

Letter to Editor
Chromosomal rearrangements of the Mixed Lineage Leukemia (MLL) gene, which result in expression of MLL fusion proteins, occur in 5–10% of both AML and ALL.1 MLL leukemia patients have very unfavorable prognoses1, 2 emphasizing the need for new therapies. The MLL fusion proteins retain the N-terminal portion of MLL fused with one of more than 70 different fusion partners, classified based on their cellular localization as nuclear and cytoplasmic proteins.3, 4 It has been proposed that molecular mechanism of transformation utilized by different MLL fusions is dependent on the type of fusion partner.4 The MLL fusions harboring nuclear fusion partners (e.g. AF4, AF9, ENL, AF10, ELL) are frequently associated with transcriptional elongation complexes leading to transcriptional activation of target genes, while cytoplasmic fusion partners of MLL (e.g. AF6, AF1p, GAS7) contain dimerization domains required for transformation.4, 5 The high diversity of MLL fusion partners raises a question whether it is possible to develop a general therapeutic strategy to block the oncogenic activity of MLL fusion proteins in a fusion partner independent manner.
The protein menin was shown to directly interact with the N-terminal fragment of MLL retained in all MLL fusion proteins, and this interaction is critical for the MLL-fusion protein mediated transformation.6, 7 Therefore, blocking the menin-MLL interaction could inhibit oncogenic activity of all MLL fusion proteins, providing a rationale for a general therapeutic strategy for MLL leukemia patients. To address this important question, we employed MI-2-2, a potent small molecule inhibitor of the menin-MLL interaction (IC50 = 46 nM), which we have previously shown to be effective in MLL leukemia cells harboring MLL-AF9 and MLL-AF4 fusion proteins.8
This study was performed to assess whether pharmacologic inhibition of the menin-MLL interaction can block oncogenic activity of a broader panel of MLL fusion proteins harboring different fusion partners and whether this may represent a general therapeutic approach for MLL leukemia. For this purpose, we generated cell lines by transforming murine bone marrow cells (BMC) with six various MLL fusions representing the major types of MLL translocations: MLL-AF9 and MLL-ENL, the most frequent fusions involving nuclear partners; MLL-CBP, representing fusion with a nuclear protein that acts through a mechanism involving histone acetyltransferase activity4; MLL-AF6, one of the most frequent cytoplasmic fusion partner, which dimerizes and may trigger RAS activation9; MLL-GAS7 and MLL-AF1P, both harbor cytoplasmic fusion partners that function through dimerization-dependent mechanisms.5
First, we found that treatment with MI-2-2 resulted in strong growth inhibition of all MLL fusion transformed cell lines, but not in control cell lines transformed with Hoxa9-Meis1 (HM-2) or E2A-HLF oncogenes (Figure 1a and Supplementary Figure 1. We then evaluated whether MI-2-2 is capable to induce differentiation of BMCs transformed with different MLL fusions. Indeed, treatment with MI-2-2 led to significant changes in morphology of these cells, consistent with monocytic differentiation (Figure 1b and Supplementary Figure 2). This was associated with a significant decrease in expression level of c-kit (CD117), a cell surface marker of hematopoietic progenitor cells (Figure 1b). In contrast, treatment of E2A-HLF and Hoxa9/Meis1 transformed cells did not affect neither morphology nor c-kit expression (Figure 2c and Supplementary Figure 2).
Figure 1.
