

Abstract/Synopsis
Obesity and its major co-morbidities, including type 2 diabetes mellitus, nonalcoholic fatty liver disease (NAFLD), obesity cardiomyopathy, and certain cancers, are major public health problems worldwide. They are responsible for substantial morbidity and mortality, to a degree that life expectancy in the United States has actually declined in recent years because of it. Obesity is the increased accumulation of fat, i.e. triglycerides (TG), which are synthesized from glycerol and long chain fatty acids (LCFA), throughout the body. Although long believed to enter cells solely by passive diffusion, it has been established over the past 30 years that LCFA enter adipocytes, hepatocytes and cardiomyocytes via specific, facilitated transport processes, and that these processes are hormonally up-regulated in obesity. Metabolism of increased cellular TG content in obesity may lead to cell-specific lipotoxicity, contributing to co-morbidities such as NAFLD and cardiomyopathy. In contrast to the popular perception, dietary control and bariatric surgery can each achieve major initial weight loss in many patients. However several mechanisms, including persistent up-regulation of LCFA transport, contribute to weight regain in the large majority of patients. Better understanding of these transport processes and their regulation may be a key to successful future strategies to treat obesity and NAFLD.
Keywords: Facilitated transport, Leptin, Lipotoxicity, Spexin, Weight regain
Introduction
Non-alcoholic fatty liver disease (NAFLD) is frequently described as encompassing a histological spectrum from simple hepatic steatosis (SHS) or non-alcoholic fatty liver (NAFL) to SHS plus a characteristic pattern of steatohepatitis (non-alcoholic steatohepatitis, NASH), to steatosis or steatohepatitis associated with fibrosis, cirrhosis, and/or hepatocellular carcinoma (HCC) [1–5]. Its various features have widely been considered to reflect hepatic manifestations of the metabolic syndrome (MetS) [6–8]. While world-wide prevalence & incidence rates of NAFLD/NASH are not known precisely [7], it is believed to have become the world’s most prevalent liver disease [9]. NAFLD is linked to obesity and insulin resistance in Western cultures, but histologically similar NAFL and NASH both occur at lower BMIs in Asian countries, where many patients also lack the insulin resistance typical in the west [10, 11]. GWAS and other approaches to studying NAFLD genomics are helping to understand both geographic differences and an increasing number of genetic differences within the Western NAFLD population [e.g. 12,13]. This review focuses on obesity and NAFLD in the western world.
The earlier literature indicated that NAFL and NASH were part of a clinical as well as a histological continuum, in which SHS progressed to NASH, which could progress further to NASH with fibrosis, cirrhosis and/or HCC [4,5]. However, while progression from NASH to NASH with fibrosis, cirrhosis, and/or HCC is by now well-documented, the frequency of evolution from SHS to NASH is unclear [5,7]. Absent reliable surrogate markers, serial liver biopsies might be expected to be the gold standard for making this determination, but their invasiveness and issues with regard to their interpretation [5] have limited their deployment for this purpose. As of mid-2015, our literature searches identified only 16 papers, including ≤500 of a world-wide population of millions of NAFLD patients, that examined the histological evolution of steatosis, steatohepatitis, and fibrosis in NAFLD via paired liver biopsies [e.g. 14–24]. Collectively, these papers suggested that development and progression of fibrosis in NAFL was uncommon, whereas fibrosis in NASH occurred and progressed more frequently. However, the most recent studies have challenged even this, suggesting that NAFL can occasionally evolve all the way to NASH with advanced fibrosis, implying that NAFL may not be the benign entity it is often considered [23,24]. A useful editorial summarizes the current data [25]. The view of NAFLD as an evolving continuum has also been challenged by claims that NAFL and NASH are distinct disease entities rather than points on a continuum [9, 26, 27].
Fatty Acids, Triglycerides, and NAFLD Pathophysiology
Most NAFLD in the Western world evolves on a background of obesity. Obesity is the increased accumulation of fat (i.e. triglycerides, TG), synthesized from glycerol and long chain fatty acids (LCFA), throughout the body. TG are stored in large lipid droplets in adipocytes and smaller droplets in parenchymal cells, notably hepatocytes [28,29] and cardiomyocytes [30]. In obesity, metabolism of increased TG in parenchymal cells leads to cell-specific lipotoxicity. Thus this review concentrates on regulation of LCFA uptake and TG deposition and metabolism in adipocytes and hepatocytes, which are key to the understanding of NAFLD pathogenesis.
Until ~30 years ago it was believed that LCFA entered cells solely by passive diffusion. However, selective TG accumulation at specific sites in obesity clearly indicated that something other than unregulated, passive diffusion was involved.
Uptake of LCFA into hepatocytes, adipocytes and cardiac myocytes in fact consists of two distinct processes, each a function of the unbound LCFA concentration [31–36]. LCFA uptake velocity at any unbound LCFA concentration ([LCFAu]) is the arithmetic sum of a saturable and a non-saturable function of the corresponding [LCFAu], according to the equation:
where UT([LCFAu]) is the experimental measurement of uptake (pmol/sec/50,000 cells) at the stated concentration of unbound LCFA ([LCFAu]); Vmax (pmol/sec/50,000 cells) & Km (nM), are the maximal velocity of saturable LCFA uptake and [LCFAu] at ½ the maximal uptake velocity, respectively; k (ml/sec/50,000 cells) is the rate constant for nonsaturable uptake. In human studies Vmax has been shown to increase as an exponential function of patient BMI (Figure 1).
Figure 1. Relationship of Vmax for LCFA uptake by isolated human omental adipocytes to patient BMI.
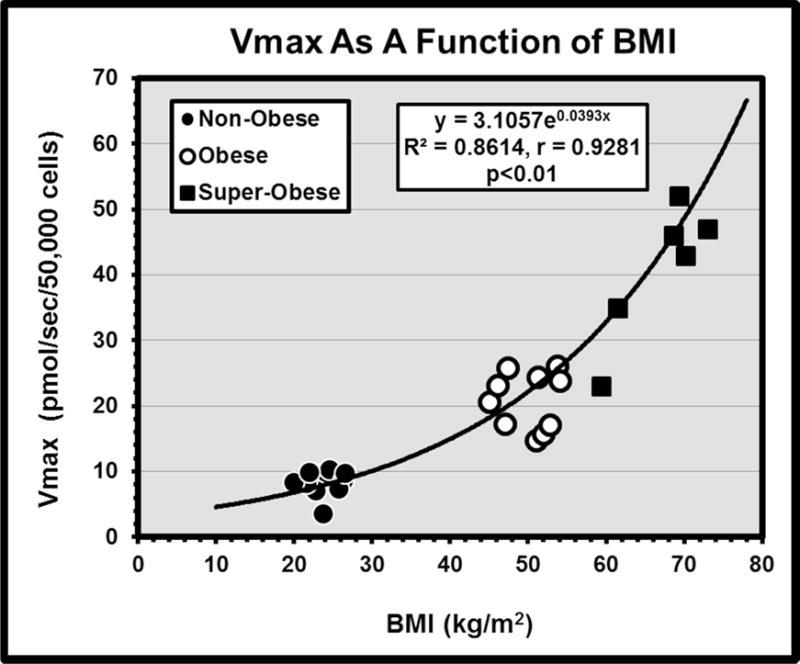
Cells were isolated from intra-operative fat biopsies obtained during clinically indicated abdominal surgical procedures in 10 non-obese patients, and during bariatric surgeries in 10 obese and 7 super-obese patients who were participants in a 2-stage bariatric surgical protocol (77). Vmax increases as an exponential function of BMI.
At normal between-meal LCFA concentrations in mammals ~95% of total cellular LCFA uptake is via the saturable pathway [34, 35]. Typical LCFA have a pKi of ~6.5, existing in plasma as a roughly equal mixture of fatty acid anions and uncharged, protonated fatty acids. Studies with the acidic LCFA-analogue α2,β2,ω3-heptaflourostearate [36] established (i) that saturable uptake reflected protein mediated, regulatable transport of LCFA anions, whereas non-saturable uptake represented passive trans-membrane diffusion of protonated LCFA, and (ii) that the rate of trans-membrane LCFA movement by the former process was ~10 times faster than by the latter [34,35]. Subsequently multiple techniques have confirmed that cellular LCFA uptake involves, at least in part, regulatable, facilitated, protein mediated transport [e.g. 37–42].
At least four proteins or protein families have been proposed as LCFA transporters: plasma membrane fatty acid binding protein (FABPpm) [43, 44]; fatty acid translocase (FAT, or CD36) [45]; the fatty acid transporting polypeptide (FATP) family [46,47], and caveolin-1[48,49]. Many reports suggest that additional transporters await discovery [50, 51].
