

Opinion statement
NRAS mutations in codons 12, 13, and 61 arise in 15–20 % of all melanomas. These alterations have been associated with aggressive clinical behavior and a poor prognosis. Until recently, there has been a paucity of promising genetically targeted therapy approaches for NRAS-mutant melanoma (and RAS-mutant malignancies in general). MEK inhibitors, particularly binimetinib, have shown activity in this cohort. Based on pre-clinical and early clinical studies, combining MEK inhibitors with agents inhibiting the cell cycling and the PI3K-AKT pathway appears to provide additional benefit. In particular, a strategy of MEK inhibition and CDK4/6 inhibition is likely to be a viable treatment option in the future, and is the most promising genetically targeted treatment strategy for NRAS-mutant melanoma developed to date. In addition, immune-based therapies have shown increasing activity in advanced melanoma and may be particularly effective in those with NRAS mutations. Combination strategies of immune and targeted therapies may also play a role in the future although clinical trials testing these approaches are in early stages.
Keywords: Melanoma, NRAS, Mutation, MEK inhibitor, CDK4, MAPK, Immunotherapy, Trametinib, Binimetinib
Introduction
Mutations in RAS occur in a large percentage of prevalent and deadly malignancies including melanoma, lung adenocarcinoma, colon cancer, pancreatic cancer, acute leukemias, and others [1]. Targeting RAS, therefore, has remained a critical priority for cancer therapy but an effective, approved treatment option has remained elusive in any cancer to date. NRAS is recurrently altered in 15–20 % of melanomas at codons 12, 13, or 61, and is the second most common oncogenic “driver” mutation in this disease. In parallel with melanoma therapy in general, there have been few treatment options for this genetic cohort in the past. More recently, the development of genetically targeted agents and immune-based therapies has yielded numerous available and potential therapeutic strategies for treating NRAS-mutant melanomas. These treatment approaches, if effective for patients with NRAS-mutant melanoma, may also have implications extending to other RAS-mutant malignancies. In this review, we will cover the clinical and pathologic characteristics of NRAS-mutant melanoma and ongoing treatment options for patients in this genetically defined cohort of melanoma (Table 1).
Table 1. Selected experimental and approved treatment options for advanced NRAS-mutant melanoma.
| Agent (reference) | NRAS specific? | Response rate | OS (median) | FDA approved? |
|---|---|---|---|---|
| Binimetinib [2••] | Yes | 20 % (6 of 30) | * | No |
| Binimetinib + LEE011 [3] | Yes | 33 % (7 of 21) | * | No |
| IL-2 [4] | No | 16 % | 11.4 months | Yes |
| Ipilimumab [5•] | No | 10.9 % | 10.1 monthsa (2nd line +) | Yes |
| Pembrolizumab [6, 7•] | No | 38 % (ipi naïve) 25 % (ipi pre-tx) |
* | Yesb |
| Nivolumab [8, 9] | No | 31 % (ipi naïve) 25 % (ipi pre-tx) |
16.8 months | No |
ipi ipilimumab, pre-tx pre-treated
Not reported
Associated with a ∼20 % 5-year survival
Epidemiology
NRAS mutations occur at a fairly consistent rate of 15– 20 % at all non-uveal sites of melanoma, including sun exposed and non-sun exposed skin, mucosal, and acral sites of origin [10, 11]. This distribution contrasts with BRAF mutations, which are more common in intermittently sun-exposed skin, and KIT mutations, which are present predominantly in mucosal and acral melanomas [12]. Also, in contrast to BRAF, NRAS mutations are rarely present in benign melanocytic nevi, with the exception of congenital nevi [13, 14]. NRAS mutations are associated with thicker primary tumors, increased mitotic rate, and lower incidence of ulceration [15]. More importantly, NRAS mutations have been generally linked to a poorer overall survival, although this has varied across studies [15, 16•, 17].
