

Abstract
Objective:
To explore clinical phenotype and characteristics of Parkinson disease (PD) at different ages at onset in recently diagnosed patients with untreated PD.
Methods:
We have analyzed baseline data from the Parkinson's Progression Markers Initiative database. Four hundred twenty-two patients with a diagnosis of PD confirmed by DaTSCAN imaging were divided into 4 groups according to age at onset (onset younger than 50 years, 50–59 years, 60–69 years, and 70 years or older) and investigated for differences in side, type and localization of symptoms, occurrence/severity of motor and nonmotor features, nigrostriatal function, and CSF biomarkers.
Results:
Older age at onset was associated with a more severe motor and nonmotor phenotype, a greater dopaminergic dysfunction on DaTSCAN, and reduction of CSF α-synuclein and total tau. The most common presentation was the combination of 2 or 3 motor symptoms (bradykinesia, resting tremor, and rigidity) with rigidity being more common in the young-onset group. In about 80% of the patients with localized onset, the arm was the most affected part of the body, with no difference across subgroups.
Conclusions:
Although the presentation of PD symptoms is similar across age subgroups, the severity of motor and nonmotor features, the impairment of striatal binding, and the levels of CSF biomarkers increase with age at onset. The variability of imaging and nonimaging biomarkers in patients with PD at different ages could hamper the results of future clinical trials.
Parkinson disease (PD) is heterogeneous in clinical manifestations1 and progression.2 The biological basis of this phenotypic variability is unknown but might be explained by the progressive and diverse accumulation of α-synuclein (α-syn) in different brain structures.3,4 It has been estimated that PD increases with age, reaching a prevalence of 2.6% in people aged 85 to 89 years.5 Although current evidence does not support the hypothesis that PD is caused by an acceleration of a natural aging process,6 age is considered its main risk factor.7 Lewy body pathology is related to age8 and, in the general population, the occurrence of mild extrapyramidal signs increases with age.8 These observations implicate a role for age in the progressive decline of nigrostriatal function.9 However, evidence supporting an influence of age on PD phenotype at its onset is lacking and conflicting.10–18 Previous studies have poorly assessed the full spectrum of motor and nonmotor symptoms and they have not evaluated the relationship between PD phenotype and imaging and nonimaging biomarkers.
The Parkinson's Progression Markers Initiative (PPMI) is an ongoing, international, multicenter, prospective study designed to discover and validate biomarkers of disease progression in newly diagnosed, drug-naive patients with PD. Here, we have analyzed baseline data from the PPMI database to explore the characteristics of patients with PD at different ages at onset.
METHODS
Population.
Demographic information, clinical characteristics, and results of clinical and biochemical tests were downloaded on August 13, 2014, from the PPMI database.19 Inclusion criteria were age 30 years or older, diagnosis of PD (based on one of the following: presence of [1] asymmetrical resting tremor or [2] asymmetrical bradykinesia or [3] at least 2 of resting tremor, bradykinesia, and rigidity), disease duration of 1 to 24 months, Hoehn and Yahr (H&Y) stage of 1 to 2, and presence of striatal dopamine transporter deficit on 123I-ioflupane SPECT imaging (DaTSCAN).
None of the patients was on antiparkinsonian medications or monoamine oxidase inhibitor inhibitors.
At enrollment, participants were assessed for clinical features and underwent DaTSCAN imaging. CSF and serum samples were collected. Healthy controls (HCs) were recruited if free of current or active neurologic disorder and had normal DaTSCAN. Details about assessments and analyses are described in the e-Methods on the Neurology® Web site at Neurology.org.
Our PD population was divided into subgroups according to the decade of their age at symptomatic onset (younger than 50 years, 50–59 years, 60–69 years, and 70 years or older). Age-related differences in clinical phenotype and imaging and nonimaging biomarkers were evaluated. This age-subgroup classification was chosen following a cluster analysis of patients with early PD.20 Age at onset was based on patients' recollection of first symptoms.
Standard protocol approvals, registrations, and patient consents.
