

Abstract
Group C orthobunyaviruses are single-stranded RNA viruses found in both South and North America. Until very recently, and despite their status as important vector-borne human pathogens, no Group C whole genome sequences containing all three segments were available in public databases. Here we report a Group C orthobunyavirus, named El Huayo virus, isolated from a pool of Culex portesi mosquitoes captured near Iquitos, Peru. Although initial metagenomic analysis yielded only a handful of reads belonging to the genus Orthobunyavirus, single contig assemblies were generated for L, M, and S segments totaling over 200,000 reads (~0.5% of sample). Given the moderately high viremia in hamsters (>107 plaque-forming units/ml) and the propensity for Cx. portesi to feed on rodents, it is possible that El Huayo virus is maintained in nature in a Culex portesi/rodent cycle. El Huayo virus was found to be most similar to Peruvian Caraparu virus isolates and constitutes a novel subclade within Group C.
Author Summary
Arthropod-borne viruses remain a significant cause of human and domestic animal disease and new viruses are constantly being discovered. RNA virus discovery and assembly remains a challenge due to highly polymorphic genomes, current lack of breadth and depth of publicly available viral genomes, and confounding factors due to host sequence and sequencing biases. We describe the discovery and genome assembly of El Huayo virus, a group C orthobunyavirus isolated from a pool of Culex portesi mosquitoes captured near Iquitos, Peru, and named for the Jardin Botanicao Arboretum El Huayo near where the Cx. portesi from which the virus was isolated were captured. Although orthobunyaviruses are not commonly associated with human disease, Group C members are widespread in Central and South America and known to cause febrile illness. The discovery, and genome assembly, of El Huayo virus may help to explain numerous dengue-like illnesses where Aedes aegypti are not commonly found.
Introduction
The Orthobunyavirus genus comprises a diverse set of viral species, represented by multiple serogroups, including: Bunyamwera, California, Group C, and Simbu [1]. Their RNA genome includes three segments (Small [S], Medium [M], and Large [L]). The L segment encodes a RNA polymerase (RdRP); the M segment encodes two glycoproteins (Gc and Gn) in addition to a non-structural protein (NS); and the S segment encodes both a nucleocapsid protein (NP or N protein) and a non-structural protein (NSs) [2, 3]. Group C viruses were first identified in Brazil around 1950. Members of the California serogroup, including La Crosse, California encephalitis, Inkoo, and Tahyna viruses, are known to cause disease in humans [4–8]. Similarly, members of the Bunyamwera serogroup, including Cache Valley and Bunyamwera viruses [9, 10], Simbu serogroup, including Akabane, Iquitos, and Schmallenberg viruses [11–13], and Group C, including Caraparu, Itaya, Marituba, and Oriboca viruses [14–16], are known to cause disease in humans or domestic animals. Because infection with Group C viruses results in a non-differentiated febrile (dengue-like) illness and the lack of available diagnostic assays for these viruses, it has been difficult to associate these viruses with human disease. However, a study by Forshey et al. [17] identified 30 cases of human illness associated with Group C orthobunyaviruses, many of them Caraparu-like, and estimated that about 2.5% of febrile illnesses in the region were due to infection with an orthobunyavirus. The goal of our study was to sample, sequence and assemble a novel member of the genus Orthobunyavirus that had been isolated from a pool of Culex portesi mosquitoes captured in Peru in order to provide further genomic insights of this potentially disease-causing virus.
Materials and Methods
Ethics statement
The animal work was approved by the USAMRIID Institutional Animal Care and Use Committee. Research was conducted under an IACUC approved protocol in compliance with the Animal Welfare Act, PHS Policy, and other Federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011.
Virus isolation
Mosquitoes were captured at Aotus monkey-baited traps as part of an enzootic dengue study conducted in the vicinity of Iquitos, Peru [18]. Mosquitoes were identified to species, pooled (up to 25 specimens/pool), frozen on dry ice, and kept at -70°C until tested for infectious virus. Mosquito pools were triturated in 2 ml of diluent [10% heat-inactivated fetal bovine serum in Medium 199 with Earle's salts, NaHCO3 and penicillin (100 U/ml), streptomycin (100 μg/ml), and nystatin (100 U/ml)]. The suspensions were clarified by centrifugation (3,000 rpm for 10 min) and tested for virus by plaque assay on Vero (African green monkey kidney, ATCC CCL81) cell monolayers. A 0.l-ml aliquot of each original mosquito suspension and a 1:100 dilution of these suspensions were inoculated into duplicate wells of Vero cell monolayers. A second overlay, containing neutral red stain, was added 2 or 6 d later. If plaques were observed, the agar was removed, and the cells washed with fresh diluent and the resulting viral suspensions aliquoted into cryovials and frozen at –70°C. An aliquot of each suspension was inoculated onto confluent monolayers of Vero cells grown in a T-25 culture flask with 5 ml of liquid cell culture medium and observed daily for evidence of cytopathology. Cell cultures showing cytopathic effects were frozen at –70°C. Later, they were thawed, the suspension clarified by centrifugation at 3,000 rpm for 5 min, and then stored as 0.5-ml aliquots at –70°C for virus identification studies. The Vero passage 2 stock of one of these viruses, PE-M-0139 (isolated from a pool of 25 Cx. portesi mosquitoes captured in June 2002), was used in these studies.