Pathogenesis
Mutations in NRAS constitutively activate intracellular signaling through a variety of pathways, most notably the RAS-RAF-MAPK and PI3K-AKT pathways. NRAS mutations activate MAPK signaling to a similar degree as BRAF mutations and rarely co-occur with mutations in the PI3K-AKT pathways, suggesting that mutant NRAS drives this pathway also [18•]. These activated signaling pathways induce cell cycle dysregulation, pro-survival pathways, and cellular proliferation. NRAS point mutations occur in codon 61 in >80 % of cases with the remaining mutations occurring in codons 12 and 13 [16•, 19]. This distribution contrasts with KRAS mutations in lung cancer or colon cancer, which occur largely in codons 12 and 13 [20, 21]. Mutations at any of these codons produce similar effects by locking NRAS into its active conformation [22]. It remains unclear whether different NRAS mutations induce distinct biologic or clinical features in melanoma.
Diagnosis
Genetic profiling of melanomas varies widely between institutions and practices. Testing platforms range from single-point mutation assays (for BRAFV600E), PCR-based tests evaluating a limited number of recurrently mutated “hotspots ” in several genes, to targeted next generation sequencing assays which may assess hundreds of genes [11, 23, 24]. Testing for BRAFV600E mutations is nearly universal in metastatic disease since three clinically active small molecule inhibitors have now been approved for BRAF V600-mutant melanoma (vemurafenib, dabrafenib, and trametinib) [25–27]. Since no therapeutic agents have been approved specifically for NRAS-mutant melanoma, mutational profiling of NRAS is not performed routinely by many clinicians. Identifying NRAS mutations may have prognostic implications and facilitate clinical trial enrollment. Most institutions that routinely perform more extensive multiplexed PCR platforms or targeted next generation sequencing assays will include at least the frequently mutated “hotspot ” codons (12, 13, 61) or exons (1 and 2) in their respective panels.
Treatment
Identifying an effective treatment option for mutant RAS is a long sought, elusive goal in cancer therapeutics since this gene drives many of the most aggressive malignancies. Previous strategies have focused on posttranslational modification of NRAS (farnesyltransferase inhibitors) whereas current approaches are largely concentrated on downstream signaling pathways. Directly targeting RAS with small molecules or small interfering RNAs (siRNAs) may also play a role in the future. Possible therapeutic approaches are outlined in (Fig. 1).
Fig. 1.
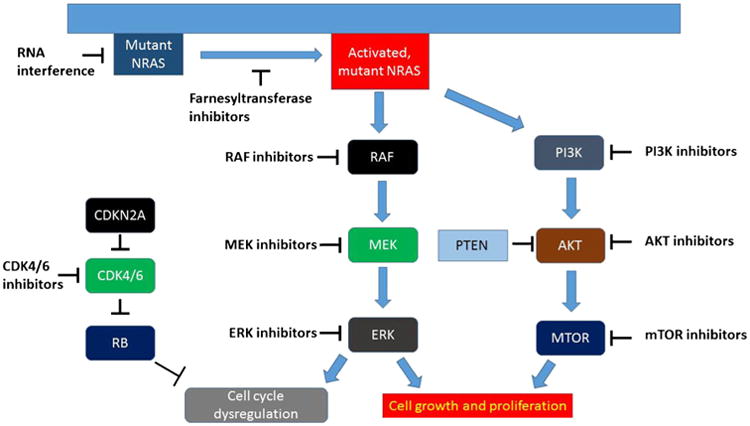
NRAS-driven cellular signalling and possible targeted therapeutic strategies.
Farnesyltransferase inhibitors (FTIs)
Farnesylation of a cysteine residue is a critical posttranslational modification of the RAS oncoprotein which occurs prior to its insertion into the cell membrane [28]. Although inhibition of this step of RAS activation showed a promising pre-clinical activity, the experience in clinical trials has generally been disappointing [29–31]. In melanoma, an FTI (R115777) was evaluated in 14 patients in a phase II clinical trial. No patients experienced a clinical response, although the NRAS mutation status of these patients was not reported [32]. Tipifarnib demonstrated efficacy in AML in occasional patients with complete remissions observed in three of 54 treated patients; the NRAS mutation status in this population is likewise unknown [33]. Further clinical trials of FTIs in melanoma are not ongoing.