The PPMI study is registered with ClinicalTrials.gov (NCT01141023). Each PPMI site received approval from an ethical committee on human experimentation before study initiation. Written informed consent for research was obtained from all individuals participating in the study.
Statistical analysis.
Continuous variables were expressed as mean ± SD and compared across subgroups with analysis of variance if normally distributed and with the Kruskal-Wallis correction when not normally distributed. Normality of data distribution was interrogated using the Kolmogorov-Smirnov test. Significance of multiple comparisons was assessed after performing a Bonferroni correction. Categorical variables were expressed as proportions and compared using a χ2 test. Correlations were interrogated using Spearman rank correlation. Data were collected and analyzed using SPSS version 19.0 (IBM Corp., Armonk, NY). Statistical significance was accepted at p < 0.05.
RESULTS
Patient characteristics.
Table 1 shows demographic and clinical characteristics of the PD population. We studied 422 patients with PD, 276 male (65.4%), with a mean age of 61.6 ± 9.7 years and mean years of education of 15.5 ± 3.0. Three hundred seventy-five patients (88.8%) were right-handed, 76 patients (8.8%) were left-handed, and 10 patients (2.4%) were ambidextrous. One hundred two patients (24.2%) had a family history of PD. Mean PD duration was 6.6 ± 6.5 months. Mean H&Y stage was 1.56 ± 0.51 and Movement Disorder Society–Unified Parkinson's Disease Rating Scale (MDS-UPDRS) total mean score was 32.4 ± 13.2. Fifty-eight patients had age at onset younger than 50 years, 117 had age at onset between 50 and 59 years, 168 between 60 and 69 years, and 79 had age at onset of 70 years or older. There were no differences for years of education, family history of PD, and PD duration. Table e-1 shows characteristics of the control population.
Table 1.
Characteristics of all patients with PD and at different ages at onset

Side predominantly affected at onset.
Disease onset was asymmetrical in 403 patients (97.8%) and symmetrical in 9 (2.2%). In patients with asymmetrical onset, 234 (56.8%) had the dominant side predominantly affected and 169 (41%) the nondominant side. In subgroups with older age at onset, the number of patients with the dominant side more affected and symmetrical onset was higher compared to younger subgroups (p = 0.002) (table 2).
Table 2.
Predominantly affected side and localization of motor symptoms in all patients with Parkinson disease and at different ages at onset

Localization of motor symptoms.
MDS-UPDRS III subitems were analyzed to identify involvement of specific parts of the body. In 191 patients (45.3%), the disease predominantly affected a specific part of the body (head-predominant, arm-predominant, or leg-predominant; see e-Methods for details about this classification). Of these patients, 162 (85%) had arm-predominant symptoms, 15 (8%) had head-predominant symptoms, and 14 (7.3%) had leg-predominant symptoms. Most patients had arm-predominant symptoms (80%–86%) (table 2).
Symptoms at time of diagnosis.
Fifty-two patients (12.4%) reported only one motor symptom (resting tremor [40 patients, 76.9%], bradykinesia [10 patients, 19.2%], or rigidity [2 patients, 3.8%]), 151 (35.9%) reported 2 motor symptoms, 200 (47.5%) reported 3 motor symptoms, and 18 (4.3%) reported all 4 motor symptoms of PD (table 3). Overall the percentage of patients with 1, 2, 3, and 4 motor symptoms at time of diagnosis was similar across age subgroups (p = 0.5). In the subgroup with age at onset younger than 50 years, 94.5% (n = 54) of the patients reported rigidity and this rate was higher compared to other age-at-onset subgroups (p = 0.003). The percentages of patients reporting bradykinesia, resting tremor, and postural instability rate were similar at different ages at onset (p = 0.626, p = 0.130, and p = 0.839, respectively) (table 3).
Table 3.
