

Abstract
Background and Purpose
Airway microvascular leak (MVL) involves the extravasation of proteins from post‐capillary venules into surrounding tissue. MVL is a cardinal sign of inflammation and an important feature of airway inflammatory diseases such as asthma. PGE2, a product of COX‐mediated metabolism of arachidonic acid, binds to four receptors, termed EP1–4. PGE2 has a wide variety of effects within the airway, including modulation of inflammation, sensory nerve activation and airway tone. However, the effect of PGE2 on airway MVL and the receptor/s that mediate this have not been described.
Experimental Approach
Evans Blue dye was used as a marker of airway MVL, and selective EP receptor agonists and antagonists were used alongside EP receptor‐deficient mice to define the receptor subtype involved.
Key Results
PGE2 induced significant airway MVL in mice and guinea pigs. A significant reduction in PGE2‐induced MVL was demonstrated in Ptger2 −/− and Ptger4 −/− mice and in wild‐type mice pretreated simultaneously with EP2 (PF‐04418948) and EP4 (ER‐819762) receptor antagonists. In a model of allergic asthma, an increase in airway levels of PGE2 was associated with a rise in MVL; this change was absent in Ptger2 −/− and Ptger4 −/− mice.
Conclusions and Implications
PGE2 is a key mediator produced by the lung and has widespread effects according to the EP receptor activated. Airway MVL represents a response to injury and under ‘disease’ conditions is a prominent feature of airway inflammation. The data presented highlight a key role for EP2 and EP4 receptors in MVL induced by PGE2.
Abbreviations
- BAFL
bronchiolar lavage fluid
- COPD
chronic obstructive pulmonary disease
- IPA
intrapulmonary airways
- MVL
microvascular leakage
- OVA
ovalbumin
Tables of Links
| LIGANDS | |
|---|---|
| 5‐HT | ONO‐AE1‐259 |
| Diclofenac | ONO‐AE1‐329 |
| ER‐819762 | PGE2 |
| Evans blue dye | PF‐04418948 |
| ONO‐AE‐248 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Airway microvascular leakage (MVL) and plasma exudation represent classical features in the pathogenesis of various respiratory diseases, including asthma and chronic obstructive pulmonary disease (COPD) (Paredi and Barnes, 2009). The bronchial microvasculature has a multitude of important functions that are essential for maintaining pulmonary homeostasis, but during an inflammatory response this barrier can be disrupted allowing fluid and large macromolecules to move into the surrounding tissues through interendothelial gaps. Many mediators (e.g. cysteinyl leukotrienes, histamine, bradykinin, 5‐HT and cytokines) are capable of inducing this effect when released in response to an inflammatory insult in the airway, where they can act upon the endothelium of post‐capillary venules to open these intercellular gaps. This effect causes plasma to ‘leak’ out into extravascular sites because of hydrostatic pressure gradients (Olivenstein et al., 1997; Reynolds et al., 2002; Greiff et al., 2003). This phenomenon is a very distinctive feature of acute inflammation but is also observed in more chronic diseases such as asthma (Laitinen et al., 1987; Li and Wilson, 1997; Innes et al., 2009; Khor et al., 2009) and COPD (Hill et al., 1999; Minakata et al., 2005; Bessa et al., 2012).
PGE2 is an endogenous lipid eicosanoid synthesized by COX‐mediated metabolism of free arachidonic acid. It is produced in a variety of cells, including airway smooth muscle, epithelial cells, alveolar macrophages and pulmonary endothelial cells (Meyrick et al., 1989; Widdicombe et al., 1989). It exerts its biological effects via activation of four cell‐surface GPCRs, termed EP1–4, encoded for by the genes Ptger1–4 (Coleman et al., 1994). Increased levels of PGE2 have been reported in bronchiolar lavage fluid (BALF) and plasma of asthma patients (Brightling et al., 2000; Birring et al., 2003; Long et al., 2004; Sastre et al., 2008), allergen‐challenged mice (Herrerias et al., 2009) and enhanced levels in the exhaled breath condensate of COPD patients (Montuschi et al., 2003; Chen et al., 2008; Antczak et al., 2012).
Previous work from our lab, and others, has demonstrated that the bronchodilator effects of PGE2 occur through the activation of the EP4 receptor (Buckley et al., 2011; Benyahia et al., 2012), whereas airway sensory nerve activation and cough appear to be via the activation of the EP3 receptor (Maher et al., 2009). However, the effect of PGE2 and the EP receptors on airway MVL has not been extensively investigated. To investigate this, Evans Blue dye was used as a marker of MVL, a method that has been used for decades to quantify vascular permeability in various tissues and a variety of species (Miles and Miles, 1952; Evans et al., 1987; Rogers et al., 1988; Baluk et al., 1999; Reynolds et al., 2002; Xie et al., 2003; Zhuang et al., 2011). First, we established the responses to PGE2 in both mouse and guinea pig airways. Then using a pharmacological approach and EP receptor‐deficient mice, we provided substantial evidence that both the EP2 and EP4 receptors mediate PGE2‐induced airway MVL.
Methods
Animals
Male C57BL/6 mice (20–25 g) and male Dunkin Hartley guinea pigs (250–350 g) were purchased from Harlan (Bicester, Oxon, UK). Homozygous breeding pairs of mice genetically modified to disrupt one of the following genes: Ptger1 (EP1), Ptger2 (EP2) and Ptger3 (EP3) (Ushikubi et al., 1998), had been backcrossed at least eight times onto the C57BL/6 background. Ptger4 −/− (EP4) mice do not survive on the C57BL/6 background because of patent ductus arteriosus (Segi et al., 1998), so they were backcrossed on a mixed background of 129/Ola X C57bl/6. Mice were kindly provided by Dr Shuh Narumiya, Kyoto University, and breeding colonies maintained at Imperial College, London. All animals were housed in individually ventilated cages and provided with food and water ad libitum in a controlled environment. A 12 h light–dark cycle was maintained for all animals. All studies and procedures were approved by the Imperial College, Animal Welfare and Ethical Review Body, and performed in accordance with Home Office guidelines under the Animals (Scientific Procedures) Act of 1986 and the ARRIVE guidelines (Kilkenny et al., 2010) and the editorial on reporting animal studies (McGrath and Lilley, 2015).