MEK inhibitors
Mutated NRAS triggers the MAPK signaling cascade through activation of RAF, which in turn activates MEK, triggering ERK phosphorylation and cellular proliferation. Blocking a downstream signaling partner, therefore, is an attractive therapeutic strategy. Inhibition of RAF in this setting is a challenge since both CRAF and BRAF have been implicated as critical downstream signaling partners for mutant NRAS. Inhibitors of BRAF, in fact, appear to paradoxically activate signaling in RAS-mutated malignancies and likely promote neoplastic proliferation. Efforts to inhibit MAPK signaling, therefore, have largely focused on MEK.
Early MEK inhibitors
CI-1040 was among the first MEK inhibitors evaluated in clinical trials. Although its toxicity profile was tolerable, objective responses were rare across malignancies (1 of 144 in phase I/II trials) [34, 35]. Of note, baseline phospho-ERK expression correlated with stable disease, suggesting that this class of therapies may be effective in tumors with MAPK activation.
PD-03225901 is a second-generation MEK inhibitor with higher potency and improved MAPK-blocking activity [36]. Forty-eight patients with advanced melanoma were enrolled with much greater activity; three patients experienced partial responses and ten had temporary stable disease [37]. High incidence of toxicity, particularly ocular and neurologic, limited further development of PD-03225901 [37, 38].
Selumetinib (AZD-6244; ARRY-142886) is another second-generation inhibitor of MEK1/2 with potent inhibition of cell lines and xenografts in both RAS and RAF-mutant cancers [39]. In a phase I clinical trial, 11 melanoma patients had evaluable responses, of which 1 had a partial response (with an NRAS mutation) and seven had stable disease [40]. Two subsequent melanoma trials did not show a benefit with selumetinib, however. A phase II trial in unselected melanoma patients compared selumetinib with temozolomide and reported equivalent or inferior response rates for selumetinib (5.8 vs. 9.7 %) and PFS (hazard ratio 1.07) [41]. This agent was subsequently evaluated only in BRAFV600-mutant melanoma in combination with dacarbazine compared to dacarbazine alone. Although PFS was extended (median 5.6 vs. 3.0 months), no improvement in OS was identified (13.9 vs. 10.5 months) [42]. A phase II trial in uveal melanoma identified a PFS advantage compared to chemotherapy as well [43]. No trials specifically for NRAS-mutant melanoma have been performed; development is ongoing for KRAS-mutant lung cancer, uveal melanoma, and other malignancies.
Trametinib
Trametinib is an allosteric inhibitor of MEK1/2 that has received FDA approval for the treatment of BRAFV600-mutant melanoma as a single agent and in combination with dabrafenib. This approval was based on improved overall survival in a phase III trial for trametinib compared with dacarbazine in BRAF-mutant melanoma [26]. The trametinib/dabrafenib combination approval was based on a randomized phase II trial which demonstrated improved PFS and response rates for the dabrafenib and trametinib compared to single-agent dabrafenib [44]. The phase I study of trametinib included a limited number of patients with NRAS-mutant melanoma. Of seven treated patients, two experienced stable disease (29 %) including one patient who remained on treatment for approximately 8 months [45•]. Given the more robust data in the BRAF-mutant population, no additional studies were performed for the NRAS-mutant cohort. Potential combination strategies with trametinib may play a role in the future (see combination therapy below).
Binimetinib (MEK162)
Binimetinib, an allosteric MEK1/2 inhibitor, has been the first agent to show robust activity specifically in NRAS-mutant melanoma. An open-label phase II trial enrolled 71 patients with advanced melanoma including 30 with NRAS mutations [2••]. Of these 30 patients, six demonstrated a partial response (20 %) and another 13 (43 %) had temporary stable disease. Median PFS for this cohort was 3.7 months (95 % CI 2.5–5.4 months). Of note, the PFS and response rate appeared to be very similar for the NRAS and BRAF cohorts in this study. Based on these results, a randomized phase III trial is ongoing comparing binimetinib to chemotherapy in NRAS-mutant melanoma (NCT01763164). Combination strategies are also being explored with binimetinib.