Number, rate, and severity of motor signs at different ages at onset
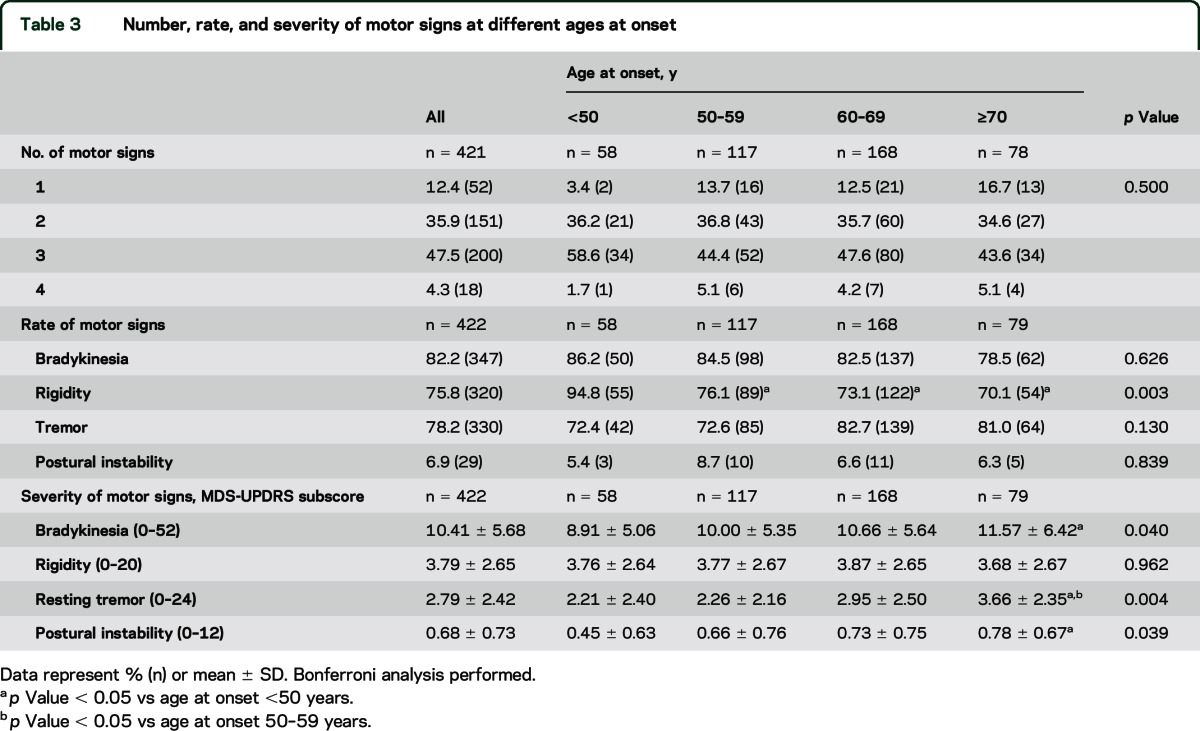
Motor features.
Table 1 shows details of motor features. The mean H&Y stage for the whole cohort of patients was 1.56 ± 0.51. They had an MDS-UPDRS Part II mean score of 5.9 ± 4.20 and an MDS-UPDRS Part III motor mean score of 20.9 ± 8.9. Seventy-five patients (17.8%) showed a tremor-dominant subtype, 329 (78%) an akinetic-rigid subtype, and 18 (4.3%) a mixed subtype. Details regarding definition of motor subtypes are provided in the e-Methods. MDS-UPDRS subscore for bradykinesia was 10.41 ± 5.68 (range 0–52), 3.79 ± 2.65 for rigidity (range 0–20), 2.79 ± 2.42 for resting tremor (range 0–24), and 0.68 ± 0.73 for postural instability (range 0–12) (table 3).
H&Y stage and MDS-UPDRS III score was higher in the subgroups with older age at onset (p = 0.001 and p = 0.005, respectively). However, no differences were found for MDS-UPDRS Part II and motor subtypes (p = 0.181 and p = 0.533, respectively) (table 1). Bradykinesia, resting tremor, and postural instability scores were more severe in the subgroup with older age at onset than in the younger subgroups (p = 0.040, p = 0.004, and p = 0.039, respectively), whereas severity of rigidity was not different across age subgroups (p = 0.962) (table 3).