Sequencing
Total RNA from the Vero passage 2-cell culture supernatant was reverse transcribed using random hexamers, and the resulting cDNA was amplified using multiple displacement amplification. A sequencing library was prepared using the Nextera XT protocol, and sequenced on an Illumina HiSeq 2500 instrument. An initial HiSeq run of 47,871,860 reads was supplemented with a second HiSeq run of 204,323,558 reads, yielding 252,195,418 total 100bp paired-end reads (NCBI BioProject PRJNA290192).
Metagenomic analysis
Initial analysis of the metagenomic sample involved a de novo assembly and taxonomic classification approach via MetAMOS [19], IDBA_UD [20], Kraken [21] and Krona [22]. However, initial inspection of the classified contigs and unassembled reads provided a convoluted picture of sample constituents, with only two reads classified as a member of the genus Orthobunyavirus (S1 Fig). LMAT [23](v1.2.3) was run on the dataset, only 5 reads were assigned to the genus Orthobunyavirus.
Quality trimming and adapter removal
The reads were adapter clipped and quality trimmed using ea-utils, part of MetAMOS [19] (fastq-mcf command, default parameters) using the Nextera XT adapter sequence CTGTCTCTTATACACATCT.
Targeted assembly and orthobunyavirus identification
To complement the de novo approach, putative orthobunyavirus reads were recruited to a diverse set of orthobunyavirus genomes via blastn [24](e-value 0.1, word size 7) using a custom orthobunyavirus database (Caraparu, Zungarococha, Oropouche viruses, containing L, M and S segments) downloaded from RefSeq [25]. The reference-based strategy filtered the nearly 50 million reads down to 234,280 paired-end reads (0.5% of the sample); blast did not report any read alignments to existing S segment sequences. Assembly of the recruited subset was performed with IDBA-UD (—pre_correction—num_threads 8—step 10); assembly was also attempted with SOAPdenovo [26] and Velvet-SC [27], but these produced fragmented assemblies.
The assembly was inspected for misassemblies by mapping all recruited reads back to the assembled contigs using Bowtie 2 [28]; a total of 121,901 reads mapped to the L segment (1762X avg. coverage) and 29,599 reads mapped to the M segment (617X avg. coverage). Coverage plots of the read mappings were visualized in IGV [29]. One junction in the assembled M segment was found to lack read support and was not consistent with related M segments (S4 Fig red arrow). A second round of recruitment was performed, including reads from the full assembly covering the region containing the erroneous deletion. The misassembled region was corrected after including these additional reads and rerunning IDBA_UD, resulting in consistent read support across both L and M segments.
Full dataset de novo assembly
In addition, a full de novo assembly of the 50 million reads was performed (IDBA-UD [20], default parameters), resulting in 340,327 total contigs. Contigs assembled with the full HiSeq dataset were screened against the Human genome (hg19) and Green Monkey (BioProject PRJNA215854) draft sequence to identify host sequence and misassembled contigs. The recruited assembly was compared to the IDBA-UD [20] assembly on the full dataset using NUCmer [30].
Sequence classification of S, M, and L segments
Orthobunyavirus (L and M) contigs were identified using both blastx and HMMER. An exhaustive search for the 900-1000bp S segment was performed, without success, using HMMER [31] (HMM profile http://pfam.xfam.org/family/PF00952, against all contigs using hmmpress and hmmscan).
Detection of S segment
As we failed to detect the S segment in the initial HiSeq run, we performed an additional sequencing run to facilitate the detection of the S segment of this novel Orthobunyavirus isolated from a pool of mosquitoes captured near Iquitos, Peru. The additional run provided over 200 million 100bp reads. All 200 million reads were first assembled with SOAPdenovo2 [32], and HMMER [31] (hmmsearch–E 1000 –cpu 4 HMM pfamseq) was used to align the 8,359,463 assembled contigs (translated to all 6-frames) against the nucleocaspid and non-structural protein HMMs. A single 608bp contig was shown to have significant hit (blastx, e-value = 2e-121) to Caraparu FMD0783 nucleocaspid protein (AGW82160.1), aligning at 89% aa identity across its entire length.
Detection of terminal hairpin sequences
Based on known conserved terminal hairpin sequences found in the UTR regions in Orthobunyavirus genomes [33, 34], we searched for terminal hairpin sequences (AGTAGTGTGCT) near both 5’and 3’ends in the L, M, and S segments (within the first 300 nt) using BLAST [24] (e-value = 10, word size = 7), to determine if the assembly was complete on both ends.