Other MEK inhibitors
RO4987655 (CH4987655) is a MEK inhibitor recently assessed in phase I trials. Eight patients with NRAS-mutant melanoma were treated with one PR (13 %) and two additional cases of stable disease [46].
Cobimetinib (GDC-0973) has not been evaluated for the NRAS cohort. In combination with vemurafenib, however, promising activity was observed in BRAFV600-mutant melanoma with an objective response rate of 87 % and a median PFS of 13.7 months for patients not previously treated with a BRAF inhibitor [47].
Other MEK inhibitors in development: An intriguing structural and functional study suggested that experimental MEK inhibitors with superior pre-clinical activity in KRAS-mutant cancers (GCD-0623 and G-573) form a hydrogen bond with MEK to prevent feedback phosphorylation by wild-type BRAF [48]. By contrast, cobimetinib, which is more effective in BRAF-mutant pre-clinical models, inhibits activated MEK. This suggests that through distinct modes of action, RAS and RAF-specific MEK inhibitors may be utilized in the future.
MEK inhibitor-based combinations
Although the clinical activity of single-agent binimetinib is unprecedented for a RAS-targeted therapy, the relatively suboptimal response rate and PFS has led to interest in MEK inhibitor-based combination strategies. Mutant NRAS activates multiple cell signaling pathways, likely necessitating such an approach. Two particular pathways of interest are CDK4/Rb and PI3K/AKT.
MEK + CDK4 inhibition: An inducible mouse model of NRAS-mutant melanoma demonstrated that MEK inhibition was sufficient to cause apoptosis but not cell cyle arrest. In contrast, complete NRAS extinction not only induced apoptosis but also drove arrest of the cell cycle [49•]. Network modeling demonstrated that CDK4 was a critical driver of these differential results; subsequent combination of MEK inhibition (trametinib) with CDK4/6 inhibition (palbociclib) caused tumor regression. In addition, alterations in genes causing cell cycle dysregulation (CDKN2A, CDK4, CCND1, others) occur with high frequency in melanoma [18•, 50]. Pre-clinical cell line data suggests that alterations in these genes predict for sensitivity to CDK4 inhibition [51, 52]. Based on these observations, two phase I/II trials particularly focusing on NRAS-mutant melanomas are ongoing (binimetinib + LEE011; NCT01781572 and trametinib + palbociclib; NCT01781572). Early results from the binimetinib + LEE011 study were presented in July 2014 with promising activity [3]. Among the first 21 patients treated, seven experienced a partial response (33 %) and another 11 (52 %) experiencing stable disease. Only three patients (14 %) had primary progressive disease. Several clinically significant toxicities have occurred (including high-grade creatine phosphokinase (CPK) elevations and a single fatal case of cardiomyopathy) and dose finding is ongoing.
MEK + PI3K/AKT inhibition: Combined targeting of MAPK and PI3K/AKT signaling has a significant rationale as dual inhibition was required to restrain tumor growth in NRAS-mutant models [53, 54]. Several early-phase studies for advanced cancers are underway co-targeting MEK and PI3K/AKT. These include the combination of trametinib and an AKT inhibitor (uprosertib; GSK2141795) in BRAF wild-type melanoma (NCT01941927) and in AML (NCT01907815). Binimetinib and various PI3K/AKT pathway inhibitors are also being assessed in early-stage trials (NCT01363232, NCT01337765, NCT01449058) [55].
Other MEK inhibitor combinations have pre-clinical support as well. Adding an ERK inhibitor to MEK inhibition increased apoptosis, suppressed CCND1 signaling, and delayed resistance in NRAS-mutant cell lines [56]. Augmenting MEK inhibitor-induced apoptosis with WNT signaling stimulation is another potential strategy with pre-clinical support [57].