Nonmotor features.
Table 4 shows details of nonmotor symptoms analysis. The global nonmotor burden as measured by the MDS-UPDRS Part I was similar among the 4 subgroups (p = 0.516) (table 1). However, when rated with specific scales, patients with an older age at onset showed a greater impairment of autonomic (Scales for Outcomes in Parkinson's Disease–Autonomic [SCOPA-AUT], p < 0.0001), olfactory (University of Pennsylvania Smell Identification Test [UPSIT], p < 0.0001), and cognitive function (Montreal Cognitive Assessment [MoCA], p < 0.0001; Benton Judgment of Line Orientation Score, p = 0.018; Hopkins Verbal Learning Test [HVLT] Delayed Recognition False Alarms, p = 0.032; HVLT Delayed Recognition Hits, p = 0.001; HVLT Immediate Recall, p < 0.0001; Semantic Fluency Test score, p < 0.0001; Symbol Digit Modalities Test score, p < 0.0001). Likewise, the number of patients with impaired autonomic, olfactory, and cognitive functions was higher in the subgroups with older age at onset (p < 0.0001, for SCOPA-AUT, p < 0.0001 for UPSIT, and p < 0.0001 for MoCA, respectively; data not shown). No differences were found in terms of severity of disability (activities of daily living, p = 0.489), depression (Geriatric Depression Scale, p = 0.890), anxiety (State-Trait Anxiety Inventory [STAI] total, p = 0.639; STAI state subscore, p = 0.948; STAI anxiety subscore, p = 0.486), impulsive control disorders (Questionnaire for Impulsive-Compulsive Disorders in Parkinson's Disease score, p = 0.673), and sleep problems (Epworth Sleepiness Scale, p = 0.980; REM Sleep Behavior Disorder Screening Questionnaire, p = 0.941).
Table 4.
Nonmotor features at different ages at onset

Striatal binding of 123I-ioflupane (DaTSCAN).
Table 5 shows DaTSCAN findings. Data are presented as reduction in 123I-ioflupane binding compared to the mean of the HCs (see details in e-Methods). In all patients, striatal uptake of 123I-ioflupane was asymmetrical with one side more affected than the other. In each patient, we identify the caudate and putamen with the largest reduction of 123I-ioflupane binding (referred to below as the most affected caudate and most affected putamen). The contralateral striatal nuclei were defined as the least affected caudate and putamen. Each nucleus was considered abnormally reduced when reduction was greater than 2 SDs below mean value of HCs for that region. Across the whole population of patients, the mean percentage reduction in 123I-ioflupane binding was 68.5% ± 12.4% for the most affected putamen, 53.8% ± 17.4% for the least affected putamen, 39% ± 18.2% for the most affected caudate, and 26.6% ± 20.1% for the least affected caudate. Three hundred ninety patients had an abnormal most affected putamen (93.5%), 262 patients an abnormal least affected putamen (62.5%), 197 patients had an abnormal most affected caudate (47%), and 93 patients had an abnormal least affected caudate (22.2%) (table 5). Percentage reductions in 123I-ioflupane binding for the most affected putamen were similar across age subgroups (p = 0.926) as were the number of patients with abnormal reductions (p = 0.123). For the most affected caudate, 123I-ioflupane binding was lower in older age subgroups (p = 0.048); however, the number of patients with abnormal caudate reduction was similar across the age subgroups (p = 0.229). In the least affected putamen and caudate, 123I-ioflupane binding was lower in older compared with younger subgroups (p = 0.006 for putamen and p = 0.0003 for caudate) and the number of patients with abnormal tracer uptake in these regions was higher in older subgroups (p = 0.004 for putamen and p = 0.047 for caudate) (table 5).
Table 5.
DaTSCAN at different ages at onset

CSF and serum biomarkers.
