess the rat recognition memory: validation of the method. Behav Brain Res. 2011;222:151–157. doi: 10.1016/j.bbr.2011.03.032. [DOI] [PubMed] [Google Scholar]
- 36.Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NE, Lebois EP, Xiang Z, Sheffler DJ, Cho HP, Davis AA, Nemirovsky NE, Mennenga SE, Camp BW, Bimonte-Nelson HA, Bode J, Italiano K, Morrison R, Daniels JS, Niswender CM, Olive MF, Lindsley CW, Jones CK, Conn PJ. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J Neurosci. 2012;32:8532–8544. doi: 10.1523/JNEUROSCI.0337-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galloway CR, Lebois EP, Shagarabi SL, Hernandez NA, Manns JR. Effects of selective activation of M1 and M4 muscarinic receptors on object recognition memory performance in rats. Pharmacology. 2014;93:57–64. doi: 10.1159/000357682. [DOI] [PubMed] [Google Scholar]
- 38.Uslaner JM, Eddins D, Puri V, Cannon CE, Sutcliffe J, Chew CS, Pearson M, Vivian JA, Chang RK, Ray WJ, Kuduk SD, Wittmann M. The muscarinic M1 receptor positive allosteric modulator PQCA improves cognitive measures in rat, cynomolgus macaque, and rhesus macaque. Psychopharmacology (Berl) 2013;225:21–30. doi: 10.1007/s00213-012-2788-8. [DOI] [PubMed] [Google Scholar]
- 39.Ghoshal A, Rook JM, Dickerson JW, Roop GN, Morrison RD, Jalan-Sakrikar N, Lamsal A, Noetzel MJ, Poslusney MS, Wood MR, Melancon BJ, Stauffer SR, Xiang Z, Daniels JS, Niswender CM, Jones CK, Lindsley CW, Conn PJ. Potentiation of M Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 41.Bartko SJ, Romberg C, White B, Wess J, Bussey TJ, Saksida LM. Intact attentional processing but abnormal responding in M1 muscarinic receptor-deficient mice using an automated touchscreen method. Neuropharmacology. 2011;61:1366–1378. doi: 10.1016/j.neuropharm.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ragozzino ME, Artis S, Singh A, Twose TM, Beck JE, Messer WS., Jr The selective M1 muscarinic cholinergic agonist CDD-0102A enhances working memory and cognitive flexibility. J Pharmacol Exp Ther. 2012;340:588–594. doi: 10.1124/jpet.111.187625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morton AJ, Skillings E, Bussey TJ, Saksida LM. Measuring cognitive deficits in disabled mice using an automated interactive touchscreen system. Nat Methods. 2006;3:767. doi: 10.1038/nmeth1006-767. [DOI] [PubMed] [Google Scholar]
- 44.Jazbec S, Pantelis C, Robbins T, Weickert T, Weinberger DR, Goldberg TE. Intra-dimensional/extra-dimensional set-shifting performance in schizophrenia: impact of distractors. Schizophr Res. 2007;89:339–349. doi: 10.1016/j.schres.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 45.Nagahara AH, Bernot T, Tuszynski MH. Age-related cognitive deficits in rhesus monkeys mirror human deficits on an automated test battery. Neurobiol Aging. 2010;31:1020–1031. doi: 10.1016/j.neurobiolaging.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci. 2001;21:5239–5250. doi: 10.1523/JNEUROSCI.21-14-05239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2002;6:51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- 48.Comte M, Schon D, Coull JT, Reynaud E, Khalfa S, Belzeaux R, Ibrahim EC, Guedj E, Blin O, Weinberger DR, Fakra E. Dissociating Bottom-Up and Top-Down Mechanisms in the Cortico-Limbic System during Emotion Processing. Cereb Cortex. 2014 doi: 10.1093/cercor/bhu185. [DOI] [PubMed] [Google Scholar]
- 49.Waskom ML, Kumaran D, Gordon AM, Rissman J, Wagner AD. Frontoparietal representations of task context support the flexible control of goal-directed cognition. J Neurosci. 2014;34:10743–10755. doi: 10.1523/JNEUROSCI.5282-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, Christopoulos A. A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J Biol Chem. 2012;287:650–659. doi: 10.1074/jbc.M111.314278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood S, Sage JR, Shuman T, Anagnostaras SG. Psychostimulants and cognition: a continuum of behavioral and cognitive activation. Pharmacol Rev. 2013;66:193–221. doi: 10.1124/pr.112.007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnsten AF. Catecholamine influences on dorsolateral prefrontal cortical networks. Biol Psychiatry. 2011;69:e89–99. doi: 10.1016/j.biopsych.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009;30:148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davie BJ, Christopoulos A, Scammells PJ. Development of M1 mAChR allosteric and bitopic ligands: prospective therapeutics for the treatment of cognitive deficits. ACS Chem Neurosci. 2013;4:1026–1048. doi: 10.1021/cn400086m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Koppen CJ, Kaiser B. Regulation of muscarinic acetylcholine receptor signaling. Pharmacol Ther. 2003;98:197–220. doi: 10.1016/s0163-7258(03)00032-9. [DOI] [PubMed] [Google Scholar]
- 56.Markram H, Segal M. Long-lasting facilitation of excitatory postsynaptic potentials in the rat hippocampus by acetylcholine. J Physiol. 1990;427:381–393. doi: 10.1113/jphysiol.1990.sp018177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang XL, Sullivan JA, Moskal JR, Stanton PK. A NMDA receptor glycine site partial agonist, GLYX-13, simultaneously enhances LTP and reduces LTD at Schaffer collateral-CA1 synapses in hippocampus. Neuropharmacology. 2008;55:1238–1250. doi: 10.1016/j.neuropharm.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 59.Brigman JL, Feyder M, Saksida LM, Bussey TJ, Mishina M, Holmes A. Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn Mem. 2008;15:50–54. doi: 10.1101/lm.777308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Darvas M, Palmiter RD. Specific contributions of N-methyl-d-aspartate receptors in the dorsal striatum to cognitive flexibility. Neuroscience. 2015;284:934–942. doi: 10.1016/j.neuroscience.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bubser M, Bridges TM, Dencker D, Gould RW, Grannan M, Noetzel MJ, Lamsal A, Niswender CM, Daniels JS, Poslusney MS, Melancon BJ, Tarr JC, Byers FW, Wess J, Duggan ME, Dunlop J, Wood MW, Brandon NJ, Wood MR, Lindsley CW, Conn PJ, Jones CK. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem Neurosci. 2014;5:920–942. doi: 10.1021/cn500128b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Talpos JC, Fletcher AC, Circelli C, Tricklebank MD, Dix SL. The pharmacological sensitivity of a touchscreen-based visual discrimination task in the rat using simple and perceptually challenging stimuli. Psychopharmacology (Berl) 2012;221:437–449. doi: 10.1007/s00213-011-2590-z. [DOI] [PubMed] [Google Scholar]
- 63.Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G. Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handb Exp Pharmacol. 2012:267–295. doi: 10.1007/978-3-642-25758-2_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- 65.Di L, Umland JP, Chang G, Huang Y, Lin Z, Scott DO, Troutman MD, Liston TE. Species independence in brain tissue binding using brain homogenates. Drug Metab Dispos. 2011;39:1270–1277. doi: 10.1124/dmd.111.038778. [DOI] [PubMed] [Google Scholar]
- 66.Richardson NR, Roberts DC. Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J Neurosci Methods. 1996;66:1–11. doi: 10.1016/0165-0270(95)00153-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.