FABPpm, the first LCFA transporter to be identified, proved similar or identical to the mitochondrial isoform of aspartate aminotransferase (mAspAT) [52]. Molecular modeling, site-directed mutagenesis, transfection, and intracellular trafficking studies proved that mAspAT contained a hydrophobic cleft of proper size to be an LCFA binding site, and that it was capable of facilitating cellular LCFA uptake [53].
For most metabolic processes, LCFA are activated by esterification to an acyl-CoA before they are utilized [54]. LCFAs can be activated by three related protein families: the long chain acyl-CoA synthetases (ACSLs), FATPs, and the “bubblegum” family [55]. Among FATPs, some that are, themselves, candidate LCFA transporters [46, 47] also have coenzyme A (CoA) enzymatic activity [56–58], leading to several different models of how FATPs might facilitate LCFA uptake [55]. (A) FATPs could be classical plasma membrane transporters [47]. (B) Since FATPs, either individually or in recently described heteromultimers (see below), have both transport and esterification activity, the driving force for LCFA transport could come from their enzyme activity via vectorial acylation [58]. (C) FATPs are enzymes, and could enhance uptake indirectly by depleting intracellular LCFA [59].
LCFA uptake into HepG2 cells was found to be mediated by a hetero-tetrameric protein complex comprised of FABPpm, Cav-1, CD36, and the Ca++-independent membrane phospholipase A2 (iPLA2β) [60]. Blocking iPLA2β with a bile acid-phospholipid conjugate dissociated the complex and inhibited LCFA uptake. Use of the same bile acid conjugate in a mouse hepatocyte cell line that exhibited both steatosis and inflammation decreased LCFA uptake by 56.5% and essentially abolished both steatosis and inflammation. Its role as a potential therapy for NASH is under investigation.
LCFA uptake clearly involves specific transport processes the regulation of which, in adipocytes & hepatocytes, is a key element in the pathogenesis of obesity per se and NAFLD. This recognition has driven a steady increase in Pub-Med citations about “FATTY ACIDS & OBESITY” from <100/year from the 1960s to mid-1980s to ≥1,200/year by 2014.
LCFA Transport in Specific Disease Models
At least in the Western world, there is a strong association between obesity and NAFLD. As just described, alterations in adipose tissue LCFA sequestration play an important role in the pathogenesis of obesity, and analogous alterations in hepatocellular LCFA uptake play a similar role in the excess hepatic TG accumulation characterizing SHS. However, in contrast to adipocyte LCFA transport, which has been extensively studied in both rodent and readily available human fat samples, the limited availability of appropriate samples of human liver has largely restricted studies of hepatocellular LCFA uptake to animal tissues.
LCFA uptake by adipocytes, hepatocytes, and cardiac myocytes from rodent models of obesity and obesity-associated fatty liver and cardiomyopathy has been extensively studied, starting with obese, leptin-receptor deficient Zucker fatty (fa/fa) and Zucker diabetic fatty (ZDF) rats and extending to analogous mouse models [61–64]. The Vmax for saturable LCFA uptake was dramatically increased in adipocytes from the fa/fa and ZDF animals compared with non-obese Zucker heterozygous (fa/+) or wild-type Wistar strains (Figure 2). Furthermore, Vmax in rat adipocytes was highly correlated with mRNA expression for the LCFA transporter FABPpm In contrast, there were only minor differences among rat groups in the Vmax for LCFA uptake into hepatocytes and cardiac myocytes (Figure 2). Initially surprising, this became an instructive finding, as described below.
Figure 2. Computed values of the Vmax for saturable [3H]-oleate uptake by isolated hepatocytes, cardiac myocytes, and adipocytes from four groups of adult male Zucker rats: +/+, fa/+, fa/fa, and ZDF.
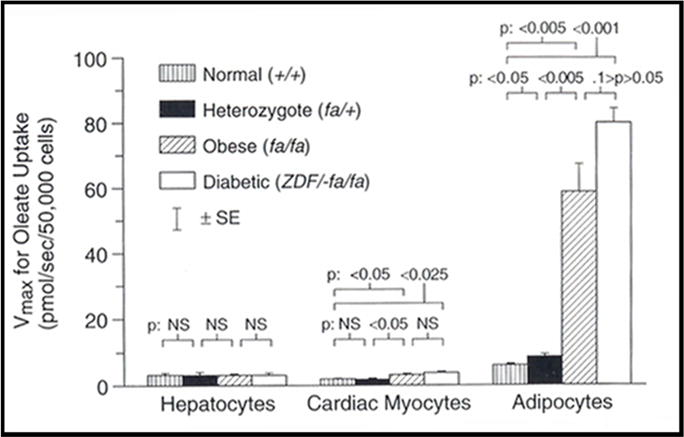
Saturable adipocyte LCFA uptake is appreciably increased compared to normal controls in the obese fa/fa and ZDF animals, despite their defective leptin signaling. There is little change in hepatocyte LCFA uptake in the different groups. Very similar findings have been reported in hepatocytes from obese, leptin signaling-deficient ob/ob & db/db mice, compared to C57BL6J control mice rendered obese by high fat diets. (From Berk PD, Zhou SL, Bradbury M, Stump D, Kiang CL, Isola LM. Regulated membrane transport of free fatty acids in adipocytes: role in obesity and non-insulin dependent diabetes mellitus. Trans Am Clin Climatol Assoc 1997; 108:26–40, with permission.)
While short-term regulation of adipocyte LCFA uptake in response to meals remains to be better defined, its chronic up-regulation characterizes all studied rodent models of obesity, as well as obese human subjects. Thus, the Vmax for adipocyte LCFA uptake is markedly increased in genetically obese (ob/ob), diabetic (db/db), fat (fat) and tubby (tub) mice; in both Wistar and Sprague Dawley rats and C57BL6/J mice on high fat diets [65], and in omental [66, 67] and subcutaneous [67] adipocytes from obese bariatric surgery patients.
In some circumstances, up-regulation of adipocyte LCFA uptake precedes onset of obesity [61] whereas down-regulation precedes weight loss [68]. These data support the hypothesis that up-regulation of adipocyte LCFA uptake contributes to both regulation of adiposity and the pathogenesis of obesity. Further observations of consistent, tissue-specific up-regulation of adipocyte LCFA uptake in association with weight gain have led to speculation that this could also contribute to and regulate LCFA partitioning. Partitioning might serve a protective function. By sequestering LCFA & TG in large droplets and protecting them from oxidative processes, adipocytes serve as a buffer. They protect downstream non-adipose cells such as pancreatic β-cells, cardiac myocytes, skeletal muscle cells, and hepatocytes from the lipotoxic consequences of their excessive LCFA & TG accumulation and metabolism to lipotoxic species, including diacyl-glycerols, ceramides, reactive oxygen species (ROS) and cholesterol [69,70].
Several diverse lines of evidence thus suggest that regulation of adipocyte LCFA uptake may have a role in controlling body adiposity [e.g. 61, 63–68, 71]. While primary genetic defects in obesity models [63,64,71] can be expressed either in the CNS (db mouse, Zucker fatty rat) [72,73] or peripheral tissues (ob mouse) [74], all such defects, rodent models of dietary obesity, and typical examples of human obesity result in selective up-regulation of facilitated adipocyte LCFA uptake. This suggests that such regulation may represent a final common pathway for control of adiposity resulting from diverse primary causes in multiple mammalian species including man (Figure 3).
Figure 3. Regulation of adipocyte LCFA uptake appears to regulate body adiposity.
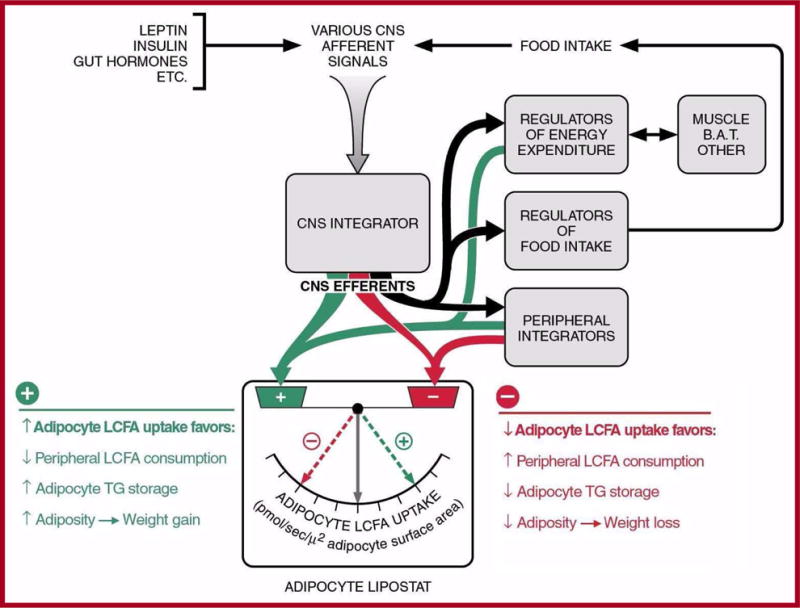
All well-studied genetic and dietary animal models of obesity, as well as obese human subjects, exhibit selective up-regulation of facilitated LCFA by adipocytes. This suggests that regulation of adipocyte LCFA uptake represents a final, common pathway for control of body adiposity resulting from a diversity of primary causes. From Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis 2004; 8: 639–671, with permission.