Table e-2 shows characteristics of CSF and serum biomarkers. Across the whole population, the levels of CSF α-syn, total tau (t-tau), and tau phosphorylated at Thr181 (p-tau181) were reduced in the patients compared with HCs, while levels of CSF β-amyloid 1–42 (Aβ1–42) and serum urate were not different between groups (table e-2). In HCs, all biomarkers increased with the age (Spearman correlation [ρ] = 0.159, p = 0.029 for α-syn; ρ = 0.354, p = 0.030 for t-tau; ρ = 0.158, p = 0.030 for p-tau181; and ρ = 0.182, p = 0.008 for serum urate) except for Aβ1–42 (ρ = −0.012, p = 0.871). In patients with PD, only α-syn (ρ = 0.124, p = 0.012) and t-tau (ρ = 0.269, p < 0.0001) increased with the age but not p-tau181 (ρ = 0.054, p = 0.277), Aβ1–42 (ρ = 0.159, p = 0.029), or serum urate (ρ = 0.068, p = 0.166). In older age subgroups, the reduction (from HCs) of α-syn and t-tau were greater compared with younger age subgroups (table e-2 and figure e-1).
DISCUSSION
The main strength of this study is that the clinical features of PD were evaluated in a large population of untreated newly diagnosed patients with a prospective design and a simultaneous evaluation of imaging and nonimaging biomarkers.
We found that older age at onset was associated with a more severe motor and nonmotor PD phenotype, a greater impairment of dopaminergic function on DaTSCAN, and a greater reduction of CSF α-syn and t-tau levels. With the exception of rigidity, which was similar across age groups, the severity of motor impairment (H&Y stage, MDS-UPDRS total and Part III scores, bradykinesia, resting tremor, and postural instability scores) was greater in the oldest age subgroup (70 years or older) compared with younger subgroups. In addition, the number of patients with symmetrical disease at onset was higher in the oldest (70 years or older) compared with younger age subgroups suggesting a more widespread degeneration. Similarly, the oldest age subgroups also presented more severe nonmotor symptoms (autonomic, olfactory, and cognitive dysfunctions) and greater reduction of striatal 123I-ioflupane binding on DaTSCAN compared with younger subgroups. Of note, while 123I-ioflupane uptake in the most affected putamen was similar across the age subgroups, uptake in the most affected caudate, least affected putamen, and least affected caudate was lower in older compared with younger subgroups, suggesting a more widespread involvement of striatal structures in elderly patients.
Finally, levels of CSF α-syn and t-tau were reduced in patients with PD, as previously suggested in the preliminary analysis of the PPMI population,21 and this reduction was greater in older age subgroups compared with younger age subgroups. A greater reduction in CSF α-syn and t-tau in patients with older onset could be explained by a higher accumulation of these proteins in the neurons.
Considering that these patients had similar disease duration, our findings of a more severe phenotype and more widespread involvement of striatal structures in the older patients could be attributable to superimposition of PD pathology on the natural aging process. A direct contribution of the aging process to the progressive neurodegeneration of PD or a decrease of compensatory mechanisms in older patients could also have occurred.
Moreover, our data suggest that the heterogeneity of PD at different ages at onset could be reflected by various impairments of imaging and nonimaging biomarkers. This variability in biomarkers in patients with PD with different ages at onset should be taken in account in future clinical trials to test neuroprotective drugs. The recruitment of patients with homogeneous age at onset will reduce this variability, resulting in an increased statistical power with smaller sample sizes. Follow-up observations of the PPMI population will clarify the role of age at onset on disease progression and whether there is an interaction with these biomarkers.
Recently, there has been a great effort to identify and formally delineate PD subtypes.22,23 So far, none of the proposed subtype classifications appears sufficiently robust to warrant formal delineation.22 Our findings suggest that in future classifications, early-onset and late-onset subtypes should also be considered.