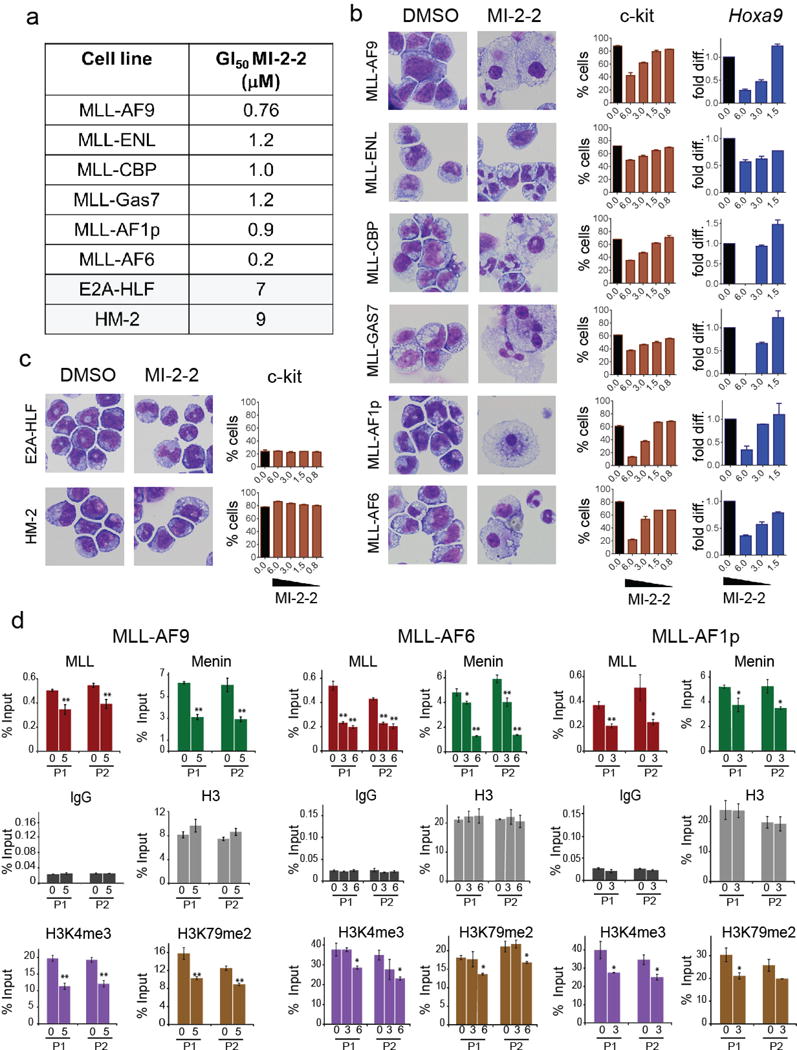
Cellular activity of MI-2-2 menin-MLL inhibitor in murine bone marrow cells transformed with various MLL fusions or control oncogenes. (a) Cell growth inhibition in MLL leukemia and control (transformed with E2A-HLF or Hoxa9/Meis1, HM-2) cell lines upon 10 days of treatment with MI-2-2. GI50 values were assessed based on cell counts performed for viable cells using trypan blue staining upon treatment with various concentrations of MI-2-2, (mean ± SD, n = 2). (b) Treatment with MI-2-2 induces differentiation, reduces c-kit and Hoxa9 expression in MLL leukemia cell lines. Left panel: Wright-Giemsa stained cytospins of MLL leukemia cells harboring different MLL fusions upon 10 days of treatment with 6 μM of MI-2-2 or DMSO. Middle panel: percentage of c-kit positive cells upon 10 days of treatment with DMSO (black) or various concentrations of MI-2-2 (brown) in MLL leukemia cells as determined by c-kit antibody staining (Biolegend, 105812) and flow cytometry analysis (cell lines the same as in the cytospin pictures). Mean values from duplicate samples ± SD are shown. MI-2-2 doses are shown in μM. Right panel: downregulation of Hoxa9 expression in various MLL leukemia cell lines upon treatment with MI-2-2 for 6 days. Total RNA was isolated and gene transcript levels were determined by real-time qRT-PCR. Transcript levels were normalized to β-actin and relative expression levels were calculated for each dose of the compound (blue) relative to DMSO (black). MI-2-2 doses are shown in μM. Mean values from duplicate samples ± s.d. are shown relative to DMSO samples. (c) Treatment with MI-2-2 does not induce differentiation or c-kit expression in control cell lines; c-kit is presented as a percentage of viable cells (mean ± SD, n = 2). Experimental conditions the same as in (b), Wright-Giemsa stained cytospins are shown for cell lines treated for 10 days with 6 μM of MI-2-2. (d) Chromatin immunoprecipitation (ChiP) experiment performed in MLL-AF9, MLL-AF6 and MLL-AF1p transformed murine bone marrow cells upon 3 (MLL-AF9) or 2 (MLL-AF6 and MLL-AF1p) days of treatment with MI-2-2 (MI-2-2 concentrations are provided in μM) or DMSO (mean ± SD, n = 2). ChIP experiment was performed using the manufacturer’s protocol (Millipore-Magna ChIP A/G). Antibodies against menin (Bethyl, A300-105A), MLL (Milipore, 05-764), histone H3 (Abcam, ab1791), H3K79 dimethylation (Abcam, ab3594), H3K4 trimethylation (Abcam, ab8580) and IgG (Milipore, PP64B) were used. Real-time PCR was performed on the precipitated DNAs with TaqMan fluorescent labeling with primers and qPCR probes (Hoxa9 primer probe set 1, P1; and Hoxa9 primer probe set 2, P2; primers sequences as described previously15). The p-values were calculated with 2-way ANOVA, * p < 0.05, ** p < 0.01. No statistical method was used to predetermine sample size.