Sequence alignment and phylogenetic reconstruction
Amino acid (aa) sequences were aligned using MUSCLE [35] (default parameters), back translated to the original nucleotide sequences, edited to trim sequences from both ends that could not be reliably aligned, and then realigned with MUSCLE. Phylogenetic trees were subsequently reconstructed for both a global set of 101 orthobunyavirus genomes (L segment) and also on six Group C orthobunyavirus genomes (L and M segments), with FastTree2 [36]. Default parameters were used, and bootstrap support was determined by resampling the site likelihoods 1000 times and applying Shimodaira-Hasegawa test [37].
Ability of El Huayo virus to replicate in a vertebrate host
To determine the potential for El Huayo virus to replicate in a vertebrate host, we inoculated three Syrian hamsters intraperitoneally with 0.2 ml of a suspension containing 106.5 PFU/ml (105.8 PFU/hamster) of the Vero passage 2 stock of El Huayo virus. The hamsters were anesthetized daily and three mosquitoes were allowed to take a blood meal from each of the hamsters. These engorged mosquitoes were then triturated individually in 1 ml of diluent and tested for infectious virus by plaque assay on Vero cells as described above. Hamsters were observed for 21 days for signs of illness.
Results
El Huayo virus sequencing
The El Huayo assembly yielded three contigs (Table 1) with alignments to orthobunyavirus sequences, with best hits for all three segments to Peruvian Caraparu strains [38]. We were unable to identify the known terminal hairpin sequences in the UTR regions, suggesting incomplete assembly of the segments and/or increased divergence in the known conserved region. The de novo assembly of the L and M segments with the first HiSeq dataset was more fragmented than the recruitment approach (95 contigs vs. 2 contigs) with >95% of aligned de novo contigs identical to the recruited assembly. However, the recruitment approach significantly reduced depth of coverage (50-fold average reduction in coverage for both segments), with a more dramatic effect on the M segment (100-fold) compared to L segment (5-fold), due to the high level of divergence from the reference strain. Differences between the two assemblies were investigated further with dnadiff [39]; the full de novo assembly had multiple small insertions with respect to the reference-recruited assembly. These insertions were found to have high identity hits to Rhesus macaque and Green monkey genomes, yet were lacking from both Caraparu genomes and the reference-recruited assembly. Closer inspection of these insertions identified them as retroviral sequences and contained within likely misassembled contigs (S3 Fig).
Table 1. Assembly statistics.
‘all’ indicates assembly generated from the full HiSeq dataset; ‘rec’ indicates assembly generated from the recruited subset; Cov indicates the combined coverage of each segment for both HiSeq runs.
| Segment | # Contigs (all) | # Contigs (rec) | Cov | Length (nt) |
|---|---|---|---|---|
| L | 65 | 1 | 1792X | 6,746 bp |
| M | 30 | 1 | 667X | 4,721 bp |
| S | 1 | NA | 25X | 608 bp |
Phylogenetic analysis of the L segment suggests that this virus is closely related to Caraparu viruses comprising Group C orthobunyaviruses (Fig 1). We placed it within the Group C phylogeny, consistent with previously published phylogenetic relationships of orthobunyaviruses isolated from Peru [1]. El Huayo virus therefore appears to represent a novel, previously uncharacterized subclade of Group C viruses.
Fig 1. Maximum-likelihood phylogenetic placement (FastTree2) of El Huayo virus (Segment L).

Strains colored in blue represent Group C orthobunyavirus genomes. Nodes with low bootstrap support (less than 0.8, Shimodaira- Hasegawa) are colored red. The strain in bold and indicated by the arrow indicates El Huayo virus, the novel strain sequenced in this study.
Orthobunyaviruses are known to have high rates of reassortment [40], and although both L and M segments were most closely related to Caraparu virus (Fig 2, Table 2), there is increased polymorphism observed in M relative to L compared to other orthobunyavirus genomes. In addition, Caraparu virus strain FMD0783 was found to be the most similar (nt/aa) to both the M and S segments, while strain IQD5973 (from Iquitos, Peru) was the most similar (nt/aa) to segment L.
Fig 2. Group C phylogeny of orthobunyaviruses listed in Table 2, for both segment M and L.

S segment not shown due to partial assembly (608 nt out of 1000–1100 nt). Nodes with low bootstrap support (less than 0.8, Shimodaira- Hasegawa) are colored red.
Table 2. Nucleotide and amino acid similarity to the most closely related Group C orthobunyavirus genomes.