Obesity has serious consequences. Available treatments, including diet and life style modifications and bariatric surgery, can achieve significant weight loss in many patients. However, post treatment weight regain is very common, often to levels exceeding the pretreatment value within 5 years [75, 76]. Despite both extensive theoretical and clinical studies in man and the recognized prolonged post-operative persistence of weight gain-promoting hormonal patterns; increased insulin sensitivity, rates of glucose transport, and LPL activity; and other metabolic abnormalities [75,76], these patients are often considered treatment failures, for which they may be blamed. However, in a recent study [67], [3H]-LCFA uptake kinetics were determined in adipocytes isolated from intra-operative omental and subcutaneous fat biopsies from 10 non-obese (NO), 10 obese (O) and 10 super-obese (SO) patients. The O and SO patients were undergoing bariatric surgical operations (sleeve gastrectomies); the NO patients were having other, clinically indicated, non-bariatric surgeries. By non-linear regression, Vmax for LCFA uptake by omental adipocytes increased exponentially as a function of BMI (r = 0.93, p < 0.01) in the 3 groups, from 5.1±0.95 to 21.3±3.20 to 68.7±9.45 pmol/sec/50,000 cells (Figure 1). Results in subcutaneous adipocytes were very similar. The SO patients returned for second biopsies 16±2 months later, after losing 113±13 lbs. Their mean BMI had fallen from 62.6±2.8 to 44.4±2.5 kg/m2 (p<0.01), and was now similar to the O group. However, Vmax (42.1±6.4 pmol/sec/50,000 cells) in the now-weight-reduced SO group remained >2X up-regulated (p<0.01) from that predicted for their new BMI by the original BMI:Vmax regression. As up-regulation of LCFA uptake strongly predicts weight gain, this suggests a biologic process rather than failure of patient will power as contributing to any weight regain [76,77]. Interestingly, these SO patients were a subset of a larger cohort of 2,458, treated mainly with either Roux-en-Y gastric bypass or laparoscopic gastric banding in the LABS-2 protocol. Most patients achieved maximum weight loss in the first post-operative year, based on which they were assigned to one of five 5 weight trajectory groups. By the end of year 3 all weight trajectory groups showed some degree of weight regain [78].
Lipotoxicity, Leptin and Spexin
In severe obesity, excessive LCFA availability can exceed the LCFA storage capacity of adipose tissue. This leads to ectopic accumulation of LCFA, TG, and their lipotoxic metabolites in non-adipose cells, and can result in the cellular injury now designated as “lipotoxicity”. Many obesity co-morbidities result from disordered LCFA disposition and/or lipotoxicity.
Leptin is widely considered the body’s principal liporegulatory hormone, exerting profound anti-steatotic effects [79–81]. When caloric intake chronically exceeds energy expenditure, TGs accumulate throughout the body. This is amplified by the hyperinsulinemia associated with the excess caloric intake, which also up-regulates enzymes involved in lipogenesis. Intermediates in the lipogenic pathway inhibit LCFA oxidation, further increasing the LCFA available for storage as TG. That much of this increase is restricted to adipose tissue reflects the fact that, as adipose tissue TG increases, it stimulates an increased release of leptin [82–84]. In some situations, leptin functions as an insulin counter-regulatory hormone [85], exerting an anti-lipogenic, pro-oxidative program on peripheral, non-adipose cells [80, 81]. In one rodent model of diet-induced obesity, when total body fat had increased 150-fold, with a significant weight regain and increased plasma LCFA, the lipid content of pancreatic islets, liver, heart, and skeletal muscle had increased by no more than 10-fold [79]. With still further increases in total body fat, these effects of leptin diminish despite markedly increased plasma leptin concentrations, indicating a state of leptin resistance. The mechanisms underlying leptin resistance remain somewhat unclear [86], but may involve inhibition of brain leptin uptake by circulating n-3 polyunsaturated fatty acids [87]. A total lack of leptin effect occurs in various lipodystrophies, as well as in situations in which leptin signaling is abolished due either to an absence of leptin (ob/ob mouse) [74] or an absence or functional mutations of its receptor (db/db mouse, Zucker fa/fa rat) [72, 73]. The result is a progressive, generalized steatosis, with ectopic accumulation of TG in non-adipose tissues. The increased ectopic TG pool exchanges with an increased intracellular pool of LCFA. These enter into non-oxidative pathways, of which the most studied, but by no means the only one [88], leads to the accumulation of ceramides and other metabolites [70, 89,90]. These cause extensive tissue damage and apoptosis, resulting e.g. in T2DM, cardiomyopathy, and liver injury, that can be ameliorated to varying degrees when there is a functioning leptin receptor by administration of recombinant leptin [91]. In rodents, lipotoxicity leads to several components of the rodent equivalent of MetSyn. While evidence that true lipotoxic disease occurs in man is limited, leptin resistance is characteristic of human obesity, and may permit TG accumulation in ectopic sites. Further studies of this phenomenon in man are clearly indicated [89,90].
Spexin
Probing whole human genome microarrays containing 55K genes & expressed sequence tags (ESTs) led to identification of ~3,500 exhibiting significant differences in expression between obese and non-obese human fat. Of these, the gene most extensively regulated was initially identified only as Ch12orf39. Its mRNA was under-expressed 14.9-fold in obese vs non-obese fat, in parallel with a similar decrease in the levels of its circulating gene product, which was subsequently recognized as being identical to spexin, a novel peptide identified by Mirabeau et al. in 2007 using Markov modeling (92). Its regulation relative to BMI, and other observations, led to studies of a possible role in weight control. In mice with diet-induced obesity (DIO), spexin administration reduced food intake in the absence of generalized taste aversive effects or evident toxicity, increased energy expenditure (locomotor activity), and decreased the respiratory exchange ratio, favoring burning of fat compared to carbohydrate [93]. Similar effects were found in rats. These results are believed to be mainly centrally mediated. In addition, spexin directly and selectively inhibits LCFA uptake by rodent adipocytes, further contributing to weight loss. In sera from non-obese, obese, and super-obese patients, spexin concentrations exhibited a negative, non-linear correlation with leptin (r = −0.64, p<0.01). Spexin concentrations were also significantly negatively correlated with the Vmax for omental adipocyte LCFA uptake in the same patient (r = −0.71, p<0.01), whereas leptin was strongly positively correlated with Vmax (r =+0.81, p<0.01). These and other data indicate that spexin and leptin may play important, antagonistic roles in regulating adipocyte LCFA uptake (Figure 4), which other studies suggest regulates overall adiposity.
Figure 4. Relationships between plasma levels of Spexin (Peptide A) and leptin and adipocyte LCFA uptake.
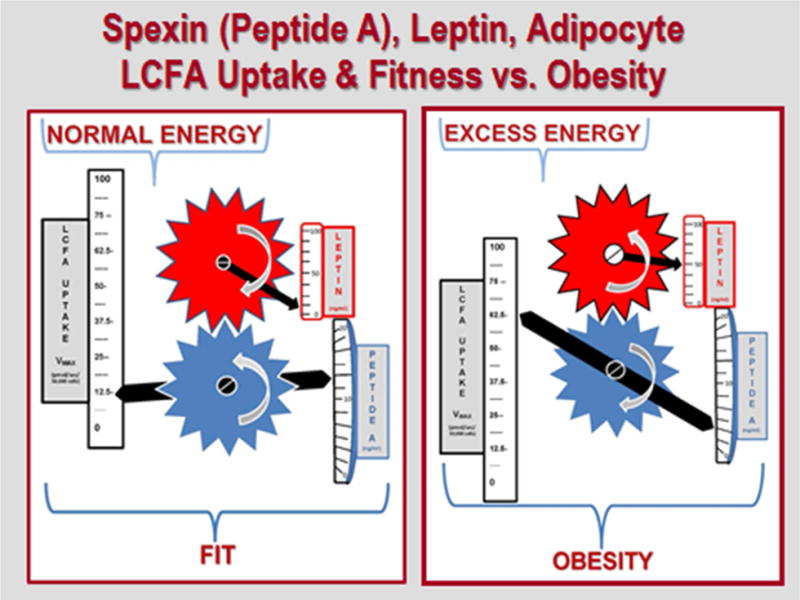
Left-hand panel illustrates these relationships in the presence of normal energy balance and physical fitness: low levels of leptin and LCFA uptake Vmax, high evels of Spexin. Right hand panel reflects the situation in the presence of excessive energy, leading to obesity: higher levels of leptin and increased LCFA uptake Vmax, low levels of spexin. The interlocking gears illustrate the strong, negative correlation between plasma Spexin and leptin concentrations, and their respective relationship to the Vmax for adipocyte LCFA uptake.