Figure 2.
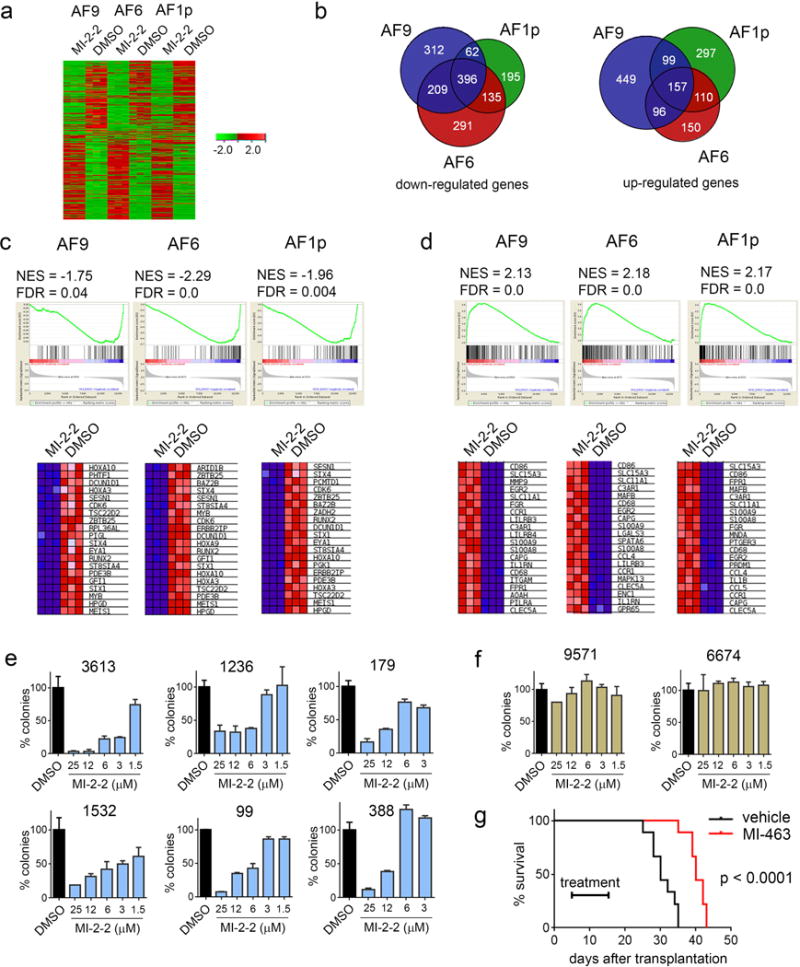
Analysis of gene expression in MLL-AF9, MLL-AF1p and MLL-AF6 cell lines upon treatment with MI-2-2. Cells were treated using DMSO or 6 μM MI-2-2 (in triplicates) for 6 days and gene expression was analyzed using RNA-seq. RNA was isolated from cells, amplified, and quality was assessed using the TapeStation (Agilent). Samples with RINs (RNA Integrity Numbers) of 8 or greater were prepped using the Illumina TruSeq mRNA kit (Illumina). RNA was converted to mRNA using a polyA purification. cDNA library was created using reverse transcriptase, barcoded and sequenced using 4 samples per lane on a HiSeq 2000 (Illumina) in High Output mode. Sequenced reads were aligned to mouse reference genome using Bowtie and Tophat (version 2.0.3). Differential gene expression analysis was done using program Cuffdiff. (a) Hitmap showing genes (2958 in total) that experience more than 2-fold change and adjusted p values < 0.05 in at least one of the three cell lines. Green and red colors correspond to down and up-regulates genes, respectively. (b) Overlap between genes down- and upregulated upon treatment of three MLL fusion transformed cell lines with MI-2-2. (c) GSEA analysis of genes downregulated upon treatment with MI-2-2 demonstrating enrichment to MLL-AF9 target genes.12 Gene set enrichment analysis (GSEA) was performed using www.broadinstitute.org/gsea software. NES – normalized enrichment score; FDR – false discovery rate. The heat maps show genes comprising the leading edge of the GESA plot. (d) GSEA analysis of genes upregulated upon treatment of MLL leukemia cell lines with MI-2-2 demonstrating enrichment of genes underexpressed in leukemic stem cells, defined as CD34+CD38− cells.13 (e, f) Colony counts in the methylcellulose colony forming assay performed upon 14 days of treatment with MI-2-2 in the primary patient samples with MLL leukemia (e) and in control AML primary patient samples without MLL translocations (f). Colony counts are normalized to DMSO treated samples (mean ± SD for duplicate samples are shown). MLL leukemia patients samples: 3613 (MLL-AF9), 1236 (MLL-AF9), 179 (MLL-ENL), 1532 (MLL-p300), 99 (MLL-AF6), 388 (MLL-AF6). Detailed description of the patient samples is provided in Supplementary Table 2. Patient-derived AML samples were collected in accordance with the guidelines and approval of institutional review boards at University of Pennsylvania Hospital and University of Michigan Hospital and with informed consent of each patient. (g) Kaplan-Meier survival curves of vehicle and MI-463 (45 mg/kg, b.i.d., p.o.) treated C57BL/6 female mice (6–8 weeks) transplanted with 1 × 105 syngeneic MLL-AF6 leukemic cells isolated from primary recipient mice (n = 9 mice per group). Time of treatment is indicated on the graph. P value (p < 0.0001) was calculated using Log-rank (Mantel-Cox) test. No statistical method was used to predetermine sample size. No specific method was used for randomization, and no exclusion of animals was applied in data analysis. The investigators were not blinded to the group allocation and when assessing the outcome. Animal experiments were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine (ULAM).