Values in parenthesis indicate % identity calculated on segment M aligned regions without the highly polymorphic region located between positions 1500–2500.
| Segment | Virus | Description | Location | Length (nt) | nt | aa |
|---|---|---|---|---|---|---|
| L | Brazoran | Houston | USA | 6911 bp | 64% | 81% |
| L | Caraparu | IQD5973 | Peru | 6794 bp | 70% | 84% |
| L | Caraparu | BeAn3994 | Brazil | 6855 bp | 69% | 84% |
| L | Caraparu | FVB0426 | Bolivia | 6850 bp | 69% | 83% |
| L | Caraparu | FMD0783 | Peru | 6849 bp | 69% | 84% |
| L | Marituba | BeAn15 | Brazil | 6894 bp | 69% | 83% |
| L | Zungarococha | IQE7620 | Peru | 6936 bp | 68% | 83% |
| M | Brazoran | Houston | USA | 4659 bp | 62% | 49% (70%) |
| M | Caraparu | IQD5973 | Peru | 4290 bp | 64% | 53% (73%) |
| M | Caraparu | BeAn3994 | Brazil | 4290 bp | 63% | 53% (70%) |
| M | Caraparu | FVB0426 | Bolivia | 4352 bp | 63% | 53% (72%) |
| M | Caraparu | FMD0783 | Peru | 4349 bp | 64% | 53% (74%) |
| M | Marituba | BeAn15 | Brazil | 4305 bp | 62% | 51% (71%) |
| M | Zungarococha | IQE7620 | Peru | 4538 bp | 62% | 52% (71%) |
| S | Brazoran | Houston | USA | 1672 bp | — | — |
| S | Caraparu | IQD5973 | Peru | 1090 bp | 80% | 87% |
| S | Caraparu | BeAn3994 | Brazil | 1048 bp | 82% | 89% |
| S | Caraparu | FVB0426 | Bolivia | 1102 bp | 81% | 87% |
| S | Caraparu | FMD0783 | Peru | 1068 bp | 82% | 89% |
Ability of El Huayo virus to replicate in a vertebrate host
El Huayo virus replicated to moderate titers in Syrian hamsters, with peak viremias of about 107.2 PFU/ml occurring on day 3 after infection (Fig 3, Table 3). None of the hamsters displayed signs of illness, and all were well 21 days after infection.
Fig 3. Viremia titers in Syrian hamsters by day after infection with El Huayo virus.

Error bars indicate standard error.
Table 3. Replication of El Huayo virus in Syrian hamsters.
| Hamster number | ||||
|---|---|---|---|---|
| Day after infection | 1 | 2 | 3 | Mean (Std. Dev.) |
| 1 | 5.8* | 5.7 | 3.8 | 5.3 (1.0) |
| 2 | 6.5 | 6.4 | 6.0 | 6.3 (0.5) |
| 3 | 7.3 | 7.2 | 7.1 | 7.2 (0.2) |
| 4 | 5.9 | 4.7 | 7.0 | 5.8 (1.1) |
*Log10 plaque-forming units of virus per ml of blood
Discussion
This is the first report of El Huayo virus, a novel member of the Group C orthobunyaviruses. Although rarely associated with human disease in nature, Group C viruses are known to cause febrile illness [13, 41]. The lack of reported cases is almost certainly due to a lack of diagnostic assays available for this group, and members of this group may be responsible for much of the dengue-like illnesses reported in areas of South and Central America where Aedes aegypti are not common [42]. In fact, Forshey et al. [17] estimated that about 2.5% of febrile illnesses in the region were due to infection with an orthobunyavirus, but were misdiagnosed as dengue.
Culex portesi, the species from which El Huayo virus was isolated, is a common species known to preferentially feed on rodents and marsupials [43, 44] and numerous viruses, including Caraparu-like viruses have been isolated from this species [45–47]. The ability of El Huayo virus to replicate to fairly high titer in hamsters indicates that like many other Group C virus, rodents may be involved in the natural maintenance cycle for this virus [48]. Thus, the natural cycle for El Huayo virus appears to be between Cx. portesi and rodents in the Amazon Basin region.
Because these viruses have a segmented genome, and because genetic reassortment has been demonstrated in this family/genus [49], the orthobunyaviruses are an ideal model for studying the evolution of novel viruses by genetic reassortment. How reassortment affects disease in humans and the ability of these viruses to replicate in vector species are key open questions.
In our initial comparative analysis, the best matches in our reference database shared ~60–80% nucleotide identity and 70–90% identity at the amino acid level with the (translated) novel S, M and L segment sequences, respectively. Given the low sequence identity of segment M relative to segment L, segment M might represent a novel reassortment; the region from 1500-2500bp contains a dramatic reduction in similarity to all known segment M strains available in RefSeq.
High divergence relative to existing genomes is a challenge for homology detection methods; sensitivity must be increased to detect divergent matches, but the increase in sensitivity also leads to potential misclassifications. Sensitive profile alignment methods based on hidden Markov models can detect protein domain signatures in cases where extreme divergence makes other methods infeasible [18], such as in the case of the highly divergent S segment recently reported for Brazoran virus [26] which was double the size of previously published orthobunyavirus S segments. Its S segment contained no known homology to existing segment S proteins; however, similar to what we report here, it did have conserved orthobunyavirus domains that were detected via InterProScan [27]. While insufficient sequencing depth in our initial HiSeq run prohibited detection of the S segment, adding another HiSeq run allowed for the detection of this small viral segment. This result highlights that lower abundance sequences in environment samples may often be missed, and sequencing depth is still an important tool for uncovering low abundance novel viruses from metagenomic samples.