Other MAPK-directed therapies
While MEK inhibitors are the most developed, additional strategies targeting the MAPK pathway are being evaluated in various stages of development. These include RAF inhibitors (alone and in combination), ERK inhibitors, and other more experimental strategies.
Sorafenib, a multitargeted kinase inhibitor, and tivantinib, a MET inhibitor, were assessed in eight patients with NRAS-mutant melanoma [58]. Two patients experienced a complete or partial response and another two had stable disease as best response.
RAF-265, an inhibitor of BRAF and VEGFR2, demonstrated pre-clinical efficacy in NRAS-mutant and NRAS/BRAF wild-type patient-derived xenografts [59]. In 71 patients, the objective response rate was 16 %for BRAF mutant melanoma patients and 13 % for BRAF wild-type (NRAS status not reported) [60].
Inhibitors of ERK, the final step of canonical MAPK signaling, have also generated interest in both NRAS and BRAF-mutant melanoma. SCH772984 has demonstrated efficacy in cell lines with NRAS, KRAS, and BRAF mutations as well as in models of BRAF inhibitor-resistant melanoma [61•]. Additionally, ERK inhibition in combination with PI3K/AKT inhibitors appeared effective against BRAF inhibitor-resistant cell lines, including those with secondary NRAS mutations [62].
An intriguing recent study identified a scaffolding protein, IQGAP1, that appears to be essential for MAPK signaling in both RAS and RAF driven tumors [63]. A peptide disrupting IQGAP1-ERK interactions was effective in cell lines and mouse models of RAS-mutant malignancies.
Other targeted strategies
Direct targeting of NRAS by RNA interference (RNAi) to block protein translation is an effective technique in laboratory models, particularly cell lines, but has not yet been implemented in clinical practice [64]. Challenges include intravascular degradation, intracellular trafficking, and potential off target effects [65]. An early-stage clinical trial demonstrated intratumoral presence of small interfering RNAs (siRNAs) with a nanoparticle delivery system and decreased expression of the target (RRM2) [66].
Heat shock protein-90 (HSP90) inhibitors may have a role in single agent or combination therapy through inhibition of multiple down-stream targets of RAS. The HSP90 inhibitor XL888 had promising activity in NRAS-mutant melanoma cell lines [67].
Combination inhibition of NF-KB and AKT by a small molecule inhibitor BI-69-A11 also was effective in NRAS-mutant mouse models [68].
Immune-based therapies
Immune-based therapies are the other cornerstone of melanoma therapeutics and are used regardless of tumor genotype. Immune therapies are generally used as first-line therapy, particularly for patients with BRAFV600 wild-type melanoma (including NRAS mutant).
High-dose interleukin-2 (IL-2) has been used for many years and causes durable responses in 5–8 % of treated patients [4, 69]. Acute toxicities, however, are severe and limit therapy to young and otherwise healthy patients [70]. Despite the drawbacks, IL-2 is still a potential therapeutic strategy for patients who qualify and adds another treatment option.
Ipilimumab is a monoclonal antibody to cytotoxic T lymphocyte antigen-4 (CTLA4) which activates an antitumor immune response by removing a key negative T cell regulator. Ipilimumab improved overall survival in pre-treated melanoma patients compared to a peptide vaccine, and in combination with dacarbazine as first-line therapy when compared to chemotherapy alone [5•, 71]. This agent is associated with durable benefit, with approximately 20 % of patients surviving 3–5 years [72, 73]. Toxicities are related to aberrant immune activation and include colitis, hepatitis, endocrinopathies, and skin rash.