General methodology for measuring microvascular leak
Apart for the data generated in the various mouse knockout line, animals were randomised as regards the treatment groups. Animals were anaesthetized with i.p. urethane (2 g·kg−1; 200 μL of a 25% solution given at two sites, followed by a further 50–100 μL as required). Mice breathe spontaneously under urethane and so were therefore not artificially ventilated. Depth of anaesthesia was assessed using the pedal and corneal reflex. Once surgical anaesthesia was reached, the jugular veins were then exposed following a midline incision over the thorax. The administration of substances i.v. was achieved by passing the injection needle through the pectoralis major as this helped prevent bleeding on withdrawal. Animals received Evans Blue dye (20 mg·kg−1) followed by the specific mediator or its vehicle 1 min later (4 mL·kg−1). The doses and timings used were those shown to be effective in previous studies within the group and from the literature (Hele et al., 2001; Belvisi et al., 2009). Thirty minutes after vehicle or compound administration, the animals were killed by an overdose of anaesthesthetic (pentobarbitone 200 mg kg−1, i.p.), the thoracic cavity was opened and a small incision was made in the left ventricle and also the left atrium of the heart. A cannula was then inserted into the left ventricle and the systemic circulation perfused with sterile saline (0.9%) at a pressure of approximately 100 mmHg. This was continued until the perfusate ran clear (2–5 min). The purpose of this was to remove any intravascular dye. The heart, lungs and oesophagus were removed en bloc and then the trachea and lungs separated from the heart and oesophagus. The oesophagus and bladder were taken for the initial studies and used as non‐airway, reference tissues. The trachea was isolated by cutting just above the bifurcation of the bronchi and the larynx removed. The parenchyma was then carefully scraped off using a scalpel to reveal the intrapulmonary airways (IPA). The trachea, the bronchi and IPA, the oesophagus and the bladder were then all weighed, and the wet tissue weight was recorded. Each tissue was then incubated in 120 μL of formamide at 37.5°C for at least 18 h to facilitate the extraction of Evans Blue dye. The concentration of Evans Blue extracted from each tissue was determined by light absorbance at 620 nm using a spectrophotomer; 100 μL of formamide was removed from each Eppendorf and pipetted into a 96‐well plate alongside a standard curve of Evans Blue in formamide (0, 0.3125, 0.625, 1.25, 2.5, 5, 10 and 20 μg·mL−1). The concentration was then calculated by interpolation from the standard curve and expressed as ng·mg−1 of tissue. End points were assessed by a different operator than the experimental part of the study.
Experimental design
PGE2‐induced airway microvascular leak
A dose–response curve to PGE2 was established where male C57BL/6 mice were given PGE2 (0.1, 0.3, 1, 3 or 10 mg·kg−1 at 4 mL·kg−1) and 5‐HT (10 mg·kg−1 at 4 mL·kg−1) as a positive control or vehicle (1% ethanol in saline). Thirty minutes after administration, Evans Blue extravasation was measured. A non‐selective COX inhibitor, diclofenac (30 mg·kg−1 in 10 mL·kg−1) (Mitchell et al., 1993), or vehicle (0.5% methylcellulose, 0.2% Tween 80 in water) was administered orally 60 min prior to PGE2 administration to block endogenous prostanoids that may influence the baseline or PGE2‐induced response. Having assessed the effect of i.v. PGE2, administration via topical delivery was then investigated to mimic what might happen in airway disease. Here, mice were anaesthetized and then dosed intra‐nasally with either vehicle (1% ethanol in saline) or PGE2 (3 mg·kg−1) in 50 μL. Once surgical anaesthesia was reached, mice were then given Evans Blue and killed 30 min later.
Guinea pigs were chosen as a second (larger) species to assess PGE2‐induced airway MVL as they more closely resemble humans in terms of their respiratory anatomy (lobing and branching) and physiology (mediator release). Male Dunkin–Hartley guinea pigs (300–600 g) were anaesthetized with urethane (2 g·kg−1 i.p. of a 25% solution). Once surgical anaesthesia was reached, animals received Evans Blue (20 mg·mL−1 at 1 mL·kg−1 i.v.) and then 1 min later vehicle (1% ethanol in saline), PGE2 (3 mg·kg−1) or 5‐HT (1 mg·kg−1), all i.v. at 1 mL kg−1; 5 min after 5‐HT or 30 min after PGE2, the extravasation of Evans Blue was measured.
Identifying the EP receptor mediating PGE2‐induced airway microvascular leak: EP receptor‐deficient mice
The same MVL protocol was used as before with a single submaximal dose of PGE2 (3 mg·kg−1) or vehicle (1% ethanol in saline) used. Additionally, the response to 5‐HT in EP receptor knockouts (ptger2 −/− and ptger4 −/−) and their wild types was investigated to ensure that their lack of response to PGE2 was not due to an overall disruption in MVL. A submaximal dose of 5‐HT (1 mg·kg−1) was used as a known inducer of MVL.
Identifying the EP receptor mediating PGE2‐induced airway microvascular leak: selective EP receptor agonists
To parallel the effects seen in the EP receptor‐deficient mice, the response to selective EP agonists was studied. Following Evans Blue administration, animals were given selective EP receptor agonists (Okada et al., 2000; Suzawa et al., 2000; Cao et al., 2002): ONO‐D1‐004 (EP1), ONO‐AE1‐259 (EP2), ONO‐AE‐248 (EP3), ONO‐AE1‐329 (EP4) or PGE2 (3 mg·kg−1) i.v. at 4 mL·kg−1 or vehicle (1% ethanol in saline).
Inhibition of EP2‐ and EP4‐induced microvascular leak using selective EP2 and EP4 receptor antagonists
Selective receptor antagonists were used to confirm data generated with selective agonists. Initially, a dose response to ONO‐AE1‐259 (EP2 agonist) was performed to establish a suitable dose to use. The doses used were 0.1, 0.3, 1 and 3 mg·kg−1 (i.v. at 4 mL·kg−1). The EP2 antagonist, PF‐04418948, was used in subsequent studies and was dosed at 3, 10, 30 or 100 mg·kg−1 (10 mL·kg−1 i.p.). A vehicle control was also included (0.5% methylcellulose, 0.2% Tween 80 in saline). Mice were dosed with PF‐04418948 (af Forselles et al., 2011) or vehicle 1 h before ONO‐AE1‐259 administration (3 mg·kg−1 in 4 mL·kg−1 i.v.). Next, a dose–response curve to ONO‐AE1‐329 (EP4 agonist) was carried out using 0.1, 0.3, 1 and 3 mg·kg−1 (i.v. at 4 mL·kg−1). The EP4 antagonist, ER‐819762 (Chen et al., 2010), was used in subsequent studies and was dosed at 3, 10, 30 or 100 mg·kg−1 (10 mL·kg−1 i.p.). A vehicle control was also included (0.5% methylcellulose, 0.2% Tween 80 in saline). Mice were dosed with ER‐819762 or vehicle 1 h before ONO‐AE1‐329 administration (3 mg·kg−1 in 4 mL·kg−1 i.v.).
Inhibition of PGE2‐induced microvascular leak by EP2 and EP4 receptor antagonists
Mice were dosed with PF‐04418948 (10 mg·kg−1 at 10 mL·kg−1 i.p.) or ER‐819762 (30 mg·kg−1 at 10 mL·kg−1 i.p.) or vehicle (0.5% methylcellulose, 0.2% Tween 80 in saline) 1 h before PGE2 administration (3 mg·kg−1 at 4 mL·kg−1 i.v.). Additionally, in a separate study, mice were dosed with both PF‐04418948 and ER‐819762 simultaneously 1 h before PGE2.