Overexpression of HOXA genes represents a hallmark of MLL-rearranged leukemias,10 and menin is required for Hoxa9 and Meis1 expression.6 Indeed, treatment with MI-2-2 resulted in a marked downregulation of Hoxa9 and Meis1expression in cells expressing different MLL fusions (Figure 1b and Supplementary Figure 3), besides MLL-CBP transformed cells where pronounced cell death was observed. The effect of MI-2-2 was stronger on Meis1 expression and consistent between all MLL leukemia cell lines (Supplementary Figure 3). Importantly, phenotypic and gene expression changes observed in a panel of cells expressing different MLL fusions were consistent with inhibition of menin interactions with various MLL fusion proteins induced by MI-2-2 (Supplementary Figure 4).8
To better understand the mechanism how MI-2-2 blocks oncogenic activity of different MLL fusions we selected one cell line transformed with nuclear fusion partner (MLL-AF9) and two with cytoplasmic fusion partners (MLL-AF6 and MLL-AF1p). It was proposed that Hoxa9 activation by MLL fusions encompassing nuclear fusion partners involves assembly of multi-protein complexes, which results in epigenetic reprograming by increasing the H3K79 and H3K4 methylation.4 On the other hand, the mechanism of Hoxa9 activation by MLL fusions involving cytoplasmic fusion partners is much less understood.4 To characterize how MI-2-2 downregulates Hoxa9 expression, we performed co-immunoprecipitation (ChiP) experiments. First, we found that treatment with MI-2-2 strongly reduced binding of both menin and MLL fusions to the Hoxa9 loci in all three MLL leukemia cell lines (Figure 1d and Supplementary Figure 5). In addition, MI-2-2 significantly reduced methylation level of H3K79 and H3K4 at Hoxa9 promoter, consistent with decreased transcriptional activation of Hoxa9 (Figure 1d and Supplementary Figure 5). These data suggest that MI-2-2 down-regulates Hoxa9 according to a similar mechanism despite different complexes formed by these MLL fusions.4
To explore genome-wide transcriptome analysis upon inhibition of the menin-MLL interaction we performed RNA-seq analysis in MLL-AF9, MLL-AF6 and MLL-AF1p transformed cells. First, we found that treatment with MI-2-2 results in a very similar pattern of gene expression changes in these cell lines (Figure 2a). The expression level of the downstream targets of MLL fusion proteins, such as Hoxa9, Meis1, Hoxa10, Hoxa5 and Hoxa11, was significantly reduced in all cell lines upon treatment with MI-2-2 (Supplementary Table 1). In addition, other genes implicated in MLL leukemias, CDK6, Eya1, Six1 and Six4,11 were also strongly downregulated. Gene Set Enrichment Analysis (GSEA) demonstrated strong enrichment for direct target genes of MLL-AF912 in all cell lines (Figure 2c). Overall, these results demonstrate that MI-2-2 is reversing gene expression signature in MLL rearranged cells in a fusion partner independent manner.
One of the most pronounced phenotypic changes observed upon treatment with MI-2-2 is a marked differentiation of MLL leukemia cells (Figure 1b). Consistent with this finding, MI-2-2 up-regulates genes associated with differentiation (e.g. CD14, MNDA), Supplementary Table 1, and GSEA demonstrates strong enrichment for genes repressed in leukemia stem cells (CD34+CD38− cells), Figure 2d.13 These effects were observed in all MLL leukemia cell lines tested, supporting a similar mechanism of action for the menin-MLL inhibitor despite different MLL fusion proteins expressed.