Obesity-Related Liver Disease: Hepatic Steatosis and Steatohepatitis
Hepatic Steatosis
Simple hepatic steatosis (SHS) and other stages of NAFLD commonly accompany obesity. Well over half of all obese patients have some form of NAFLD; ~ 25% have NASH, with or without significant hepatic fibrosis [e.g. 7–9, 94].
Mechanisms of Hepatic Steatosis
The basis for the development of insulin resistance and SHS in the setting of obesity is clear (Figure 5). Not every case of SHS results from obesity. SHS can potentially result from many different processes. Virtually all of those listed in Figure 6 reportedly play a role in one or another model of hepatic steatosis, increasing the hepatocyte TG pool either by ultimately increasing TG input or decreasing output from the pool. Very few studies have assessed several of these processes simultaneously, especially in man, so that their relative contributions remain largely unclear.
Figure 5. The road to insulin resistance and hepatic steatosis in obesity.
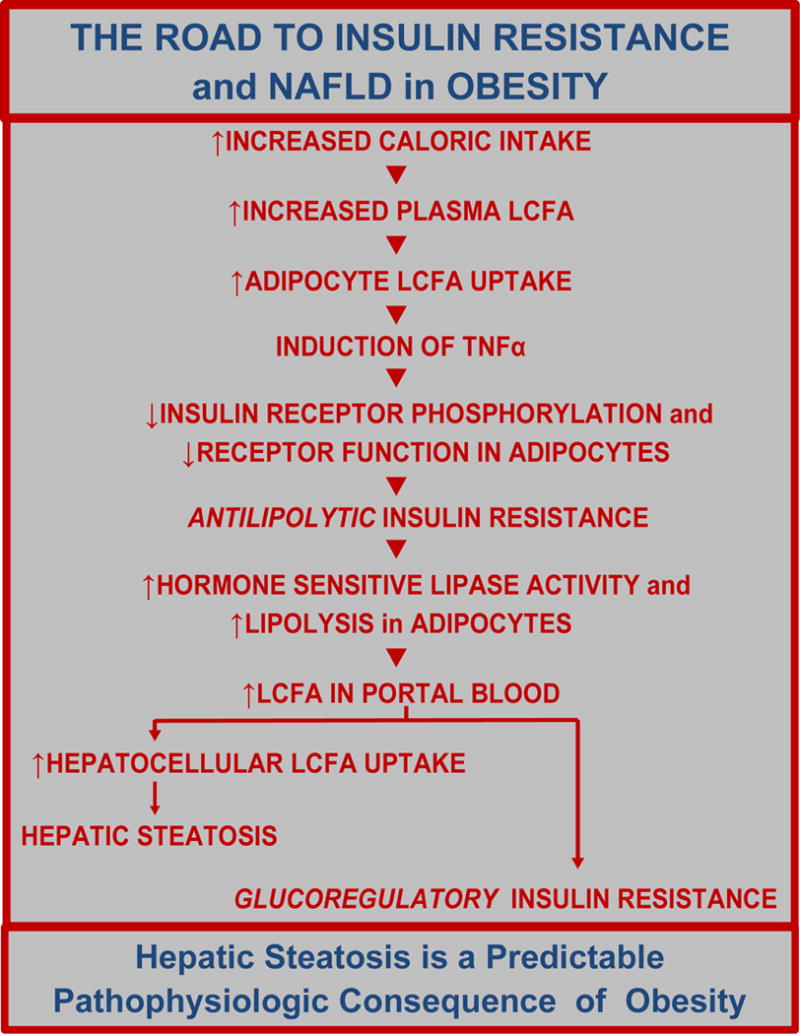
Ten discrete and identifible steps leading to insulin resistance and hepatic steatosis are initiated by an increase in caloric intake and consequent increase in the plasma concentration of LCFA. Adapted from Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis 2004; 8: 639-671, with permission.
Figure 6. Processes that could contribute to the increased hepatic triglyceride content that characterizes hepatic steatosis and NASH.
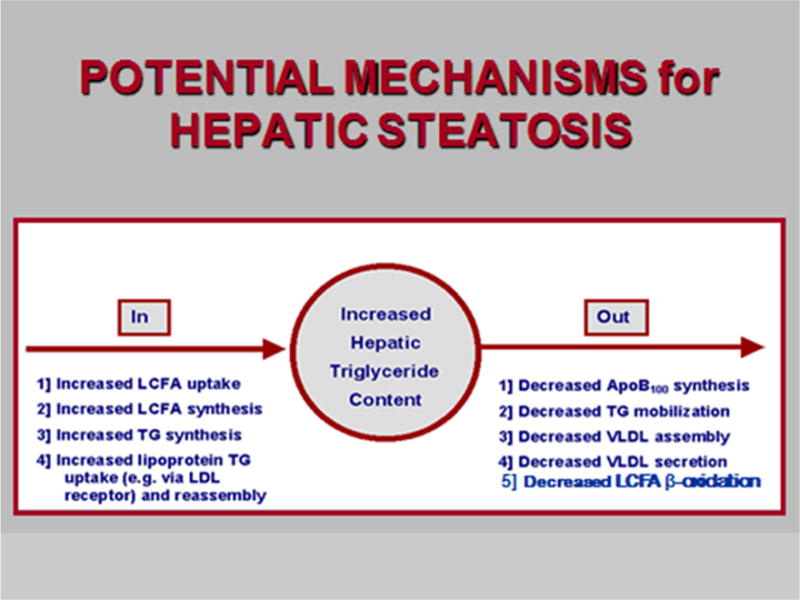
Those on the left potentially contribute to an increased input to the hepatocellular pool of triglycerides. Those on the right increase hepatic trigyceride content by decreasing normal levels of triglyceride output, principally in VLDL. From Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis 2004; 8: 639-671 with permission.
Increased hepatocellular LCFA uptake is a major contributor to both obesity- and EtOH-related hepatic steatosis in rat and mouse models and human-derived HepG2 cells [28, 95, 96]. In a mouse study [28] involving C57BL6J controls; similar mice on a high fat diet or consuming 10, 14 or 18% EtOH, designated the functional leptin signaling groups (FLSGs); and leptin signaling deficient homozygous ob/ob and db/db animals, the Vmax’s for hepatocellular LCFA uptake were significantly increased in all FLSG’s, but not in the ob/ob and db/db animals, which had the heaviest, greasiest livers. There was a highly significant linear correlation between the Vmax for hepatocellular LCFA uptake and hepatic TG content in the FLSGs, but hepatic TG content in the ob/ob and db/db animals was well above the FLSG regression line suggesting that a significant part of the hepatic TG content in these latter animals was not derived from LCFA uptake [28].
In FLSGs, despite variably increased expression of single transporter genes in individual EtOH & HFD groups, the mean expression ratio for FABPpm, FATPs 1, 2, 4, & 5, & CD36 in each group was highly correlated with both Vmax for hepatocellular LCFA uptake and hepatic TG. Vmax was also highly correlated with the corresponding expression ratios for Srebp-1c (r =0.99) and NfkB.(r=0.94). Increased hepatic TGs in ob/ob & db/db mice did not relate to hepatic LCFA uptake, but instead were highly correlated with increased expression of lipogenic enzymes involved in LCFA synthesis (SCD-1, FASN). Thus, hepatic TG is seemingly regulated by the same factors that control a wide array of hepatic lipid metabolic pathways.
Individual FABPpm, Slc27a2, and Slc27a4, Slc27a5 and CD36 gene expression ratios were significantly up-regulated in a dose-dependent fashion in the EtOH groups, and the mean values for their expression ratios in the different groups were closely correlated with the Vmax for hepatocellular LCFA uptake. The Vmax data, in turn, were highly correlated with hepatic TG content. However, these expression ratios were not significantly increased in any of the obesity groups (HFD, ob/ob, or db/db), whether or not they had functional leptin signaling. CD36 was the most widely upregulated individual transporter, being significantly increased in the 14% and 18% EtOH, HFD, ob/ob, and db/db groups. These data suggest that regulation of hepatic LCFA transporter expression & participation of individual transporters in LCFA uptake are far more complex than previously believed. Correlations between transcription factor expression & mean expression of multiple transporter genes suggests possible regulatory interaction, and may support reports postulating that a complex of FABPpm, CAV-1, CD36 & FATP4, and, possibly, FATP5 mediates hepatic LCFA uptake [55,60].