Identifying the EP receptors involved in ovalbumin‐induced microvascular leak: EP receptor‐deficient mice
Mice were sensitized with ovalbumin (OVA) (10 μg per mouse, 100 μL i.p.) on days 0 and 14 and on days 24, 25 and 26 mice were intranasally challenged with saline or OVA (50 μg in 50 μL) under isoflurane anaesthesia. PGE2 levels in the BALF were measured using LC‐MS/MS following lipid extraction as follows: 5 ng PGE2‐d4 was added to samples before extraction, as an internal standard. Lipids were extracted by adding a solvent mixture (1 mol·L−1 acetic acid, isopropyl alcohol, hexane (2:20:30, v v−1v−1)) to the sample at a ratio of 2.5 to 1 mL, vortexing and then adding 2.5 mL of hexane. After vortexing and centrifugation, lipids were recovered in the upper hexane layer. The samples were then re‐extracted by addition of an equal volume of hexane. The combined hexane layers were dried and analysed for free or esterified PGs using LC‐MS/MS. Lipids were separated on a C18 Spherisorb ODS2, 5 μm, 150 × 4.6 mm column (Waters, Hertfordshire, UK) using a gradient of 50–90% B over 10 min (A, water : acetonitrile : acetic acid, 75:25:0.1; B, methanol : acetonitrile : acetic acid, 60:40:0.1) with a flow rate of 1 mL·min−1. Products were quantified by LC‐MS/MS electrospray ionization on a Sciex 4000 Q‐Trap using parent‐to‐daughter transitions of m/z 351.2 [M − H]− to m/z 271 for PGE2 and m/z 355.2 to 275.3 for PGE2‐d4 with declustering potential of −55 and collision energy of −26 V. Products were identified and quantified using standards run in parallel under the same conditions.
Based on these data, MVL was measured at 2 and 24 h after the final intranasal challenge. In a subsequent study, allergy‐induced MVL was compared in wild‐type and EP receptor knockout (ptger2 −/− and ptger4 −/−) mice.
Compounds and materials
The EP2 receptor antagonist, PF‐04418948, was a gift from Nick Pullen, Pfizer (Kent, UK). The EP4 antagonist, ER‐819762, was a gift from Eisai (Hertfordshire, UK). The EP1 receptor agonist (ONO‐D1‐004), the EP2 receptor agonist (ONO‐AE1‐259), the EP3 receptor agonist (ONO‐AE‐248) and the EP4 receptor agonist (ONO‐AE1‐329) were gifts from Ono Pharmaceuticals (Osaka, Japan). PGE2 was purchased from Cayman Europe (Tallinn, Estonia). Both antagonists were made up in 0.5% methylcellulose, 0.2% Tween 80 in saline (or water for p.o. administration). PGE2 and all agonists were made up in 1% ethanol in saline. All other chemicals and reagents were from Sigma Aldrich (Poole, UK).
Data analysis and statistical procedures
All data were analysed using GraphPad Prism 5. Data are expressed as mean ± SEM. Multiple measurements were analysed by one‐way ANOVA; non‐parametric Kruskal–Wallis test and post‐hoc comparisons were performed by Dunn's multiple comparison test, comparing selected columns to a control. Additionally, an unpaired t‐test and/or non‐parametric Mann–Whitney test, with two‐tailed P values, was carried out, where appropriate, to determine statistical significance between two groups. Differences were considered statistically significant if P < 0.05. The figures have been graphically presented on a range‐specific axis. The data and statistical analysis comply with British Journal of Pharmacology guidelines (Curtis et al., 2015).
Results
PGE2‐induced airway microvascular leak
PGE2 (0.1, 0.3, 1, 3, 10 mg·kg−1, i.v.) induced dose‐dependent MVL in both the trachea and the bronchi and IPA (Fig. 1A, B). From this, a submaximal dose of 3 mg·kg−1 was chosen. No statistically significant increase in MVL was detected in either the bladder or oesophagus after PGE2 challenge [vehicle = 58.1 ± 20.3 vs. PGE2 = 46.5 ± 21.2, vehicle = 25.4 ± 15.1 vs. PGE2 = 32.2 ± 8, Evans Blue (ng·mg−1 of tissue), respectively]. Diclofenac, a nonselective COX inhibitor, was administered prior to PGE2 administration to block the production and release of endogenous prostanoids to ensure that they were not influencing baseline or PGE2‐induced airway leak. The data indicate that diclofenac had no effect on baseline or PGE2‐induced leak in the trachea or bronchi and IPA (Fig. 1C, D). In both the vehicle‐ and diclofenac‐treated groups, PGE2 significantly increased airway MVL. Having established that i.v. PGE2 induced MVL in murine airways, we wanted to examine whether such a response could be replicated via topical administration direct to the airways. Therefore, a single intranasal dose of PGE2 (3 mg·kg−1) was administered and EB leak measured after 30 min. Topical PGE2 induced a similar MVL response to that seen with i.v administration (Fig. 2A, B). Last, to determine whether the effect seen with PGE2 was exclusive to the mouse, a submaximal dose of PGE2 was administered to guinea pigs alongside 5‐HT, acting as a positive control. PGE2 (3 mg·kg−1 i.v.) induced significant MVL in the trachea, bronchi and IPAs was comparable with 5‐HT (Fig. 2C–E).
Figure 1.
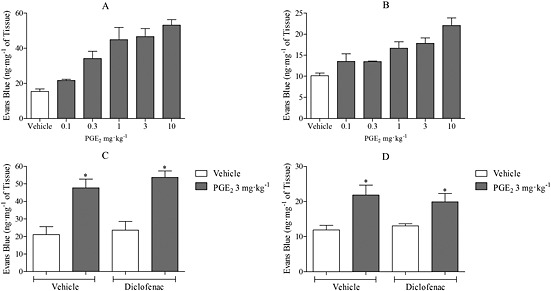
The effect of PGE2 on MVL in the trachea and IPA of C57BL/6 mice. (A) Dose–response to PGE2 (0.1–10 mg·kg−1 i.v., n = 3–4) after 30 min (A: trachea, B: bronchi and IPA). Effect of diclofenac (30 mg·kg−1 in 10 mL·kg−1 p.o.; 1 h) on MVL in vehicle and PGE2 (3 mg·kg−1 i.v.; 30 min) treated mice (n = 4) (C: trachea, D: bronchi and IPA). Data expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of tissue). *P < 0.05 indicates significance of treatment groups from vehicle control.
Figure 2.
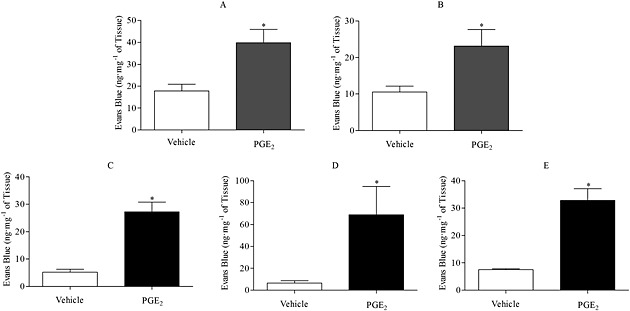
Effect of an intranasal dose of PGE2 (3 mg·kg−1 intranasally, n = 6) on MVL after 30 min into the trachea (A) or bronchi and IPA (B) of mice. Effect of PGE2 (3 mg·kg−1 i.v.) on MVL into the upper trachea (C), lower trachea (D) or bronchi and IPA (E) of Dunkin–Hartley guinea pigs. Data expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of tissue). *P < 0.05 indicates significance of treatment groups from vehicle control.
PGE2‐induced airway microvascular leak in EP receptor‐deficient mice: role for EP2 and EP4 receptors
The effect of PGE2 (3 mg·kg−1) on MVL was compared in wild type and mice deficient in individual EP receptors (Ptger1–4 −/−). The data demonstrated a significant increase in MVL in wild‐type, Ptger1 −/− and Ptger3 −/− mice in response to PGE2 (Fig. 3A and C). However, a substantial and significant reduction in PGE2‐induced MVL was shown in Ptger2 −/− and Ptger4 −/− mice (Fig. 3A–D). This was apparent in the trachea and the bronchi and IPA. To determine whether their lack of response to PGE2 was not due to an overall disruption in MVL, 5‐HT was administered to Ptger2 −/− and Ptger4 −/− mice. Here, 5‐HT produced a significant increase in MVL in the trachea and in the bronchi and IPA, with no difference seen between the EP receptor‐deficient mice and the wild types (Fig. 4).