Agents targeting the programmed cell death-1 receptor and its ligand (PD-1/PD-L1) appear still more promising. Nivolumab (BMS-936558) and pembrolizumab (MK-3475) are associated with objective response rates in the 25–40 % range with many appearing durable [6, 7•, 74•, 75]. Furthermore, immune-related adverse events occur much less commonly than with ipilimumab. Pembrolizumab recently received FDA approval for treatment of patients who previously progressed on ipilimumab and BRAF inhibitors (if applicable). Clinical trials comparing these agents with ipilimumab as well as numerous combination therapy trials are ongoing.
Although these therapies are used irrespective of genotype, some retrospective data suggests that patients with NRAS-mutant melanoma may have higher response rates to immune-based therapies. Joseph and colleagues observed nearly a 50 % response rate to IL-2 among a small NRAS-mutated cohort [76•]. Our group saw a similar effect among patients treated with newer immune checkpoint inhibitors, particularly for those treated with anti-PD-1/PD-L1 [77]. The underlying mechanism for this finding is not clear and prospective validation is needed.
Combined immune and targeted therapy strategies may also hold promise. Several clinical trials are ongoing for the BRAF-mutant population combining immune checkpoint inhibitors and BRAF and/or MEK inhibitors. Early phase studies are also combining MEK inhibitors with anti-PD-1 or anti-PD-L1 across a variety of solid tumors. Concerns to such an approach include aberrant T cell activation (toxicity) or dampening the immune response (lack of efficacy), highlighting the need for rational and carefully conducted clinical trials [78, 79].
Conclusions
NRAS-mutant melanoma is a common subtype of this disease with a poor prognosis. No approved therapies specifically targeting NRAS have been approved. However, newer targeted therapy strategies, particularly MEK inhibitor monotherapy and combinations, will likely provide effective treatment strategies in the near future. These therapies may also have an impact in other RAS-mutant malignancies. Immune-based therapies, while not genotype-specific, are also incredibly promising and appear at least as effective in the NRAS-mutant cohort compared to other melanoma populations.
Acknowledgments
Funding sources: Douglas B. Johnson was supported by NIH grant K12 CA 0906525.
Footnotes
Compliance with ethics guidelines: Conflict of interest: Douglas B. Johnson and Igor Puzanov declare that they have no conflict of interest.
Human and animal rights and informed consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 2••.Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–56. doi: 10.1016/S1470-2045(13)70024-X. This is the first clinical trial to show consistent activity of any genetically targeted therapy in NRAS mutant melanoma. [DOI] [PubMed] [Google Scholar]
- 3.Sosman JA, Kittaneh M, Lolkema MPJ, Postow MA, Schwartz G, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity. J Clin Oncol. 2014;32:9009. Abstract. [Google Scholar]
- 4.Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–16. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 5•.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. Demonstrates an overall survival benefit of ipilimumab in unselected melanoma populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (Anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma:a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–17. doi: 10.1016/S0140-6736(14)60958-2. Large clinical trial demonstrating the benefit of pembrolizumab in patients who progressed on ipilimumab. [DOI] [PubMed] [Google Scholar]
- 8.Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–30. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber JS, Kudchadkar RR, Yu B, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31:4311–8. doi: 10.1200/JCO.2013.51.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 11.Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012;7:e35309. doi: 10.1371/journal.pone.0035309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–6. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 13.Charbel C, Fontaine RH, Malouf GG, et al. NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Investig Dermatol. 2014;134:1067–74. doi: 10.1038/jid.2013.429. [DOI] [PubMed] [Google Scholar]
- 14.Poynter JN, Elder JT, Fullen DR, et al. BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res. 2006;16:267–73. doi: 10.1097/01.cmr.0000222600.73179.f3. [DOI] [PubMed] [Google Scholar]
- 15.Devitt B, Liu W, Salemi R, et al. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011;24:666–72. doi: 10.1111/j.1755-148X.2011.00873.x. [DOI] [PubMed] [Google Scholar]