Steatohepatitis
HS is the most common form of NAFLD and is, in its earliest stages, largely reversible. In fact, despite some controversy, the most common fates of SHS are either regression, maintenance of status quo, or progression to NASH.
Mechanisms Leading to NASH
In NASH, inflammatory processes are super-imposed on SHS, resulting in a well-described histologic picture [97–99] similar to that of alcoholic hepatitis. Although a “two-hit” model of the progression from SHS to NASH, in which the first “hit” is the development of SHS [100] had become widely accepted two decades ago, it has subsequently become clear that the second “hit” is, itself, multifactorial [101]. In fact, rather than a sequential series of hits, many of the now multiple “hits” are actually pathways, that act on the increased TG mass more or less simultaneously, and in recent widely accepted models involve oxidative stress, resulting in hepatocellular injury and apoptosis; inflammation & cytokine cascades [e.g. 102–105]; stellate cell activation; fibrosis; and, ultimately, progression to cirrhosis.
Hepatocellular carcinoma (HCC) is an increasingly recognized outcome of NASH [106, 107], evolving not infrequently and almost uniquely in a non-cirrhotic liver [108].
Insulin Resistance
Early SHS may develop in the absence of insulin resistance (IR). However, a critical step in the progression of NAFLD is the virtually universal development of IR, which plays an important role in adipocyte LCFA disposition. Under normal circumstances adipocytes are intermittent importers of LCFA, sequestering them post-prandially as TG and then releasing them via lipolysis as required to meet metabolic needs. IR, by de-repressing adipocyte hormone sensitive lipase, converts these cells into virtually continuous net LCFA exporters. LCFA released from the intra-abdominal, visceral fat depots enter the portal vein and are translocated directly to the liver. Hepatocellular LCFA oxidation at both mitochondrial and extra-mitochondrial sites is a major source of the reactive oxygen species (ROS) that initiate the processes of hepatocellular injury that characterize steatohepatitis (Figure 7). In particular, ROS lead to mitochondrial injury, including both morphologic changes and defective ATP repletion; lipid peroxidation with production of malondialdehyde (MDA) and hydroxynonenal (HNE); activation of the FAS system and the release of specific cytokines, in particular TNFα, TGFβ, and IL-6 [69]. Together these result in several of the characteristic features of NASH recognizable histologically: apoptotic cell death, development of Mallory’s hyaline, polymorphonuclear leukocyte infiltration, and fibrosis. ROS generated from ethanol oxidation lead to many of the same metabolites, which may explain histologic similarities between alcoholic and nonalcoholic steatohepatitis. However, since alcoholic steatohepatitis also develops on a background of simple steatosis, oxidation of fatty acids may also contribute to that condition. Although numerous processes have been identified as contributing to the development of NASH [109], all typically operate against a background of SHS, which is the sine qua non of NAFLD.
Figure 7. Consequences of increased hepatocellular uptake and oxidation of LCFA in obesity.
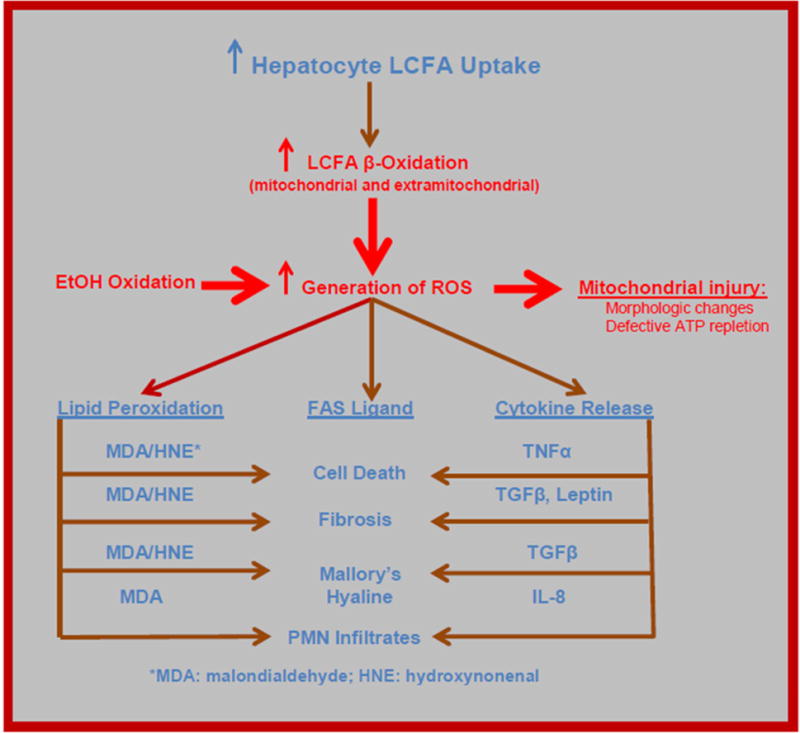
Many of the features of NASH follow logically from the increase in hepatocellular LCFA uptake and subsequent increase in the generation of reactive oxygen species (ROS) by mitochondrial and extra-mitochondrial LCFA oxidation. These, in turn, lead to generation of intracellular mediators such as MDA, HNE, TNFα, TGFβ, leptin, and IL8, which – in turn – cause several of the characteristic histologic features of NASH. ROS resulting from EtOH oxidation lead to generation of some of the same mediators, potentially explaining the histologic similarity between NASH and alcoholic hepatitis. Adapted from Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis 2004; 8:639-671, with permission.
QUO VADIS?
NAFLD has sparked a literature explosion. From mid 1995 to mid 2015, more than 8,200 articles about NAFLD entered major databases. Despite improved understanding of its pathophysiology, the prevalences of NAFLD and NASH continue to increase world-wide, and the long-term effectiveness of most currently available therapies is limited [110,111], in part due to the frequency of weight regain. Additional reviews about current and future therapies will be found in articles by Rustgi and Terrault in this issue of Clinics in Liver Disease. Although several drugs are under advanced clinical development and testing for patients with early-stage disease, as well as those with advanced liver fibrosis [112, 113], there are currently no USFDA approved therapeutic agents for the treatment of NAFLD and NASH.
Our ultimate goal should be development of effective prevention strategies and therapies for NAFLD. Clearly, we are not there yet.
Key Points.
Obesity is the increased accumulation of fat, i.e. triglycerides (TG), which is synthesized from glycerol and long chain fatty acids (LCFA), throughout the body.
LCFA enter adipocytes, hepatocytes and cardiomyocytes via specific, facilitated transport processes, which are regulated in obesity at least in part by insulin, leptin and spexin.
In obesity, metabolism of the increased cellular TG content may lead to cell-specific lipotoxicity, contributing to co-morbidities such as NAFLD and cardiomyopathy.
Dietary control and bariatric surgery can achieve major weight loss in many patients, but persistent up-regulation of LCFA transport contributes to weight regain.
Better understanding of these transport processes and their regulation may be a key to successful future strategies for treatment of obesity and NAFLD.
Acknowledgments
Supported by Grant # 5U01DK066667-11 REVISED, Bariatric Surgery: Outcomes & Impact on Pathophysiology, from the National Institute of Diabetes and Digestive and Kidney Diseases of the NATIONAL INSTITUTES OF HEALTH.
Abbreviations
- ACSL
long chain acyl-CoA synthetase
- FABPpm
plasma membrane fatty acid binding protein
- FAT
fatty acid translocase, or CD36
- FATP
the fatty acid transporting polypeptide (FATP) family
- HCC
hepatocellular carcinoma
- IR
insulin resistance
- LCFA
long chain fatty acids
- MetSyn
metabolic syndrome
- NAFLD
nonalcoholic fatty liver disease
- NAFL
nonalcoholic fatty liver
- NASH
nonalcoholic steatohepatitis
- ROS
reactive oxygen species
- SHS
simple hepatic steatosis (equivalent to NAFL)
- TG
triglycerides
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have nothing to disclose.
Contributor Information
Paul D Berk, Department of Medicine, Divisions of Digestive & Liver Diseases and of Preventive Medicine Columbia College of Physicians and Surgeons and Columbia University Medical Center New York, NY 10032.
Elizabeth C Verna, Department of Medicine, Division of Digestive & Liver Diseases, Columbia College of Physicians and Surgeons and Columbia University Medical Center, New York, NY 10032.