Figure 3.
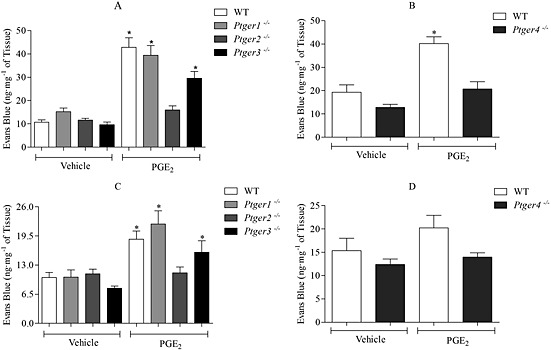
Investigating the responses to PGE2 (3 mg·kg−1 i.v., n = 6) in wild‐type (WT) and EP1–4 receptor‐deficient mice (A and B) trachea and (C and D) bronchi and IPAs. EP1–3 receptor‐deficient mice are bred on a C57BL/6 background, and EP4 receptor‐deficient mice are bred on a mixed background of 129/Ola X C57BL/6. Data expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of tissue). *P < 0.05 indicates significance from vehicle control.
Figure 4.
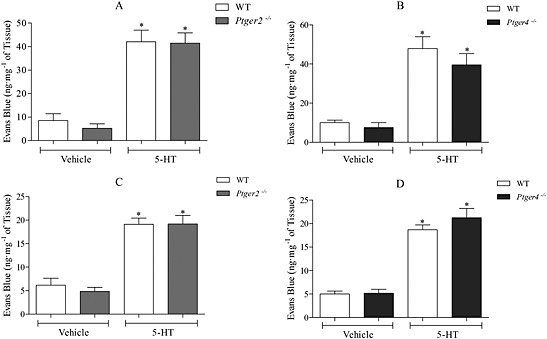
Effect of 5‐HT (1 mg·kg−1 i.v., n = 6) on MVL in wild‐type (WT) and EP2 or EP4 receptor‐deficient mice (A and B) trachea and (C and D) bronchi and IPA. EP2 receptor‐deficient mice were bred on a C57BL/6 background, and EP4 receptor‐deficient mice were bred on a mixed background of 129/Ola X C57BL/6. Data expressed as mean ± SEM of the concentration of Evans blue dye (ng·mg−1 of tissue). *P < 0.05 indicates a significant increase from vehicle control.
Selective EP2 and EP4 receptor agonists induce airway microvascular leak
Having shown an inhibition in PGE2‐induced MVL in EP2 and EP4 receptor‐deficient mice, selective EP receptor agonists were investigated. ONO‐AE1‐259 (EP2) and ONO‐AE1‐329 (EP4) both significantly increased MVL in both the trachea and bronchi and IPA of mice, while ONO‐D1‐004 (EP1) and ONO‐AE‐248 (EP3) had little or no effect on MVL in either trachea or bronchi and IPA (Fig. 5).
Figure 5.
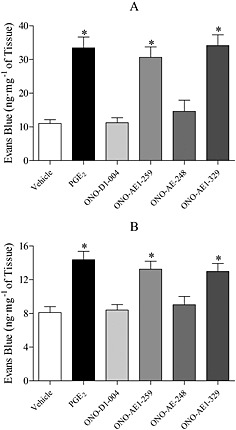
Effect of PGE2, ONO‐D1‐004 (EP1), ONO‐AE1‐259 (EP2), ONO‐AE‐248 (EP3) and ONO‐AE1‐329 (EP4) (3 mg·kg−1 i.v.; 30 min) on MVL into the trachea (A) and bronchi and IPA (B) of C57BL/6 mice. Data expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of tissue), n = 6. *P < 0.05 indicates a significant increase from vehicle control.
Inhibition of EP2‐ and EP4‐induced microvascular leak using selective EP2 and EP4 receptor antagonists
The EP2 agonist (ONO‐AE1‐259) produced a dose‐dependent increase in MVL. Similar to previous studies 3 mg·kg−1 induced a significant increase in MVL in the trachea (data not shown). This dose was therefore used in subsequent antagonist studies. The EP2 receptor antagonist, PF‐04418948 (i.p.), evoked a dose‐dependent inhibition with a dose of 30 mg·kg−1 showing a significant inhibition of EP2‐induced MVL (Fig. 6A, B). ONO‐AE1‐329 (EP4 agonist) produced a dose‐dependent increase in MVL with a significant increase in airway MVL at 3 mg·kg−1 (data not shown). The EP4 receptor antagonist, ER‐819762 (i.p.), was able to significantly inhibit MVL induced by ONO‐AE1‐329 (Fig. 6C, D). PF‐04418948 (30 mg·kg−1 i.p.) and ER‐819762 (10 mg·kg−1 i.p.) were selected for further study.
Figure 6.
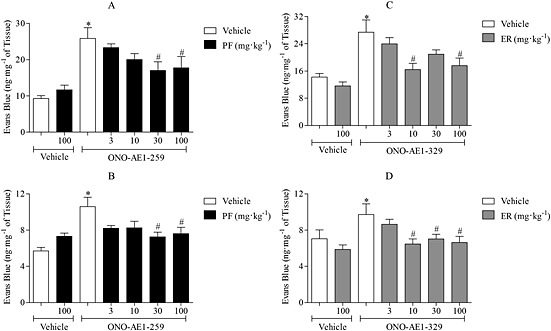
Inhibition of EP2‐ and EP4‐induced MVL using selective EP receptor antagonists. Effect of the EP2 receptor antagonist, PF‐04418948 (PF; 3, 10, 30 and 100 mg·kg−1 i.p.; 1 h), on ONO‐AE1‐259‐induced MVL (3 mg·kg−1 i.v.) in trachea (A) and bronchi/IPA (B). The effect of the EP4 receptor antagonist, ER‐819762 (3, 10, 30 and 100 mg·kg−1 i.p.; 1 h), on ONO‐AE1‐329‐induced MVL (3 mg·kg−1 i.v.) in trachea (C) and bronchi and IPA (D). Data expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of tissue), n = 6. *P < 0.05 indicates a significant increase from vehicle control. #P < 0.05 indicates a significant decrease from treatment control.
Inhibition of PGE2‐induced microvascular leak by EP2 and EP4 receptor antagonists
The effect of PF‐04418948 and ER‐819762 on PGE2‐induced MVL in mouse airways was investigated. Initially, each antagonist was dosed separately; however, as demonstrated in Fig. 7A and B, neither antagonist alone inhibited PGE2‐induced airway MVL. When both antagonists were given simultaneously, there was a significant reduction in PGE2‐induced MVL in the trachea and in the bronchi and IPA (Fig. 7C, D).
Figure 7.