- 16•.Jakob JA, Bassett RL, Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–23. doi: 10.1002/cncr.26724. Establishes prognostic significance of NRAS mutations in advanced melanoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlino MS, Haydu LE, Kakavand H, et al. Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br J Cancer. 2014;111:292–9. doi: 10.1038/bjc.2014.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18•.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–63. doi: 10.1016/j.cell.2012.06.024. Large next generation sequencing study that defines much of the landscape of genetic alterations in melanoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 21.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fedorenko IV, Gibney GT, Smalley KS. NRAS mutant melanoma: biological behavior and future strategies for therapeutic management. Oncogene. 2012;32(25):3009–18. doi: 10.1038/onc.2012.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halait H, Demartin K, Shah S, et al. Analytical performance of a real-time PCR-based assay for V600 mutations in the BRAF gene, used as the companion diagnostic test for the novel BRAF inhibitor vemurafenib in metastatic melanoma. Diagn Mol Pathol. 2012;21:1–8. doi: 10.1097/PDM.0b013e31823b216f. [DOI] [PubMed] [Google Scholar]
- 24.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 27.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 28.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6:541–55. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 29.Kohl NE, Wilson FR, Mosser SD, et al. Protein farnesyltransferase inhibitors block the growth of RAS-dependent tumors in nude mice. Proc Natl Acad Sci U S A. 1994;91:9141–5. doi: 10.1073/pnas.91.19.9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao S, Cunningham D, de Gramont A, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950–7. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 31.Van Cutsem E, van de Velde H, Karasek P, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–8. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 32.Gajewski TF, Salama AK, Niedzwiecki D, et al. Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104) J Transl Med. 2012;10:246. doi: 10.1186/1479-5876-10-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirschbaum MH, Synold T, Stein AS, et al. A phase 1 trial dose-escalation study of tipifarnib on a week-on, week-off schedule in relapsed, refractory or high-risk myeloid leukemia. Leukemia. 2011;25:1543–7. doi: 10.1038/leu.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23:5281–93. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- 35.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 36.Brown AP, Carlson TC, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59:671–9. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- 37.LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–37. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 38.Haura EB, Ricart AD, Larson TG, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16:2450–7. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- 39.Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 40.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robert C, Dummer R, Gutzmer R, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14:733–40. doi: 10.1016/S1470-2045(13)70237-7. [DOI] [PubMed] [Google Scholar]
- 43.Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311:2397–405. doi: 10.1001/jama.2014.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45•.Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–9. doi: 10.1016/S1470-2045(12)70269-3. Phase I study of trametinib, shows a small amount of activity in the NRAS population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zimmer L, Barlesi F, Martinez-Garcia M, et al. Phase I expansion and pharmacodynamic study of the oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS-RAF mutations. Clin Cancer Res. 2014;20(16):4251–61. doi: 10.1158/1078-0432.CCR-14-0341. [DOI] [PubMed] [Google Scholar]
- 47.Ribas A, Gonzalez R, Pavlick A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol. 2014;15:954–65. doi: 10.1016/S1470-2045(14)70301-8. [DOI] [PubMed] [Google Scholar]
- 48.Hatzivassiliou G, Haling JR, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature. 2013;501:232–6. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 49•.Kwong LN, Costello JC, Liu H, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–10. doi: 10.1038/nm.2941. Shows the pre-clinical rationale for co-targeting MEK and CDK4/6 in NRAS mutant melanoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–14. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res. 2013;19:5320–8. doi: 10.1158/1078-0432.CCR-13-0259. [DOI] [PubMed] [Google Scholar]
- 52.Young RJ, Waldeck K, Martin C, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27(4):590–600. doi: 10.1111/pcmr.12228. [DOI] [PubMed] [Google Scholar]