References
- 1.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 2.Diehl AM, Goodman Z, Ishak KG. Alcohol-like liver disease in non-alcoholics. Gastroenterol. 1988;95:1056–1062. [PubMed] [Google Scholar]
- 3.Clain DJ, Lefkowitch JH. Fatty liver disease in morbid obesity. Gastroenterol Clin N Amer. 1987;16:239–252. [PubMed] [Google Scholar]
- 4.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ, American Gastroenterological Association; American Association for the Study of Liver Diseases; American College of Gastroenterology The diagnosis and management of nonalcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroentl. 2012 Jun;142(7):1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Burt AD, Lackner C, Tiniakos DG. Diagnosis and assessment of NAFLD: definitions and histopathological classification. Semin Liv Dis. 2015 Aug;35(3):207–220. doi: 10.1055/s-0035-1562942. [DOI] [PubMed] [Google Scholar]
- 6.Vanni E, Bugianesi E, Kotronen A, De Minicis S, Yki-Järvinen H, Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa. Dig Liver Dis. 2010;42(5):320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 7.Satapathy SK, Sanyal AJ. Epidemiology and Natural History of Non-Alcoholic Fatty Liver Disease. Semin Liv Dis. 2015 Aug;35(3):221–235. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- 8.Vanni E, Marengo A, Mezzabotta L, Bugianesi E. Systemic complications of nonalcoholic fatty liver disease: when the liver is not an innocent bystander. Semin Liv Dis. 2015 Aug;35(3):236–249. doi: 10.1055/s-0035-1562944. [DOI] [PubMed] [Google Scholar]
- 9.Moore JB. Non-alcoholic fatty liver disease: the hepatic consequence of obesity and the metabolic syndrome. Proc Nutrition Soc. 2010;69:211–220. doi: 10.1017/S0029665110000030. [DOI] [PubMed] [Google Scholar]
- 10.Farrell GC, Wong V, W-S V, Chitturi S. NAFLD in Asia- as common and as important as in the West. Nat Rev Gastroenterol Hepatol. 2013;10:307–318. doi: 10.1038/nrgastro.2013.34. [DOI] [PubMed] [Google Scholar]
- 11.Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10:686–690. doi: 10.1038/nrgastro.2013.171. [DOI] [PubMed] [Google Scholar]
- 12.Speliotes EK, 801 collaborators Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011 Mar;7(3):e1001324. doi: 10.1371/journal.pgen.1001324. Epub 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anstee QM, Day CP. The genetics of nonalcoholic fatty liver disease; spotlight on PNPLA3 & TM6SF2. Semin Liv Dis. 2015 Aug;35(5):270–290. doi: 10.1055/s-0035-1562947. [DOI] [PubMed] [Google Scholar]
- 14.Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatol. 1990 Jan;11(1):74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 15.Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatol. 1995 Dec;22(6):1714–1719. [PubMed] [Google Scholar]
- 16.Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol. 2003;98:2042–2047. doi: 10.1111/j.1572-0241.2003.07659.x. [DOI] [PubMed] [Google Scholar]
- 17.Fassio E, Alvarez E, Dominguez N, Landeira G, Longo C. Natural history of nonalcoholic steatohepatitis: a longitudinal study of repeat liver biopsies. Hepatol. 2004;40(4):820–826. doi: 10.1002/hep.20410. [DOI] [PubMed] [Google Scholar]
- 18.Adams LA, Sanderson S, Lindor K, Angulo P. The histological course of nonalcoholic fatty liver: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005 Jan;42(1):132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatol. 2006 Oct;44(4):865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 20.Merriman RB, Ferrell LD, Patti MG, Weston SR, Pabst MS, Aouizerat BE, Bass NM. Correlation of paired liver biopsies in morbidly obese patients with suspected nonalcoholic fatty liver disease. Hepatol. 2006 Oct;44(4):874–80. doi: 10.1002/hep.21346. [DOI] [PubMed] [Google Scholar]
- 21.Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, Chim AM, Yu J, Sung JJ, Chan HL. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010 Jul;59(7):969–974. doi: 10.1136/gut.2009.205088. [DOI] [PubMed] [Google Scholar]
- 22.Pais R, Charlotte F, Fedchuk L, Bedossa P, Lebray P, Poynard T, Ratziu V, LIDO Study Group A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J Hepatol. 2013 Sep;59(3):550–556. doi: 10.1016/j.jhep.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 23.McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol. 2015 May;62(5):1148–1155. doi: 10.1016/j.jhep.2014.11.034. [DOI] [PubMed] [Google Scholar]
- 24.Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs. nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015 Apr;13(4):643–654. doi: 10.1016/j.cgh.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison SA. Nonalcoholic fatty liver disease and fibrosis progression: the Good, the Bad, and the Unknown. Clin Gastroenterol Hepatol. 2015;13(4):655–657. doi: 10.1016/j.cgh.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 26.Yilmaz Y. Review article: is non-alcoholic fatty liver disease a spectrum, or are steatosis and non-alcoholic steatohepatitis distinct conditions? Aliment Pharmacol Ther. 2012;36:815–823. doi: 10.1111/apt.12046. [DOI] [PubMed] [Google Scholar]
- 27.Fielding C, Angulo P. Hepatic steatosis and steatohepatitis: are they really two distinct entities? Curr Hepatol Rep. 2014;13(2):151–158. doi: 10.1007/s11901-014-0227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge F, Zhou S, Hu C, Lobdell H, IV, Berk PD. Increased insulin- and leptin-regulated fatty acid uptake plays a key causal role in hepatic steatosis in mice with intact leptin signaling, but not in those lacking leptin or its receptor. American Journal of Physiology: Hepatic & Gastrointestinal Physiology. 2010;299:855–866. doi: 10.1152/ajpgi.00434.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge F, Lobdell H, IV, Zhou S, Hu C, Berk PD. Digital analysis of hepatic sections accurately quantitates triglycerides and selected properties of lipid droplets. Exp Biol Med. 2010 Nov 1;235(11):1282–1286. doi: 10.1258/ebm.2010.010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge F, Hu C, Hyodo E, Arai K, Zhou S, Lobdell H, IV, Walewski JL, Homma S, Berk PD. Cardiomyocyte triglyceride accumulation and reduced ventricular function occurs in mice with obesity and hepatic steatosis from many causes and reflects increased long chain fatty acid uptake and de novo fatty acid synthesis. J Obes. 2012 doi: 10.1155/2012/205648. #205648. Epub 2011 Nov 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stremmel W, Berk PD. Hepatocellular influx of [14C]oleate reflects membrane transport rather than intracellular metabolism or binding. Proc Natl Acad Sci U S A. 1986;83(10):3086–3090. doi: 10.1073/pnas.83.10.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwieterman W, Sorrentino D, Potter BJ, et al. Uptake of oleate by isolated rat adipocytes is mediated by a 40-kDa plasma membrane fatty acid binding protein closely related to that in liver and gut. Proc Natl Acad Sci U S A. 1988;85(2):359–363. doi: 10.1073/pnas.85.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stump DD, Nunes RM, Sorrentino D, Isola LM, Berk PD. Characteristics of oleate binding to liver plasma membranes and its uptake by isolated hepatocytes. J Hepatol. 1992;16(3):304–315. doi: 10.1016/s0168-8278(05)80661-0. [DOI] [PubMed] [Google Scholar]
- 34.Berk PD, Stump DD. Mechanisms of cellular uptake of long chain free fatty acids. Mol Cell Biochem. 1999;192(1–2):17–31. [PubMed] [Google Scholar]
- 35.Stump DD, Fan X, Berk PD. Oleic acid uptake and binding by rat adipocytes define dual pathways for cellular fatty acid uptake. J Lipid Res. 2001;42(4):509–520. [PubMed] [Google Scholar]
- 36.Schmider W, Fahr A, Blum HE, Kurz G. Transport of heptafluorostearate across model membranes: membrane transport of long-chain fatty acid anions. J Lipid Res. 2000;41(5):775–787. [PubMed] [Google Scholar]
- 37.Abumrad NA, Park JH, Park CR. Permeation of long-chain fatty acid into adipocytes: kinetics, specificity, and evidence for involvement of a membrane protein. J Biol Chem. 1984;259(14):8945–8953. [PubMed] [Google Scholar]
- 38.Weisiger RA, Fitz JG, Scharschmidt BF. Hepatic oleate uptake: electrochemical driving forces in intact rat liver. J Clin Invest. 1989;83(2):411–420. doi: 10.1172/JCI113899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glatz JF, van Nieuwenhoven FA, Luiken JJ, Schaap FG, van der Vusse GJ. Role of membrane-associated and cytoplasmic fatty acid-binding proteins in cellular fatty acid metabolism. Prostaglandins Leukot Essent Fatty Acids. 1997;57(4–5):373–378. doi: 10.1016/s0952-3278(97)90413-0. [DOI] [PubMed] [Google Scholar]