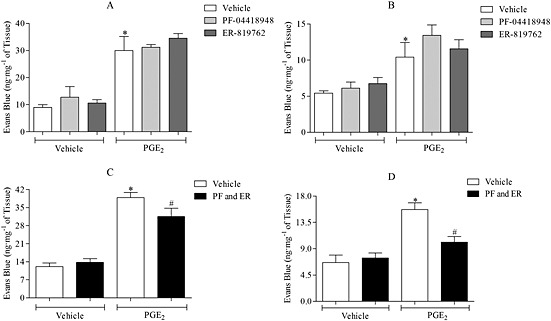
Effect of PF‐04418948 (PF; 30 mg·kg−1 i.p.; 1 h) and ER‐819762 (ER; 10 mg·kg−1 i.p.; 1 h) on PGE2‐induced MVL (3 mg·kg−1 i.v.; 30 min) in the trachea (A) and bronchi and IPA (B) of mice. In the top two graphs, each antagonist was dosed separately (n = 5–6), while in the bottom two graphs, both antagonists were given simultaneously to the same animal in trachea (C) and bronchi and IPA (D). Data expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of tissue). *P < 0.05 indicates a significant increase from vehicle control. #P < 0.05 indicates a significant decrease from treatment control.
The role of PGE2 and EP2 and EP4 receptors in asthma‐related airway microvascular leak
The effect of antigen challenge on BALF PGE2 levels in a model of allergic asthma was investigated. The antigen caused an increase in BALF PGE2 levels, with levels increased at 2 and 6 h after challenge and returning to basal by 24 h (Fig. 8A). This temporal increase coincided with a significant increase in airway MVL 2 h after challenge, but not after 24 h (Fig. 8B, C).
Figure 8.
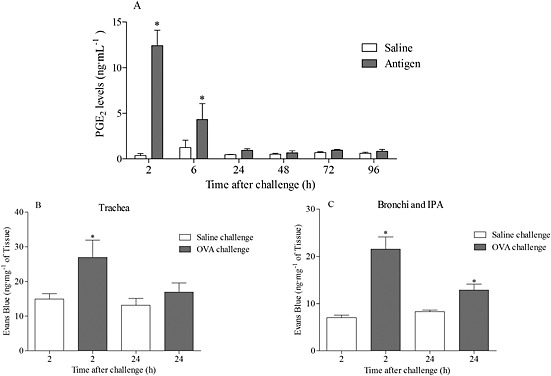
Effect of OVA challenge on BALF PGE2 levels (A) and microvascular leak into the trachea (B) and bronchi and IPA (C) of sensitized mice. Levels of BALF PGE2 were determined using LC‐MS/MS and expressed as mean ± SEM (n = 6). The airway MVL data are expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of tissue, n = 4). *P < 0.05 indicates a significant increase from vehicle control.
The data from a follow‐up study showed that the MVL in the asthma model was absent in mice missing functional EP2 or EP4 receptors, mirroring the observations made in the exogenous PGE2 studies (Fig. 9A–D).
Figure 9.
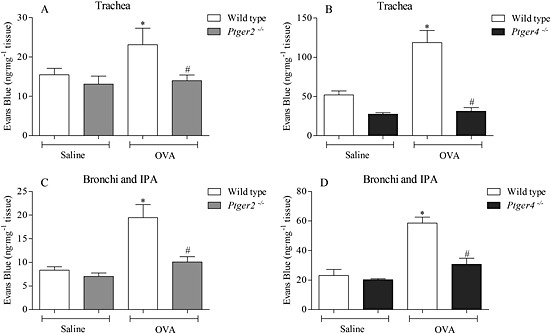
Investigating the responses to OVA challenge in sensitized, wild‐type and EP2 or EP4 receptor‐deficient mice in the (A and B) trachea and the (C and D) bronchi and IPA. Airway MVL was measured 2 h following the final intranasal antigen challenge. Airway tissue was dried before the extraction of Evans Blue dye. Data expressed as mean ± SEM of the concentration of Evans Blue dye (ng·mg−1 of dry tissue), n = 4–8. *P < 0.05 indicates a significant increase from vehicle control. #P < 0.05 indicates a significant decrease from wild‐type control.
Discussion
Airway microvascular leak is a cardinal sign of inflammation and is a prominent feature of both asthma and COPD. Various inflammatory mediators are capable of inducing this effect following an inflammatory insult to the airways. These inflammatory mediators act upon the endothelium of post‐capillary venules to open intercellular gap junctions. PGE2 is a ubiquitous eicosanoid that has been implicated in the pathogenesis of both asthma and COPD. Increased levels of PGE2 have been reported in BALF and plasma of asthma patients and the levels are enhanced in the breath condensate of COPD patients (Montuschi et al., 2003; Long et al., 2004). The purpose of this investigation was to establish the role of PGE2 in mediating airway microvascular leak and the EP receptor(s) responsible. This is an important step in understanding the biological profile of EP receptors given that PGE2 is a key mediator produced by the lung in health and in patients with lung disease.
Initial experiments were focused on investigating the effect of exogenous PGE2 in the airways of mice. PGE2 caused a dose‐dependent increase in MVL in the trachea and bronchi and IPA of mice, and in addition, blocking the production of endogenous prostanoids with the COX inhibitor diclofenac did not impact on PGE2‐induced MVL. We also observed a measurable and significant increase in MVL following an intranasal dose of PGE2, indicating that the effects we are seeing are not specific to the route of administration. Furthermore, we did not detect any MVL in non‐airway, reference tissues, oesophagus and bladder, suggesting that in our model system, i.v. administration of PGE2 mainly impacts on the airways. As the majority of research into airway MVL has been carried out in larger species, including the rat and the guinea pig, we wanted to rule out that what was being observed with PGE2 was not exclusive to the mouse. In the guinea pig, PGE2 produced a large, substantial increase in airway MVL that was comparable to that seen with 5‐HT. Both the mouse and the guinea pig produced similar profiles in the magnitude of their responses, demonstrating that the effect of PGE2 is translatable between species.
Having shown that PGE2 induced MVL in the mouse, we wanted to determine which of the EP receptors were central to this response. Differing effects of PGE2 have been demonstrated in vivo, and these, often opposing, effects can be attributed to the activation of different receptor subtypes (EP1–4) with different signalling characteristics (Breyer et al., 2001). Using mice deficient in specific prostanoid receptors, we were able to identify the EP2 and EP4 receptor/s as mediating the inflammatory actions of PGE2. 5‐HT evoked MVL in wild‐type, EP2 and EP4 knockout mice, indicating that MVL was not modulated per se in the genetically modified mice. In addition to data generated in gene‐deleted mice, pharmacological characterisation was also performed. Experiments were performed with the four agonists, ONO‐D1‐004 (EP1), ONO‐AE1‐259 (EP2), ONO‐AE‐248 (EP3) and ONO‐AE1‐329 (EP4), and the data clearly demonstrated that, similar to PGE2, the EP2 and EP4 agonists significantly increase airway MVL. This confirmed that activation of these EP receptors mediated MVL in mouse airways, while activation of EP1 and EP3 receptors produced no significant increase. Interestingly, others, using an in vitro system with human microvascular endothelial cells, have reported that PGE2 can strengthen endothelial barrier function via the EP4 receptor (Birukova et al., 2007; Konya et al., 2013). It is not clear why there is a difference, but it could be because the cell‐based system does not resemble the complexity of an in vivo model or the permeability characteristics of an intact microvascular endothelium.