Based on amino acid sequence similarity, the orthobunyavirus genome of El Huayo virus reported in this study is most closely related to Caraparu virus Peruvian strains IQD5973 and FMD0783 [38], both recently deposited in GenBank. This recent growth in publicly available Group C orthobunyavirus genomes enabled the reliable placement of our novel strain within the Group C serogroup. Prior to Huang et al. 2014, there were no complete genomes (including all three segments) from within Group C. Lack of complete genomic sequences of serogroups of interest can lead to misclassification or misidentification, evidenced by a recent study that reported that a collection of Group C genomes likely require further validation [38]. This highlights the importance of efforts to populate reference databases. There exists a vast underrepresentation of viral diversity for various clades, and of particular interest to this study, there are only a small number of South American orthobunyavirus sequences. Continuing efforts are required to fill out viral reference databases to ensure reliable identification and characterization of novel Bunyaviridae genomes.
An additional confounding factor for novel virus identification and assembly is host endogenous retroviral elements [50] (S2 Fig and S3 Fig). Aggressive assembly strategies can result in chimeric host-plus-virus assemblies in which sequence shared by both virus and host results in false joins between the two genomes; specifically, retroviral elements integrated into host genomes. We have shown that a recruitment-based strategy, even at relatively high levels of amino acid divergence, can prove useful for avoiding co-assembly of host and target virus. However, this approach requires the presence of reference strains in the database and is prone to under-recruitment of reads in highly polymorphic regions. In summary, while advances in sequencing technology allow for the discovery of novel viruses present at low abundances in a sample, care must be taken to properly address confounding factors.
Supporting Information
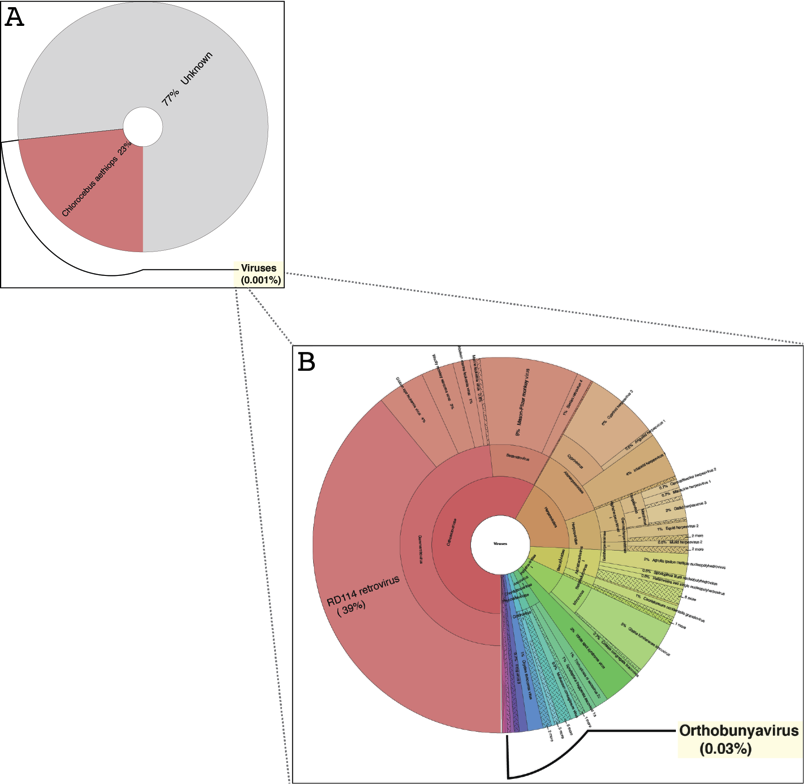
A. Krona visualization of Kraken-based classification of entire sample. Chlorocebus aethiops (Green Monkey) is the host sequence used for cell cultures; Unknown indicates 77% of the reads were unable to be classified. There were 4770 total reads (0.01%) classified as Viruses (including RNA Viruses). B. Krona visualization of Kraken-based classification of putative viral reads. Of the 4770 viral reads, only 0.03% were assigned to the genus Orthobunyavirus, representing two reads out of the nearly 50 million total (0.000003%).
(PNG)
{kind=link}
Assembly on the full dataset resulted in a handful of misassembled contigs that incorrectly joined orthobunyavirus segment L with host retrovirus elements. The horizontal black line at the top represents a region from the Green Monkey genome, the red line indicates a shared k-mer (20) between Segment L and the endogenous retroviral element, the green line represents the retrovirus and blue lines represent segment L.
(PNG)
{kind=link}
The read pileup on the right hand side of the figure corresponds to a high coverage assembly of a small region of segment L, while the read pileup at reduced coverage found on the left-hand side corresponds to Green Monkey chromosome 9 (aligns across entire length at 99% identity).