- 40.Luiken JJ, Glatz JF, Bonen A. Fatty acid transport proteins facilitate fatty acid uptake in skeletal muscle. Can J Appl Physiol. 2000;25(5):333–352. [PubMed] [Google Scholar]
- 41.Kampf JP, Kleinfeld AM. Fatty acid transport in adipocytes monitored by imaging intracellular free fatty acid levels. J Biol Chem. 2004;279(34):35775–35780. doi: 10.1074/jbc.M403630200. [DOI] [PubMed] [Google Scholar]
- 42.Kleinfeld AM, Kampf JP, Lechene C. Transport of 13C-oleate in adipocytes measured using multi-imaging mass spectrometry. J Am Soc Mass Spectrom. 2004;15(11):1572–158056. doi: 10.1016/j.jasms.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 43.Stremmel W, Strohmeyer G, Borchard F, Kochwa S, Berk PD. Isolation and partial characterization of a fatty acid binding protein in rat liver plasma membranes. Proc Natl Acad Sci U S A. 1985;82(1):4–8. doi: 10.1073/pnas.82.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bradbury MW, Berk PD. Cellular uptake of long chain free fatty acids: the structure and function of plasma membrane fatty acid binding protein. Adv Mol Cell Biol. 2004;33:47–81. [Google Scholar]
- 45.Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation: homology with human CD36. J Biol Chem. 1993;268(24):17665–17668. [PubMed] [Google Scholar]
- 46.Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79(3):427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 47.Stahl A, Gimeno RE, Tartaglia LA, Lodish HF. Fatty acid transport proteins: a current view of a growing family. Trends Endocrinol Metab. 2001;12(6):266–273. doi: 10.1016/s1043-2760(01)00427-1. [DOI] [PubMed] [Google Scholar]
- 48.Trigatti BL, Anderson RG, Gerber GE. Identification of caveolin-1 as a fatty acid binding protein. Biochem Biophys Res Commun. 1999;255(1):34–39. doi: 10.1006/bbrc.1998.0123. [DOI] [PubMed] [Google Scholar]
- 49.Pohl J, Ring A, Stremmel W. Uptake of long-chain fatty acids in HepG2 cells involves caveolae: analysis of a novel pathway. J Lipid Res. 2002;43(9):1390–1399. doi: 10.1194/jlr.m100404-jlr200. [DOI] [PubMed] [Google Scholar]
- 50.Kampf JP, Parmley D, Kleinfeld AM. Free fatty acid transport across adipocytes is mediated by an unknown membrane protein pump. Am J Physiol Endocrinol Metab. 2007;293(5):E1207–E1214. doi: 10.1152/ajpendo.00259.2007. [DOI] [PubMed] [Google Scholar]
- 51.Kampf JP, Kleinfeld AM. Is membrane transport of FFA mediated by lipid, protein, or both? An unknown protein mediates free fatty acid transport across the adipocyte plasma membrane. Physiology (Bethesda) 2007 Feb;22:7–14. doi: 10.1152/physiol.00011.2006. [DOI] [PubMed] [Google Scholar]
- 52.Berk PD, Wada H, Horio Y, Potter BJ, Sorrentino D, Zhou SL, Isola LM, Stump D, Kiang CL, Thung S. Plasma membrane fatty acid-binding protein and mitochondrial glutamic-oxaloacetic transaminase of rat liver are related. Proc Natl Acad Sci U S A. 1990;87(9):3484–3488. doi: 10.1073/pnas.87.9.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bradbury MW, Stump D, Guarnieri F, Berk PD. Molecular modeling and functional confirmation of a predicted fatty acid binding site in mitochondrial aspartate aminotransferase. J Mol Biol. 2011 Sep 23;412(3):412–22. doi: 10.1016/j.jmb.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watkins PA. Fatty acid activation. Progr Lipid Res. 1997;36:55–83. doi: 10.1016/s0163-7827(97)00004-0. [DOI] [PubMed] [Google Scholar]
- 55.Digel M, Ehehalt R, Stremmel W, Fullekrug J. Acyl-CoA synthetases: fatty acid uptake and metabolic channeling. Molec Cell Biochem. 2009;326:23–28. doi: 10.1007/s11010-008-0003-3. [DOI] [PubMed] [Google Scholar]
- 56.Hall AM, Smith AJ, Bernlohr DA. Characterization of the acyl-CoA synthetase activity of purified murine fatty acid transport protein 1. J Biol Chem. 2003;278:43008–43013. doi: 10.1074/jbc.M306575200. [DOI] [PubMed] [Google Scholar]
- 57.Hall AM, Wiczer BM, Herrmann T, Stremmel W, Bernlohr DA. Enzymatic properties of purified murine fatty acid transport protein 4 and analysis of acyl CoA synthetase activities in tissues from FATP4 null mice. J Biol Chem. 2005;280:11948–11954. doi: 10.1074/jbc.M412629200. [DOI] [PubMed] [Google Scholar]
- 58.Black PN, DiRusso CC. Transmembrane movement of exogenous long-chain fatty acids: proteins, enzymes, and vectorial esterification. Microbiol Mol Biol Rev. 2003;67:4544–4572. doi: 10.1128/MMBR.67.3.454-472.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalant D, Cianflone K. Regulation of fatty acid transport. Curr Opin Lipidol. 2004;15:309–314. doi: 10.1097/00041433-200406000-00011. [DOI] [PubMed] [Google Scholar]
- 60.Stremmel W, Staffer S, Wannhoff A, Pathil A. Plasma membrane phospholipase A2 controls hepatocellular fatty acid uptake and is responsive to pharmacologic modulation: implications for nonalcoholic steatohepatitis. FASEB J. 2014;28:3159–3170. doi: 10.1096/fj.14-249763. [DOI] [PubMed] [Google Scholar]
- 61.Berk PD, Zhou SL, Kiang CL, Stump D, Bradbury M, Isola LM. Uptake of long chain free fatty acids is selectively up-regulated in adipocytes of Zucker rats with genetic obesity and non-insulin-dependent diabetes mellitus. J Biol Chem. 1997 Mar 28;272(13):8830–8835. doi: 10.1074/jbc.272.13.8830. [DOI] [PubMed] [Google Scholar]
- 62.Chua SC, Jr, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia L, Leibel RL. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996 Feb 16;271(5251):994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 63.Wang B, Charukeshi-Chandrasekera P, Pippin JJ. Leptin- and leptin receptor-deficient rodent models: relevance for human type 2 diabetes. Curr Diabetes Revs. 2014;10:131–145. doi: 10.2174/1573399810666140508121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanches SC, Ramalho LN, Augusto MJ, da Silva DM, Ramalho FS. Nonalcoholic steatohepatitis: a search for factual animal models. Biomed Res Internat. 2015 doi: 10.1155/2015/574832. article ID 574832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berk PD, Zhou S, Kiang C, Stump DD, Fan X, Bradbury MW. Selective up-regulation of fatty acid uptake by adipocytes characterizes both genetic and diet-induced obesity in rodents. J Biol Chem. 1999 Oct 1;274(40):28626–28631. doi: 10.1074/jbc.274.40.28626. [DOI] [PubMed] [Google Scholar]
- 66.Petrescu O, Fan X, Gentileschi P, et al. Long-chain fatty acid uptake is upregulated in omental adipocytes from patients undergoing bariatric surgery for obesity. Int J Obes (Lond) 2005 Feb;29(2):196–203. doi: 10.1038/sj.ijo.0802868. [DOI] [PubMed] [Google Scholar]
- 67.Ge F, Walewski JL, Torghabeh MH, Lobdell iv H, Hu C, Zhou S, Dakin G, Pomp A, Bessler M, Schrope B, Ude-Welcome A, Inabnet WB, Feng T, Carras-Terzian E, Anglade D, Ebel FE, Berk PD. Facilitated long chain fatty acid uptake by adipocytes remains upregulated relative to BMI for more than a year after major bariatric surgical weight loss. Obesity (Silver Spring) 2016 Jan;24(1):113–22. doi: 10.1002/oby.21249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fan X, Bradbury MW, Berk PD. Leptin and insulin modulate nutrient partitioning and weight loss in ob/ob mice through regulation of long-chain fatty acid uptake by adipocytes. J Nutr. 2003 Sep;133(9):2707–2715. doi: 10.1093/jn/133.9.2707. [DOI] [PubMed] [Google Scholar]
- 69.Vacca M, Allison M, Griffin JL, Vidal-Puig A. Fatty acid and glucose sensors in hepatic lipid metabolism: implications for NAFLD. Semin Liv Dis. 2015 Aug;35(5):250–261. doi: 10.1055/s-0035-1562945. [DOI] [PubMed] [Google Scholar]