Interestingly, only a subset of genes affected by MI-2-2 (20–30% of upregulated genes and 38–50% of downregulated genes) is identical among the three cell lines tested (Figure 2b), despite the fact these cell lines were created from the same cellular background. These results suggest that transformation with different MLL fusions, such as MLL-AF9, MLL-AF6 or MLL-AF1p, leads to substantial differences between these cells. Nevertheless, all these cell lines overexpress subset of genes that are downstream targets of MLL fusions and are sensitive to inhibition of the menin-MLL interaction by small molecules.
To further support general applicability of the menin-MLL inhibitors, we tested the effect of MI-2-2 in primary samples isolated from MLL leukemia patients harboring different MLL translocations, including nuclear and cytoplasmic fusion partners. In the clonogenic assay, treatment with MI-2-2 substantially reduced the number and size of colonies in all samples harboring various MLL fusions, but not in the control AML samples (Figure 2e, 2f, Supplementary Figure 6 and Supplementary Table 2). Finally, we assessed the in vivo efficacy of the menin-MLL inhibitor MI-463, an optimized analogue of MI-2-2 with substantially improved pharmacokinetic profile,14 using a bone marrow transplantation model of murine MLL-AF6 leukemia. Treatment with MI-463 greatly improved survival (~35% survival benefit) of MLL-AF6 mice (Figure 2g), consistent with the effect we observed for this compound in MLL-AF9 leukemia mice.14
In conclusion, our studies demonstrate that small molecule inhibition of the menin-MLL interaction leads to growth arrest, differentiation and downregulation of MLL fusion target genes in leukemia cells transformed with various MLL fusions, and affects colony formation in leukemia patient samples harboring different MLL translocations. Furthermore, menin-MLL inhibitor blocks binding of both menin and MLL and/or MLL fusions to Hoxa9 loci, consistent with co-localization of these proteins to the target genes,6 and decreases H3K4 and H3K79 methylation in cell lines transformed with MLL-AF9, MLL-AF6 and MLL-AF1p, despite different protein complexes formed by these MLL fusions.4 Genome-wide analysis of gene expression revealed that MI-2-2 reverses MLL-rearranged gene signature and induced expression of genes associated with differentiation in cells harboring various MLL fusion proteins. Taken together, the menin-MLL inhibitor is capable to reverse oncogenic activity of different MLL fusions according to a mechanism that is independent on the fusion partner. Such activity remains in agreement with menin binding motif retained in all MLL fusion proteins. This is further validated by the studies with our new menin-MLL inhibitor with improved pharmacokinetics,14 which demonstrate that blocking the menin-MLL interaction markedly improves survival in animal model of MLL-AF6 leukemia. Altogether, these studies demonstrate that pharmacologic inhibition of the menin-MLL interaction has a strong potential to represent a general therapeutic approach for MLL leukemias regardless of MLL translocation, a highly desired effect due to a large variability of MLL fusion partners. These findings further support advancement of menin-MLL inhibitors as a targeted therapy for the MLL leukemia patients.