The association between handedness and first affected side at onset in PD is still controversial, as some studies have reported an association and others failed to demonstrate it.24 A recent meta-analysis24 including 4,405 patients with asymmetrical PD has shown that in about 60% of patients, the disease started on the dominant side. This finding is confirmed in the overall population of our study, where the disease was mostly asymmetrical and the dominant side was predominantly affected. However, in the youngest subgroup (younger than 50 years), it was the nondominant side that was mostly affected. It has been suggested that increased movement complexity in the dominant hand could be a risk factor for dopamine cell loss in the contralateral hemisphere via glutaminergic excitotoxicity.24 Therefore, the dominant hemisphere could be more vulnerable to dopaminergic denervation than the nondominant hemisphere in patients with PD.25 However, this theory does not explain the onset in the nondominant side in our younger patients and the symmetrical involvement in some of our older patients. Further studies are required to understand the pathophysiologic mechanisms of asymmetry in PD.
The limited data available in the literature indicate that abnormalities of the arm are first noticed in the majority (65%–80%) of patients.26,27 Similarly, in about 80% of our patients with localized onset, the arm was the most affected part of the body, with no difference across age subgroups. This is in line with our recent [18F]-dopa PET study,28 which has shown that the middle strip (intermediate) of the putamen was more affected than other striatal subregions in patients with early PD and correlated with severity of arm impairment.
Regarding the motor picture at onset, we did not find any specific feature of PD that was typical of any age subgroup, with the exception of rigidity, which was more commonly noticed by the group with youngest age at onset, as also reported previously.17,18 Most patients had a combination of 2 or more motor symptoms and we found a similar rate of tremor-predominant and akinetic-rigid patients across age-at-onset subgroups, as also indicated previously.12
There are some potential limitations in this study that require further discussion. In older patients, the initial symptoms could have been detected later than in younger patients since these symptoms could be attributed to physiologic aging rather than to a pathologic process. Thus, the disease duration in older patients could be underestimated in comparison with younger patients. However, it should be noticed that the magnitude of tracer uptake in the most affected putamen on DaTSCAN was similar across the age subgroups, suggesting that disease duration was indeed similar in the different subgroups.
Older people have a larger number of comorbidities and medications than younger patients, which might lead to higher scores on clinical scales, particularly for nonmotor symptoms. This is alleviated by the fact that comparison was made with HCs rigorously age- and sex-matched to the PD cohort. Unfortunately, the PPMI study has not included any measure of comorbidities to be used as covariate in comparisons of age subgroups. We strongly suggest that measurements of comorbidities such as the Elixhauser Index or the Romano adaptation of the Charlson Index be added to the PPMI study and to similar database studies in the future.29
Finally, about 25% of our patients had a family history of PD and it is possible that some of them were carriers of a genetic mutation. Many genetic forms have an influence on PD phenotype and therefore the presence of patients with genetic forms of PD could be a possible confounding factor in our study. However, this concern is probably mitigated by the equal distribution of family history across all age groups. In addition, genetic forms of PD associated with more severe phenotypes (including more severe and symmetrical striatal loss of dopamine transporter on DaTSCAN) have early onset, affecting young patients; therefore, the possible presence of such patients in our cohort would not change the meaningfulness of our findings.
Supplementary Material
GLOSSARY
- Aβ1–42
β-amyloid 1–42
- α-syn
α-synuclein
- HC
healthy control
- HVLT
Hopkins Verbal Learning Test
- H&Y
Hoehn and Yahr
- MDS-UPDRS
Movement Disorder Society–Unified Parkinson's Disease Rating Scale
- MoCA
Montreal Cognitive Assessment
- PD
Parkinson disease
- PPMI
Parkinson's Progression Markers Initiative
- p-tau181
tau phosphorylated at Thr181
- SCOPA-AUT
Scales for Outcomes in Parkinson's Disease–Autonomic
- STAI
State-Trait Anxiety Inventory
- t-tau
total tau
- UPSIT
University of Pennsylvania Smell Identification Test
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Pagano: study concept and design, statistical analysis and interpretation of data, and drafting of the manuscript. Prof. Ferrara: critical revision of the manuscript for important intellectual content. Prof. Brooks: critical revision of the manuscript for important intellectual content. Dr. Pavese: study concept and design, study supervision, drafting of the manuscript, and final approval of the manuscript.