- 70.Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003;14:281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 71.Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis. 2004 Aug;8(3):639–671. doi: 10.1016/j.cld.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 72.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996 Feb 15;379(6566):632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 73.Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CJ, Hess JF. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996 May;13(1):18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994 Dec 1;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 75.Maclean PS, Bergouignan A, Cornier MA, Jackman MR. Biology’s response to dieting: the impetus for weight regain. Am J Physiol Regul Integr Comp Physiol. 2011 Sep;301(3):R581–R600. doi: 10.1152/ajpregu.00755.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blomain ES, Dirhan DA, Valentino MA, Kim GW, Waldman SA. Mechanisms of weight regain following weight loss. ISRN Obes. 2013 Apr;16:2013. doi: 10.1155/2013/210524. Article ID 210524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Greenway FL. Physiological adaptations to weight loss and factors favouring weight regain. Int J Obes (Lond) 2015 Apr 21; doi: 10.1038/ijo.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Courcoulas AP, Christian NJ, Belle SH, Berk PD, Flum DR, Garcia L, Horlick M, Kalarchian MA, King WC, Mitchell JE, Patterson EJ, Pender JR, Pomp A, Pories WJ, Thirlby RC, Yanovski SZ, Wolfe BM, Longitudinal Assessment of Bariatric Surgery (LABS) Consortium Weight change and health outcomes at 3 years after bariatric surgery among individuals with severe obesity. JAMA. 2013 Dec 11;310(22):2416–2425. doi: 10.1001/jama.2013.280928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee Y, Wang MY, Kakuma T, et al. Liporegulation in diet induced obesity: the antisteatotic role of hyperleptinemia. J Biol Chem. 2001;276(8):5629–5635. doi: 10.1074/jbc.M008553200. [DOI] [PubMed] [Google Scholar]
- 80.Unger RH. Lipotoxic diseases. Annu Rev Med. 2002;53:319–336. doi: 10.1146/annurev.med.53.082901.104057. [DOI] [PubMed] [Google Scholar]
- 81.Unger RH. The physiology of cellular liporegulation. Annu Rev Physiol. 2003;65:333–347. doi: 10.1146/annurev.physiol.65.092101.142622. [DOI] [PubMed] [Google Scholar]
- 82.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269(5223):543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 83.Pelleymounter MA, Cullen MJ, Baker MB, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269(5223):540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 84.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269(5223):546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 85.Remesar X, Rafecas I, Fernandez-Lopez JA, Alemany M. Is leptin an insulin counter-regulatory hormone? FEBS Lett. 1997;402(1):9–11. doi: 10.1016/s0014-5793(96)01477-9. [DOI] [PubMed] [Google Scholar]
- 86.Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 87.Oh-I S, Shimizu H, Sato T, Uehara Y, Okada S, Mori M. Molecular mechanisms associated with leptin resistance: n-3 polyunsaturated fatty acids induce alterations in the tight junction of the brain. Cell Metab. 2005;1(5):331–341. doi: 10.1016/j.cmet.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 88.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276(18):14890–14895. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 89.Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J. 2001;15(2):312–321. doi: 10.1096/fj.00-0590. [DOI] [PubMed] [Google Scholar]
- 90.Unger RH. Minireview: weapons of lean body mass destruction—the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144(12):5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 91.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401(6748):73–76. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 92.Mirabeau O, Perlas E, Severini C, Audero E, Gascuel O, Possenti R, Birney E, Rosenthal N, Gross C. Identification of novel peptide hormones in the human proteome by hidden Markov model screening. Genome Res. 2007;17:320–327. doi: 10.1101/gr.5755407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Walewski JL, Ge F, Lobdell H, 4th, Levin N, Schwartz GJ, Vasselli JR, Pomp A, Dakin G, Berk PD. Spexin is a novel human peptide that reduces adipocyte uptake of long chain fatty acids and causes weight loss in rodents with diet-induced obesity. Obesity (Silver Spring) 2014 Jul;22(7):1643–1652. doi: 10.1002/oby.20725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haynes P, Liangpunsakul S, Chalasani N. Nonalcoholic fatty liver disease in individuals with severe obesity. Clin Liver Dis. 2004 Aug;8(3):535–547. doi: 10.1016/j.cld.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 95.Zhou SL, Gordon RE, Bradbury M, Stump D, Kiang CL, Berk PD. Ethanol up-regulates fatty acid uptake and plasma membrane expression and export of mitochondrial aspartate aminotransferase in HepG2 cells. Hepatology. 1998 Apr;27(4):1064–1074. doi: 10.1002/hep.510270423. [DOI] [PubMed] [Google Scholar]
- 96.Berk PD, Zhou S, Bradbury MW. Increased hepatocellular uptake of long chain fatty acids occurs by different mechanisms in fatty livers due to obesity or excess ethanol use, contributing to development of steatohepatitis in both settings. Trans Am Clin Climatol Assoc. 2005;116:335–344. [PMC free article] [PubMed] [Google Scholar]
- 97.Yeh MM, Brunt EM. Pathological features of fatty liver disease. Gastroenterol. 2014 Oct;147(4):754–64. doi: 10.1053/j.gastro.2014.07.056. Epub 2014 Aug 7. [DOI] [PubMed] [Google Scholar]
- 98.Kleiner DE, Berk PD, Hsu JY, Courcoulas AP, Flum D, Khandelwal S, Pender J, Pomp A, Roerig J, Machado LL, Wolfe BM, Belle SH, LABS Consortium Hepatic pathology among patients without known liver disease undergoing bariatric surgery: observations and a perspective from the Longitudinal Assessment of Bariatric Surgery (LABS) study. Semin Liver Dis. 2014;34(1):98–107. doi: 10.1055/s-0034-1371083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Neuschwander-Tetri BA, Clark JM, Bass NM, Van Natta ML, Unalp-Arida A, Tonascia J, Zein CO, Brunt EM, Kleiner DE, McCullough AJ, Sanyal AJ, Diehl AM, Lavine JE, Chalasani N, Kowdley KV, NASH Clinical Research Network Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology. 2010 Sep;52(3):913–924. doi: 10.1002/hep.23784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998 Apr;114(4):842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 101.Charlton M. Noninvasive indices of fibrosis in NAFLD: starting to think about a three-hit (at least) phenomenon. Am J Gastroenterol. 2007 Feb;102(2):409–411. doi: 10.1111/j.1572-0241.2006.01039.x. [DOI] [PubMed] [Google Scholar]
- 102.McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006 Mar;40(3 Suppl 1):S17–29. doi: 10.1097/01.mcg.0000168645.86658.22. [DOI] [PubMed] [Google Scholar]
- 103.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008 Apr 1;44(7):1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mahli H, Gores G. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liv Dis. 2008;28(4):360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Diehl A. Mechanisms of disease and progression in NAFLD. Semin Liv Dis. 2008;28(4):370–379. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- 106.Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, Musso A, De Paolis P, Capussotti L, Salizzoni M, Rizzetto M. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterol. 2002 Jul;123(1):134–140. doi: 10.1053/gast.2002.34168. [DOI] [PubMed] [Google Scholar]
- 107.Pocha C, Kolly P, Dufour J-F. Nonalcoholic fatty liver disease-related hepatocellular carcinoma – a problem of growing magnitude. Semin Liver Dis. 2015 Aug;35(3):304–317. doi: 10.1055/s-0035-1562949. [DOI] [PubMed] [Google Scholar]
- 108.Torres DM, Harrison SA. Nonalcoholic steatohepatitis and noncirrhotic hepatocellular carcinoma: fertile soil. Semin Liver Dis. 2012 Feb;32(1):30–38. doi: 10.1055/s-0032-1306424. [DOI] [PubMed] [Google Scholar]
- 109.Ponziani FR, Pecere S, Gasbarrini A, Ojetti V. Physiology and pathophysiology of liver lipid metabolism. Expert Rev Gastroenterol Hepatol. 2015 Aug;9(8):1055–67. doi: 10.1586/17474124.2015.1056156. Epub 2015 Jun 12. [DOI] [PubMed] [Google Scholar]
- 110.Nguyen V, George J. NAFLD Therapy: Dietary & Lifestyle Modification. Semin Liv Dis. 2015 Aug;35(3):jjj–kkk. doi: 10.1055/s-0035-1562950. [DOI] [PubMed] [Google Scholar]
- 111.Gawrieh S, Chalasani N. Pharmacotherapy for Non-alcoholic Fatty Liver Disease. Semin Liv Disease. 2015 Aug;35(3):338–348. doi: 10.1055/s-0035-1562951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Filozof C, Goldstein BJ, Williams RN, Sanyal A. Non-alcoholic steatohepatitis: limited available treatment options but promising drugs in development and recent progress towards a regulatory approval pathway. Drugs. 2015 Jul 23; doi: 10.1007/s40265-015-0437-3. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takahashi Y, Sugimoto K, Inui H, Fukusato T. Current pharmacological therapies for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2015 Apr 7;21(13):3777–3785. doi: 10.3748/wjg.v21.i13.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]