(PNG)
{kind=link}
The red arrow indicates the false join at positions 995-1005bp in the assembly, lacking clear read support.
(PNG)
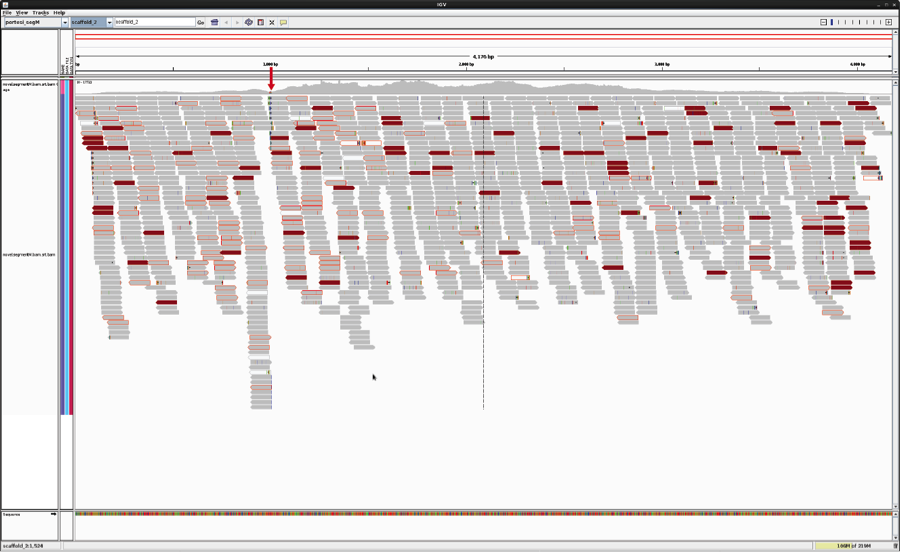{kind=link}
Acknowledgments
We thank the following people from the Peruvian Primatological Project, Dr. Enrique Montoya (for his help in setting up the field study that allowed for the isolation of El Huayo virus), Alfredo Cetraro (for daily care of the animals in that study), and Arnulfo Romaina for his technical assistance. We thank Carlos Tong, Dario Ramirez, and Clever Donayre (U.S. Naval Medical Research Unit No. 6, Callao, Peru) for their assistance in identifying the mosquitoes, and Miguel Vasquez (U.S. Naval Medical Research Unit No. 6) for his support in the establishment of the project as well as on the field collections. We also thank David Dohm, USAMRIID, for his assistance in isolating the viruses from the mosquitoes collected in Peru. We also would like to thank Brian Walenz and Sergey Koren for helpful discussions of the segment L and M assemblies. The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the DHS or S&T. In no event shall DHS, NBACC, S&T or Battelle National Biodefense Institute have any responsibility or liability for any use, misuse, inability to use, or reliance upon the information contained herein. DHS does not endorse any products or commercial services mentioned in this publication.
Data Availability
All relevant data are within the paper and its Supporting Information files. All associated WGS reads and assemblies files are available from NCBI under BioProject PRJNA290192.
Funding Statement
This work was funded under Agreement No. HSHQDC-07-C-00020 awarded by the Department of Homeland Security (DHS) Science and Technology Directorate (S&T) for the management and operation of the National Biodefense Analysis and Countermeasures Center (NBACC), a Federally Funded Research and Development Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Mores CN, Turell MJ, Dyer J, Rossi CA. Phylogenetic relationships among orthobunyaviruses isolated from mosquitoes captured in Peru. Vector borne and zoonotic diseases. 2009;9(1):25–32. 10.1089/vbz.2008.0030 [DOI] [PubMed] [Google Scholar]
- 2.Eifan S, Schnettler E, Dietrich I, Kohl A, Blomstrom AL. Non-structural proteins of arthropod-borne bunyaviruses: roles and functions. Viruses. 2013;5(10):2447–68. 10.3390/v5102447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter CT, Barr JN. Recent advances in the molecular and cellular biology of bunyaviruses. The Journal of general virology. 2011;92(Pt 11):2467–84. 10.1099/vir.0.035105-0 [DOI] [PubMed] [Google Scholar]
- 4.Pekosz A, Griot C, Stillmock K, Nathanson N, Gonzalez-Scarano F. Protection from La Crosse virus encephalitis with recombinant glycoproteins: role of neutralizing anti-G1 antibodies. Journal of virology. 1995;69(6):3475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore CG, Mitchell CJ. Aedes albopictus in the United States: ten-year presence and public health implications. Emerging infectious diseases. 1997;3(3):329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Francy DB, Karabatsos N, Wesson DM, Moore CG Jr., Lazuick JS, Niebylski ML, et al. A new arbovirus from Aedes albopictus, an Asian mosquito established in the United States. Science. 1990;250(4988):1738–40. [DOI] [PubMed] [Google Scholar]
- 7.Mitchell CJ, Niebylski ML, Smith GC, Karabatsos N, Martin D, Mutebi JP, et al. Isolation of eastern equine encephalitis virus from Aedes albopictus in Florida. Science. 1992;257(5069):526–7. [DOI] [PubMed] [Google Scholar]
- 8.Karabatsos N. Supplement to International Catalogue of Arboviruses including certain other viruses of vertebrates. The American journal of tropical medicine and hygiene. 1978;27(2 Pt 2 Suppl):372–440. [DOI] [PubMed] [Google Scholar]
- 9.Lambert AJ, Lanciotti RS. Molecular characterization of medically important viruses of the genus Orthobunyavirus. The Journal of general virology. 2008;89(Pt 10):2580–5. 10.1099/vir.0.2008/002253-0 [DOI] [PubMed] [Google Scholar]
- 10.Campbell GL, Mataczynski JD, Reisdorf ES, Powell JW, Martin DA, Lambert AJ, et al. Second human case of Cache Valley virus disease. Emerging infectious diseases. 2006;12(5):854–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Afonso A, Abrahantes JC, Conraths F, Veldhuis A, Elbers A, Roberts H, et al. The Schmallenberg virus epidemic in Europe-2011-2013. Preventive veterinary medicine. 2014. [DOI] [PubMed] [Google Scholar]