- 53.Posch C, Moslehi H, Feeney L, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci USA. 2013;110:4015–20. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jaiswal BS, Janakiraman V, Kljavin NM, et al. Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS One. 2009;4:e5717. doi: 10.1371/journal.pone.0005717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson DB, Smalley KS, Sosman JA. Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clin Cancer Res. 2014;20(16):4186–92. doi: 10.1158/1078-0432.CCR-13-3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rebecca VW, Alicea GM, Paraiso KH, et al. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res. 2014;27(6):1154–8. doi: 10.1111/pcmr.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conrad WH, Swift RD, Biechele TL, et al. Regulating the response to targeted MEK inhibition in melanoma: enhancing apoptosis in NRAS- and BRAF-mutant melanoma cells with Wnt/beta-catenin activation. Cell Cycle. 2012;11:3724–30. doi: 10.4161/cc.21645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Means-Powell JA, Adjei AA, Puzanov I, Dy GK, Goff LA, et al. Safety and efficacy of MET inhibitor tivantinib (ARQ 197) combined with sorafenib in patients (pts) with NRAS wild-type or mutant melanoma from a phase I study. J Clin Oncol. 2012;30:8519. Abstract. [Google Scholar]
- 59.Su Y, Vilgelm AE, Kelley MC, et al. RAF265 inhibits the growth of advanced human melanoma tumors. Clin Cancer Res. 2012;18:2184–98. doi: 10.1158/1078-0432.CCR-11-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharfman WH, Hodi FS, Lawrence DP, Flaherty KT, Amaravadi RK, et al. Results from the first-in-human (FIH) phase I study of the oral RAF inhibitor RAF265 administered daily to patients with advanced cutaneous melanoma. J Clin Oncol. 2011;29:8508. Abstract. [Google Scholar]
- 61•.Morris EJ, Jha S, Restaino CR, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–50. doi: 10.1158/2159-8290.CD-13-0070. Early characterization of ERK inhibitors which may have activity in both RAF and RAS mutant cancers. [DOI] [PubMed] [Google Scholar]
- 62.Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol. 2014;8:544–54. doi: 10.1016/j.molonc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jameson KL, Mazur PK, Zehnder AM, et al. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat Med. 2013;19:626–30. doi: 10.1038/nm.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eskandarpour M, Kiaii S, Zhu C, et al. Suppression of oncogenic NRAS by RNA interference induces apoptosis of human melanoma cells. Int J Cancer. 2005;115:65–73. doi: 10.1002/ijc.20873. [DOI] [PubMed] [Google Scholar]
- 65.Pecot CV, Calin GA, Coleman RL, et al. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11:59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davis ME, Zuckerman JE, Choi CH, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haarberg HE, Paraiso KH, Wood E, et al. Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Mol Cancer Ther. 2013;12:901–12. doi: 10.1158/1535-7163.MCT-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng Y, Lau E, Scortegagna M, et al. Inhibition of melanoma development in the Nras((Q61K))∷Ink4a(-/-) mouse model by the small molecule BI-69A11. Pigment Cell Melanoma Res. 2013;26:136–42. doi: 10.1111/pcmr.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–13. [PubMed] [Google Scholar]
- 70.Schwartzentruber DJ. Guidelines for the safe administration of high-dose interleukin-2. J Immunother. 2001;24:287–93. doi: 10.1097/00002371-200107000-00004. [DOI] [PubMed] [Google Scholar]
- 71.Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 72.Prieto PA, Yang JC, Sherry RM, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18:2039–47. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McDermott D, Lebbe C, Hodi FS, et al. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. Cancer Treat Rev. 2014;40(9):1056–64. doi: 10.1016/j.ctrv.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 74•.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. First large clinical trial to show activity of nivolumab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weber JS, Kudchadkar RR, Gibney GT, De Conti RC, Yu B, et al. Phase I/II trial of PD-1 antibody nivolumab with peptide vaccine in patients naive to or that failed ipilimumab. J Clin Oncol. 2013;31:9011. [Google Scholar]
- 76•.Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother. 2012;35:66–72. doi: 10.1097/CJI.0b013e3182372636. Suggests that NRAS mutations may correlate with response to immune therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson DB, Lovly CM, Flavin M, et al. NRAS mutation: a potential biomarker of clinical response to immune-based therapies in metastatic melanoma (MM) J Clin Oncol. 2013;31:9019. Abstract. [Google Scholar]
- 78.Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 79.Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]