In recent years, novel EP2 and EP4 receptor antagonists that are potent and selective for their respective receptor subtype have emerged and can be used to confirm data generated in developmental knockout mouse strains. Previously, AH6809 had been used by most investigators to examine EP2 receptor events, but this also antagonizes EP1 and DP1 receptors (Jones et al., 2009; Woodward et al., 2011). However, af Forselles et al. (2011 recently described a novel and selective EP2 receptor antagonist, PF‐04418948, which was further characterised by our laboratory (Birrell et al., 2013). In these experiments, PF‐04418948 produced a dose‐dependent inhibition of MVL induced by the EP2 agonist, ONO‐AE1‐259. In 2010, Chen et al. described a novel EP4 receptor antagonist, ER‐819762, that was highly selective for EP4 receptors in a number of different in vitro assays as well as being effective in both mouse and rat models of arthritis and chronic pain. Similarly, ER‐819762 produced a dose‐dependent inhibition of EP4‐induced MVL. However, initial experiments demonstrated that neither antagonist alone was able to inhibit PGE2‐mediated airway MVL. Interestingly, when both antagonists were administered simultaneously, at the same doses and time points, a decrease in PGE2‐induced MVL was evident, with a significant inhibition seen in the trachea and in the bronchi and IPA. This emphasized the possibility for a dual role for EP2 and EP4 receptors in mediating PGE2‐induced MVL responses. Little research has been published on the role of prostanoid receptors of the EP subtype in airway MVL, and so, the data presented herein demonstrate an EP receptor activity profile, which will aid in further understanding its role in airway inflammation.
There is, however, some information available from other organ systems implicating various EP receptors in MVL by evoking vasodilatation and increasing blood flow, an effect that is particularly evident in the skin. In this regard, both EP2 and EP4 receptor signalling have been implicated in acute skin inflammation by enhancing blood flow in the micro‐environment (Kabashima et al., 2007). It could also be possible that PGE2‐induced MVL may be caused by vasodilatation and an increase in blood flow to the airways. There is considerable evidence indicating that EP2 and EP4 receptors are pivotal in mediating the vasodepressor actions in mouse and human vascular preparations (Kennedy et al., 1999; Zhang et al., 2000; Imig et al., 2002; Davis et al., 2004). However, in the lung, potent vasodilators such as calcitonin gene‐related peptide do not produce MVL or enhance MVL produced by other agents probably because of the greater blood flow to the lung than to the skin, and so, this is less likely (Rogers et al., 1988).
It has been well‐documented that microvascular leak and oedema are prominent features of allergic airway diseases, such as asthma, with high concentrations of plasma proteins being present in airway secretions from acute asthmatic patients (Meerschaert et al., 1999; Kanazawa et al., 2002, 2000; Khor et al., 2009; Tseliou et al., 2012). Having already demonstrated that PGE2 induced airway MVL via the EP2 and EP4 receptors, its role in a murine respiratory model of airway inflammation was then investigated. An increase in PGE2 was measureable in lavage fluid 2 and 6 h but not 24 h after OVA challenge. Others have reported increases in PGE2 levels in their asthma models (Herrerias et al., 2009; Swedin et al., 2009). MVL appeared to parallel the changes in PGE2 levels in that we could detect an increase at 2 h post‐challenge but not 24 h later. Furthermore, as with the exogenous PGE2 experiments, it appears that allergen‐induced MVL is via the activity of PGE2 on EP2 and EP4 receptors.
An interesting observation from the data presented here was that both EP2 and EP4 agonists caused almost identical increases in airway MVL and, in the knockout mice, responses to PGE2 were almost completely abolished in both EP2 and EP4 receptor knockouts. Moreover, the selective receptor antagonists required simultaneous dosing to block the stimulatory effects of PGE2. This suggests that EP2 and EP4 may be functionally interdependent. A similar effect has been reported in human adipocytes and breast cancer cells with both receptors playing a role in PGE2‐induced induction of aromatase and inhibition of either receptor attenuating the effects of PGE2 (Subbaramaiah et al., 2008). One hypothesis could be that the EP2 and EP4 receptors form heterodimers, which has been illustrated in Fig. 10. The data from the knockouts indicate that without functional EP2 or EP4 receptors, PGE2 is unable to induce MVL. Furthermore, when EP2 and EP4 antagonists were administered alone, when both functional receptors are present, PGE2 still induced significant MVL. Only when both receptors were antagonized simultaneously was PGE2‐induced leak attenuated.
Figure 10.
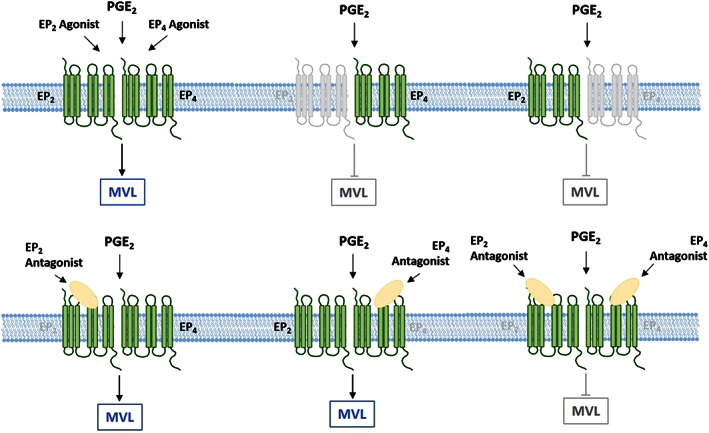
Schematic of possible EP2/4 heterodimer and how this interaction influences PGE2‐induced airway microvascular leak. (A) PGE2 and EP2 and EP4 agonists are all able to induce a significant increase in airway MVL. However, in EP2 and EP4 receptor knockouts, this response to PGE2 is absent. With either functional receptor missing, the EP2/4 heterodimer cannot form, and so, stimulation with PGE2 cannot elicit a response. (B) Pharmacological inhibition of either receptor with an antagonist does not inhibit airway MVL. When both EP2 and EP4 antagonists are administered simultaneously, both receptors are blocked, and PGE2‐induced MVL is attenuated.
There is increasing evidence that GPCRs are able to heterodimerize and that this interaction can affect the function of both receptors (McGraw et al., 2006; Wilson et al., 2007). Wilson et al. (2007 demonstrated a functionally important heterodimerization of the Tx (TP) and prostacyclin (IP) receptors in human and mouse aortic smooth muscle cells. Additionally, McGraw et al. (2006 used fluorescence microscopy in airway smooth muscle cells and BRET and co‐immunoprecipitation in a cell line to show heterodimerization between EP1 receptors and β2 adrenoceptors. However, more investigations are obviously needed to establish whether this is occurring between the EP2 and EP4 receptors in this in vivo model.
PGE2 is a key mediator produced by the lung and has widespread effects according to the EP receptor that is activated. Airway MVL represents a response to injury and under ‘disease’ conditions is a prominent feature of airway inflammation. The data presented here highlight a key role for PGE2 and the EP2 and EP4 receptors. These findings are novel and make an exciting addition to the expanding repertoire of effects mediated by EP receptor activation.
Author contributions
M. A. B., M. G. B., S. A. M., M. G. and V. O. D. devised the experiments; V. C. J., M. A. B. and M. G. B. wrote the manuscript; V. C. J., S. A. M., S. R. C. and M. G. performed the experiments.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
S. A. M. and the project consumables were funded by a project grant from the Medical Research Council (MRC, UK) (G0800195). V. J. was funded by an MRC studentship. This work was supported by the Royal Brompton and Harefield NHS Foundation Trust, NIHR Respiratory Biomedical Research Unit, London, UK. S. R. C. and V. O. D. were funded by the Wellcome Trust.