- 12.Elbers AR, Meiswinkel R, van Weezep E, Sloet van Oldruitenborgh-Oosterbaan MM, Kooi EA. Schmallenberg virus in Culicoides spp. biting midges, the Netherlands, 2011. Emerging infectious diseases. 2013;19(1):106–9. 10.3201/eid1901.121054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguilar PV, Barrett AD, Saeed MF, Watts DM, Russell K, Guevara C, et al. Iquitos virus: a novel reassortant Orthobunyavirus associated with human illness in Peru. PLoS neglected tropical diseases. 2011;5(9):e1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iversson LB, Travassos da Rosa AP, Coimbra TL, Ferreira IB, Nassar Eda S. Human disease in Ribeira Valley, Brazil caused by Caraparu, a group C arbovirus—report of a case. Revista do Instituto de Medicina Tropical de Sao Paulo. 1987;29(2):112–7. [DOI] [PubMed] [Google Scholar]
- 15.da Rosa APAT, Vasconcelos PFC, da Rosa JFSTX. An overview of arbovirology in Brazil and neighbouring countries: Citeseer; 1998.
- 16.Hontz RD, Guevara C, Halsey ES, Silvas J, Santiago FW, Widen SG, et al. Itaya virus, a Novel Orthobunyavirus Associated with Human Febrile Illness, Peru. Emerging infectious diseases. 2015;21(5):781–8. 10.3201/eid2105.141368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forshey BM, Guevara C, Laguna-Torres VA, Cespedes M, Vargas J, Gianella A, et al. Arboviral etiologies of acute febrile illnesses in Western South America, 2000–2007. PLoS neglected tropical diseases. 2010;4(8):e787 10.1371/journal.pntd.0000787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrews ES, Schoeler GB, Gozalo AS, Carbajal F, Lopez-Sifuentes V, Turell MJ. Species Diversity, Seasonal, and Spatial Distribution of Mosquitoes (Diptera: Culicidae) Captured in Aotus Monkey-Baited Traps in a Forested Site Near Iquitos, Peru. Journal of Medical Entomology. 2014;51(6):1127–35. 10.1603/ME14058 [DOI] [PubMed] [Google Scholar]
- 19.Treangen TJ, Koren S, Sommer DD, Liu B, Astrovskaya I, Ondov B, et al. MetAMOS: a modular and open source metagenomic assembly and analysis pipeline. Genome biology. 2013;14(1):R2 10.1186/gb-2013-14-1-r2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420–8. 10.1093/bioinformatics/bts174 [DOI] [PubMed] [Google Scholar]
- 21.Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome biology. 2014;15(3):R46 10.1186/gb-2014-15-3-r46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC bioinformatics. 2011;12:385 10.1186/1471-2105-12-385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ames SK, Hysom DA, Gardner SN, Lloyd GS, Gokhale MB, Allen JE. Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics. 2013;29(18):2253–60. 10.1093/bioinformatics/btt389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42(Database issue):D756–63. 10.1093/nar/gkt1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li R, Fan W, Tian G, Zhu H, He L, Cai J, et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463(7279):311–7. 10.1038/nature08696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chitsaz H, Yee-Greenbaum JL, Tesler G, Lombardo MJ, Dupont CL, Badger JH, et al. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nature biotechnology. 2011;29(10):915–21. 10.1038/nbt.1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9(4):357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14(2):178–92. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delcher AL, Phillippy A, Carlton J, Salzberg SL. MUMmer: comparative applications Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Research. 2002;30:2478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eddy SR. Accelerated Profile HMM Searches. PLoS Computational Biology. 2011;7(10):e1002195–e. 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1(1):18 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barr JN, Wertz GW. Role of the conserved nucleotide mismatch within 3'- and 5'-terminal regions of Bunyamwera virus in signaling transcription. Journal of virology. 2005;79(6):3586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowen AC, Elliott RM. Mutational analyses of the nonconserved sequences in the Bunyamwera Orthobunyavirus S segment untranslated regions. Journal of virology. 2005;79(20):12861–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32(5):1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PloS one. 2010;5(3):e9490 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Molecular biology and evolution. 1999;16:1114–6%@ 0737–4038. [Google Scholar]