Acknowledgments
This work was supported by the National Institute of Health grant R01 (1R01CA160467) to J.G., a Leukemia and Lymphoma Society (LLS) TRP grant (6116-12) and the LLS Therapy Acceleration Program to J.G., LLS Scholar (1215-14) to J.G., American Cancer Society grants (RSG-11-082-01-DMC to T.C. and RSG-13-130-01-CDD to J.G.), and the Department of Pathology, University of Michigan.
Footnotes
Conflict of Interest
Drs. Grembecka and Cierpicki receive research support from Kura Oncology. They are also receiving compensation as members of the scientific advisory board of Kura Oncology, and they have an equity ownership in the company. Other co-authors declare no potential conflict of interest.
Supplementary information is available at Leukemia’s website.
References
- 1.Marschalek R. Mechanisms of leukemogenesis by MLL fusion proteins. British Journal of Haematology. 2011;152:141–154. doi: 10.1111/j.1365-2141.2010.08459.x. [DOI] [PubMed] [Google Scholar]
- 2.Cox MC, Panetta P, Lo-Coco F, Del Poeta G, Venditti A, Maurillo L, et al. Chromosomal aberration of the 11q23 locus in acute leukemia and frequency of MLL gene translocation: results in 378 adult patients. American Journal of Clinical Pathology. 2004;122:298–306. doi: 10.1309/RX27-R8GJ-QM33-0C22. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Chen A, Yan XM, Huang G. Disordered epigenetic regulation in MLL-related leukemia. International Journal of Hematology. 2012;96:428–437. doi: 10.1007/s12185-012-1180-0. [DOI] [PubMed] [Google Scholar]
- 4.Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94:984–993. doi: 10.3324/haematol.2008.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer Cell. 2003;4:99–110. doi: 10.1016/s1535-6108(03)00188-0. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 7.Grembecka J, Belcher AM, Hartley T, Cierpicki T. Molecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemias. J Biol Chem. 2010;285:40690–40698. doi: 10.1074/jbc.M110.172783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, et al. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manara E, Baron E, Tregnago C, Aveic S, Bisio V, Bresolin S, et al. MLL-AF6 fusion oncogene sequesters AF6 into the nucleus to trigger RAS activation in myeloid leukemia. Blood. 2014;124:263–272. doi: 10.1182/blood-2013-09-525741. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nature Genetics. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 11.Wang QF, Wu G, Mi S, He F, Wu J, Dong J, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117:6895–6905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gal H, Amariglio N, Trakhtenbrot L, Jacob-Hirsh J, Margalit O, Avigdor A, et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia. 2006;20:2147–2154. doi: 10.1038/sj.leu.2404401. [DOI] [PubMed] [Google Scholar]
- 14.Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia In Vivo. Cancer Cell. 2015;27:589–602. doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]