Jones, V. C. , Birrell, M. A. , Maher, S. A. , Griffiths, M. , Grace, M. , O'Donnell, V. B. , Clark, S. R. , and Belvisi, M. G. (2016) Role of EP2 and EP4 receptors in airway microvascular leak induced by prostaglandin E2 . British Journal of Pharmacology, 173: 992–1004. doi: 10.1111/bph.13400.
References
- af Forselles KJ, Root J, Clarke T, Davey D, Aughton K, Dack K, et al. (2011). In vitro and in vivo characterization of PF‐04418948, a novel, potent and selective prostaglandin EP4 receptor antagonist. Br J Pharmacol 164: 1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015). The Concise Guide to Pharmacology 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antczak A, Ciebiada M, Pietras T, Piotrowski WJ, Kurmanowska Z, Górski P (2012). Exhaled eicosanoids and biomarkers of oxidative stress in exacerbation of chronic obstructive pulmonary disease. Arch Med Sci 2: 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Thurston G, Murphy TJ, Bunnett NW, McDonald DM (1999). Neurogenic plasma leakage in mouse airways. Br J Pharmacol 126: 522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvisi MG, Patel HJ, Freund‐Michel V, Hele DJ, Crispino N, Birrell MA (2009). Inhibitory activity of the novel CB2 receptor agonist, GW833972A, on guinea‐pig and human sensory nerve function in the airways. Br J Pharmacol 155: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyahia C, Gomez I, Kanyinda L, Boukais K, Danel C, Leséche G, et al. (2012). PGE(2) receptor (EP(4)) agonists: potent dilators of human bronchi and future asthma therapy? Pulm Pharmacol Ther 25: 115–118. [DOI] [PubMed] [Google Scholar]
- Bessa V, Loukides S, Hillas G, Delimpoura V, Simoes D, Kontogianni K, et al. (2012). Levels of angiopoietins 1 and 2 in induced sputum supernatant in patients with COPD. Cytokine 58: 455–460. [DOI] [PubMed] [Google Scholar]
- Birrell MA, Maher SA, Buckley J, Dale N, Bonvini S, Raemdonck K, et al. (2013). Selectivity profiling of the novel EP2 receptor antagonist, PF‐04418948, in functional bioassay systems: atypical affinity at the guinea pig EP2 receptor. Br J Pharmacol 168: 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birring SS, Prudon B, Carr AJ, Singh SJ, Morgan MDL, Pavord ID (2003). Development of a symptom specific health status measure for patients with chronic cough: Leicester Cough Questionnaire (LCQ). Thorax 58: 339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, et al. (2007). Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA‐ and Epac1/Rap1‐dependent Rac activation. Exp Cell Res 313: 2504–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD (2001). Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41: 661–690. [DOI] [PubMed] [Google Scholar]
- Brightling CE, Ward R, Woltmann G, Bradding P, Sheller JR, Dworski R, et al. (2000). Induced sputum inflammatory mediator concentrations in eosinophilic bronchitis and asthma. Am J Respir Crit Care Med 162: 878–882. [DOI] [PubMed] [Google Scholar]
- Buckley J, Birrell MA, Maher SA, Nials AT, Clarke DL, Belvisi MG (2011). EP4 receptor as a new target for bronchodilator therapy. Thorax 66: 1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Shayibuzhati M, Tajima T, Kitazawa T, Taneike T (2002). In vitro pharmacological characterization of the prostanoid receptor population in the non‐pregnant porcine myometrium. Eur J Pharmacol 442: 115–123. [DOI] [PubMed] [Google Scholar]
- Chen Q, Muramoto K, Masaaki N, Ding Y, Yang H, MacKey M, et al. (2010). A novel antagonist of the prostaglandin E 2 EP 4 receptor inhibits Th1 differentiation and Th17 expansion and is orally active in arthritis models. Br J Pharmacol 160: 292–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K (2008). Enhanced levels of prostaglandin E2 and matrix metalloproteinase‐2 correlate with the severity of airflow limitation in stable COPD. Respirology 13: 1014–1021. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S (1994). International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 46: 205–229. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Murdoch CE, Ali M, Purbrick S, Ravid R, Baxter GS, et al. (2004). EP4 prostanoid receptor‐mediated vasodilatation of human middle cerebral arteries. Br J Pharmacol 141: 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TW, Chung KF, Rogers DF, Barnes PJ (1987). Effect of platelet‐activating factor on airway vascular permeability: possible mechanisms. J Appl Physiol 63: 479–484. [DOI] [PubMed] [Google Scholar]
- Greiff L, Andersson M, Erjefält JS, Persson CGA, Wollmer P (2003). Airway microvascular extravasation and luminal entry of plasma. Clin Physiol Funct Imaging 23: 301–306. [DOI] [PubMed] [Google Scholar]
- Hele DJ, Birrell MA, Webber SE, Foster ML, Belvisi MG (2001). Mediator involvement in antigen‐induced bronchospasm and microvascular leakage in the airways of ovalbumin sensitized Brown Norway rats. Br J Pharmacol 132: 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrerias A, Torres R, Serra M, Marco A, Pujols L, Picado C, et al. (2009). Activity of the cyclooxygenase 2‐prostaglandin‐E prostanoid receptor pathway in mice exposed to house dust mite aeroallergens, and impact of exogenous prostaglandin E2. J Inflamm (Lond) 6: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AT, Bayley D, Stockley RA (1999). The interrelationship of sputum inflammatory markers in patients with chronic bronchitis. Am J Respir Crit Care Med 160: 893–898. [DOI] [PubMed] [Google Scholar]
- Imig JD, Breyer MD, Breyer RM (2002). Contribution of prostaglandin EP(2) receptors to renal microvascular reactivity in mice. Am J Physiol Renal Physiol 283: F415–F422. [DOI] [PubMed] [Google Scholar]
- Innes AL, Carrington SD, Thornton DJ, Kirkham S, Rousseau K, Dougherty RH, et al. (2009). Ex vivo sputum analysis reveals impairment of protease‐dependent mucus degradation by plasma proteins in acute asthma. Am J Respir Crit Care Med 180: 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Giembycz MA, Woodward DF (2009). Prostanoid receptor antagonists: development strategies and therapeutic applications. Br J Pharmacol 158: 104–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima K, Nagamachi M, Honda T, Nishigori C, Miyachi Y, Tokura Y, et al. (2007). Prostaglandin E2 is required for ultraviolet B‐induced skin inflammation via EP2 and EP4 receptors. Lab Invest 87: 49–55. [DOI] [PubMed] [Google Scholar]
- Kanazawa H, Asai K, Hirata K, Yoshikawa J (2002). Vascular involvement in exercise‐induced airway narrowing in patients with bronchial asthma. Chest 122: 166–170. [DOI] [PubMed] [Google Scholar]
- Kanazawa H, Hirata K, Yoshikawa J (2000). Role of endogenous nitric oxide in exercise‐induced airway narrowing in patients with bronchial asthma. J Allergy Clin Immunol 106: 1081–1087. [DOI] [PubMed] [Google Scholar]
- Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, et al. (1999). Salt‐sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med 5: 217–220. [DOI] [PubMed] [Google Scholar]