- 38.Forshey BM, Castillo RM, Hang J. Group C orthobunyavirus genomic sequences require validation. Journal of virology. 2014;88(5):3052–3. 10.1128/JVI.03295-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillippy AM, Schatz MC, Pop M. Genome assembly forensics: finding the elusive mis-assembly. Genome biology. 2008;9(3):R55–R. 10.1186/gb-2008-9-3-r55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yanase T, Kato T, Aizawa M, Shuto Y, Shirafuji H, Yamakawa M, et al. Genetic reassortment between Sathuperi and Shamonda viruses of the genus Orthobunyavirus in nature: implications for their genetic relationship to Schmallenberg virus. Archives of virology. 2012;157(8):1611–6. 10.1007/s00705-012-1341-8 [DOI] [PubMed] [Google Scholar]
- 41.Allard MW, Luo Y, Strain E, Li C, Keys CE, Son I, et al. High resolution clustering of Salmonella enterica serovar Montevideo strains using a next-generation sequencing approach. BMC genomics. 2012;13:32 10.1186/1471-2164-13-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.FP P APA dR. Group C bunyaviral fevers GM B, editor. Boca Raton: CRC Press; 1994. [Google Scholar]
- 43.Sirivanakarn S, Degallier N. Redescription of Culex (Melanoconion) portesi Senevet & Abonnenc, 1941, with notes on synonymy (Diptera: Culicidae). DTIC Document; 1981.
- 44.Davies JB. Attraction of Culex portesi Senevet & Abonnenc and Culex taeniopus Dyar & Knab (Diptera: Culicidae) to 20 animal species exposed in a Trinidad forest. I. Baits ranked by numbers of mosquitoes caught and engorged. Bulletin of Entomological Research. 1978;68(04):707–19%@ 1475–2670. [Google Scholar]
- 45.Auguste AJ, Adams AP, Arrigo NC, Martinez R, Travassos da Rosa AP, Adesiyun AA, et al. Isolation and characterization of sylvatic mosquito-borne viruses in Trinidad: enzootic transmission and a new potential vector of Mucambo virus. The American journal of tropical medicine and hygiene. 2010;83(6):1262–5. 10.4269/ajtmh.2010.10-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Confalonieri UE, Costa Neto C. Diversity of mosquito vectors (Diptera: culicidae) in caxiuana, para, Brazil. Interdisciplinary perspectives on infectious diseases. 2012;2012:741273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haas RA, Arron-Leeuwin AE. Arboviruses isolated from mosquitos and man in Surinam. Tropical and geographical medicine. 1975;27(4):409–12. [PubMed] [Google Scholar]
- 48.Monath TP. The arboviruses: epidemiology and ecology Volume V: CRC Press, Inc.; 1989. [Google Scholar]
- 49.Turell MJ, Saluzzo JF, Tammariello RF, Smith JF. Generation and transmission of Rift Valley fever viral reassortants by the mosquito Culex pipiens. The Journal of general virology. 1990;71 (Pt 10):2307–12. [DOI] [PubMed] [Google Scholar]
- 50.Leib-Mosch C, Brack-Werner R, Werner T, Bachmann M, Faff O, Erfle V, et al. Endogenous retroviral elements in human DNA. Cancer research. 1990;50(17 Suppl):5636S–42S. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Krona visualization of Kraken-based classification of entire sample. Chlorocebus aethiops (Green Monkey) is the host sequence used for cell cultures; Unknown indicates 77% of the reads were unable to be classified. There were 4770 total reads (0.01%) classified as Viruses (including RNA Viruses). B. Krona visualization of Kraken-based classification of putative viral reads. Of the 4770 viral reads, only 0.03% were assigned to the genus Orthobunyavirus, representing two reads out of the nearly 50 million total (0.000003%).
(PNG)
Assembly on the full dataset resulted in a handful of misassembled contigs that incorrectly joined orthobunyavirus segment L with host retrovirus elements. The horizontal black line at the top represents a region from the Green Monkey genome, the red line indicates a shared k-mer (20) between Segment L and the endogenous retroviral element, the green line represents the retrovirus and blue lines represent segment L.
(PNG)
The read pileup on the right hand side of the figure corresponds to a high coverage assembly of a small region of segment L, while the read pileup at reduced coverage found on the left-hand side corresponds to Green Monkey chromosome 9 (aligns across entire length at 99% identity).
(PNG)
The red arrow indicates the false join at positions 995-1005bp in the assembly, lacking clear read support.
(PNG)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. All associated WGS reads and assemblies files are available from NCBI under BioProject PRJNA290192.