- Khor YH, Teoh AKY, Lam SM, Mo DCQ, Weston S, Reid DW, et al. (2009). Increased vascular permeability precedes cellular inflammation as asthma control deteriorates. Clin Exp Allergy 39: 1659–1667. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill I (2010). The ARRIVE guidelines ReqartoCom 8: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konya V, Üllen A, Kampitsch N, Theiler A, Philipose S, Parzmair GP, et al. (2013). Endothelial E‐type prostanoid 4 receptors promote barrier function and inhibit neutrophil trafficking. J Allergy Clin Immunol 131: 532–540. [DOI] [PubMed] [Google Scholar]
- Laitinen LA, Laitinen A, Widdicombe J (1987). Effects of inflammatory and other mediators on airway vascular beds. Am Rev Respir Dis 135: S67–S70. [DOI] [PubMed] [Google Scholar]
- Li X, Wilson JW (1997). Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med 156: 229–233. [DOI] [PubMed] [Google Scholar]
- Long JA, Fogel‐Petrovic M, Knight DA, Thompson PJ, Upham JW (2004). Higher prostaglandin E2 production by dendritic cells from subjects with asthma compared with normal subjects. Am J Respir Crit Care Med 170: 485–491. [DOI] [PubMed] [Google Scholar]
- Maher SA, Birrell MA, Belvisi MG (2009). Prostaglandin E2 mediates cough via the EP3 receptor: implications for future disease therapy. Am J Respir Crit Care Med 180: 923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw DW, Mihlbachler KA, Schwarb MR, Rahman FF, Small KM, Almoosa KF, et al. (2006). Airway smooth muscle prostaglandin‐EP1 receptors directly modulate b2‐adrenergic receptors within a unique heterodimeric complex. J Clin Invest 116: 1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerschaert J, Kelly EA, Mosher DF, Busse WW, Jarjour NN (1999). Segmental antigen challenge increases fibronectin in bronchoalveolar lavage fluid. Am J Respir Crit Care Med 159: 619–625. [DOI] [PubMed] [Google Scholar]
- Meyrick B, Hoover R, Jones MR, Berry LC, Brigham KL (1989). In vitro effects of endotoxin on bovine and sheep lung microvascular and pulmonary artery endothelial cells. J Cell Physiol 138: 165–174. [DOI] [PubMed] [Google Scholar]
- Miles AA, Miles EM (1952). Vascular reactions to histamine, histamine‐liberator and leukotaxine in the skin of guinea‐pigs. J Physiol 118: 228–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakata Y, Nakanishi M, Hirano T, Matsunaga K, Yamagata T, Ichinose M (2005). Microvascular hyperpermeability in COPD airways. Thorax 60: 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR (1993). Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A 90: 11693–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montuschi P, Kharitonov SA, Ciabattoni G, Barnes PJ (2003). Exhaled leukotrienes and prostaglandins in COPD. Thorax 58: 585–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Hara A, Ma H, Xiao CY, Takahata O, Kohgo Y, et al. (2000). Characterization of prostanoid receptors mediating contraction of the gastric fundus and ileum: studies using mice deficient in prostanoid receptors. Br J Pharmacol 131: 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivenstein R, Du T, Xu LJ, Martin JG (1997). Microvascular leakage in the airway wall and lumen during allergen induced early and late responses in rats. Pulm Pharmacol Ther 10: 223–230. [DOI] [PubMed] [Google Scholar]
- Paredi P, Barnes PJ (2009). The airway vasculature: recent advances and clinical implications. Thorax 64: 444–450. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al., NC‐IUPHAR (2014). The IUPHAR/BPS Guide to Pharmacology: An Expert‐Driven Knowledge Base of Drug Targets and their Ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AM, Holmes MD, Scicchitano R (2002). Interleukin‐1beta and tumour necrosis factor‐alpha increase microvascular leakage in the guinea pig trachea. Respirology 7: 23–28. [DOI] [PubMed] [Google Scholar]
- Rogers DF, Belvisi MG, Aursudkij B, Evans TW, Barnes PJ (1988). Effects and interactions of sensory neuropeptides on airway microvascular leakage in guinea‐pigs. Br J Pharmacol 95: 1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre B, Fernández‐Nieto M, Mollá R, López E, Lahoz C, Sastre J, et al. (2008). Increased prostaglandin E2 levels in the airway of patients with eosinophilic bronchitis. Allergy Eur J Allergy Clin Immunol 63: 58–66. [DOI] [PubMed] [Google Scholar]
- Segi E, Sugimoto Y, Yamasaki A, Aze Y, Oida H, Nishimura T, et al. (1998). Patent ductus arteriosus and neonatal death in prostaglandin receptor EP4‐deficient mice. Biochem Biophys Res Commun 246: 7–12. [DOI] [PubMed] [Google Scholar]
- Subbaramaiah K, Hudis C, Chang SH, Hla T, Dannenberg AJ (2008). EP2 and EP4 receptors regulate aromatase expression in human adipocytes and breast cancer cells: evidence of a BRCA1 and p300 exchange. J Biol Chem 283: 3433–3444. [DOI] [PubMed] [Google Scholar]
- Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, et al. (2000). The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology 141: 1554–1559. [DOI] [PubMed] [Google Scholar]
- Swedin L, Neimert‐Andersson T, Hjoberg J, Jonasson S, VAN Hage M, Adner M, et al. (2009). Dissociation of airway inflammation and hyperresponsiveness by cyclooxgenase inhibition in allergen challenged mice. Eur Respir J 34: 200–208. [DOI] [PubMed] [Google Scholar]
- Tseliou E, Bakakos P, Kostikas K, Hillas G, Mantzouranis K, Emmanouil P, et al. (2012). Increased levels of angiopoietins 1 and 2 in sputum supernatant in severe refractory asthma. Allergy 67: 396–402. [DOI] [PubMed] [Google Scholar]
- Ushikubi F, Segi E, Sugimoto Y, Murata T, Matsuoka T, Kobayashi T, et al. (1998). Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature 395: 281–284. [DOI] [PubMed] [Google Scholar]
- Widdicombe JH, Ueki IF, Emery D, Margolskee D, Yergey J, Nadel JA (1989). Release of cyclooxygenase products from primary cultures of tracheal epithelia of dog and human. Am J Physiol 257: L361–L365. [DOI] [PubMed] [Google Scholar]
- Wilson SJ, Dowling JK, Zhao L, Carnish E, Smyth EM (2007). Regulation of thromboxane receptor trafficking through the prostacyclin receptor in vascular smooth muscle cells: role of receptor heterodimerization. Arterioscler Thromb Vasc Biol 27: 290–296. [DOI] [PubMed] [Google Scholar]
- Woodward D, Jones R, Narumiya S (2011). International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev 63: 471–538. [DOI] [PubMed] [Google Scholar]
- Xie G, Wang Y, Sharma M, Gabriel A, Mitchell J, Xing Y, et al. (2003). 5‐Hydroxytryptamine‐induced plasma extravasation in the rat knee joint is mediated by multiple prostaglandins. Inflamm Res 52: 32–38. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Guan Y, Schneider A, Brandon S, Breyer RM, Breyer MD (2000). Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertension 35: 1129–1134. [DOI] [PubMed] [Google Scholar]
- Zhuang J, Xu J, Zhang C, Xu F (2011). IL‐1b acutely increases pulmonary SP and permeability without associated changes in airway resistance and ventilation in anesthetized rats. Respir Physiol Neurobiol 175: 12–19. [DOI] [PubMed] [Google Scholar]