STUDY FUNDING
Data used in the preparation of this article were obtained from the Parkinson's Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org. PPMI is sponsored and partially funded by The Michael J. Fox Foundation for Parkinson's Research. Other funding partners include a consortium of industry players, nonprofit organizations, and private individuals. Industry partners are contributing to PPMI through financial and in-kind donations and are playing a lead role in providing feedback on study parameters through the Industry Scientific Advisory Board (ISAB). Through close interaction with the study, the ISAB is positioned to inform the selection and review of potential progression markers that could be used in clinical testing. The Medical Research Council UK pays for part of the salary for Dr. Pavese's research activity.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lewis SJ, Foltynie T, Blackwell AD, Robbins TW, Owen AM, Barker RA. Heterogeneity of Parkinson's disease in the early clinical stages using a data driven approach. J Neurol Neurosurg Psychiatry 2005;76:343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eggers C, Pedrosa DJ, Kahraman D, et al. Parkinson subtypes progress differently in clinical course and imaging pattern. PLoS One 2012;7:e46813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ. A clinico-pathological study of subtypes in Parkinson's disease. Brain 2009;132:2947–2957. [DOI] [PubMed] [Google Scholar]
- 4.Parkkinen L, O'Sullivan SS, Collins C, et al. Disentangling the relationship between Lewy bodies and nigral neuronal loss in Parkinson's disease. J Parkinsons Dis 2011;1:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson's disease: a systematic review and meta-analysis. Mov Disord 2014;29:1583–1590. [DOI] [PubMed] [Google Scholar]
- 6.Kish SJ, Shannak K, Rajput A, Deck JH, Hornykiewicz O. Aging produces a specific pattern of striatal dopamine loss: implications for the etiology of idiopathic Parkinson's disease. J Neurochem 1992;58:642–648. [DOI] [PubMed] [Google Scholar]
- 7.de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol 2006;5:525–535. [DOI] [PubMed] [Google Scholar]
- 8.Kempster PA, O'Sullivan SS, Holton JL, Revesz T, Lees AJ. Relationships between age and late progression of Parkinson's disease: a clinico-pathological study. Brain 2010;133:1755–1762. [DOI] [PubMed] [Google Scholar]
- 9.Critchley M. The neurology of old age. Lancet 1931;1:1221–1230. [Google Scholar]
- 10.Wickremaratchi MM, Ben-Shlomo Y, Morris HR. The effect of onset age on the clinical features of Parkinson's disease. Eur J Neurol 2009;16:450–456. [DOI] [PubMed] [Google Scholar]
- 11.Quinn N, Critchley P, Marsden CD. Young onset Parkinson's disease. Mov Disord 1987;2:73–91. [DOI] [PubMed] [Google Scholar]
- 12.Gomez Arevalo G, Jorge R, Garcia S, Scipioni O, Gershanik O. Clinical and pharmacological differences in early- versus late-onset Parkinson's disease. Mov Disord 1997;12:277–284. [DOI] [PubMed] [Google Scholar]
- 13.Wickremaratchi MM, Perera D, O'Loghlen C, et al. Prevalence and age of onset of Parkinson's disease in Cardiff: a community based cross sectional study and meta-analysis. J Neurol Neurosurg Psychiatry 2009;80:805–807. [DOI] [PubMed] [Google Scholar]
- 14.Schrag A, Ben-Shlomo Y, Brown R, Marsden CD, Quinn N. Young-onset Parkinson's disease revisited clinical features, natural history, and mortality. Mov Disord 1998;13:885–894. [DOI] [PubMed] [Google Scholar]
- 15.Golbe LI. Young-onset Parkinson's disease: a clinical review. Neurology 1991;41:168–173. [DOI] [PubMed] [Google Scholar]
- 16.Wickremaratchi MM, Knipe MD, Sastry BS, et al. The motor phenotype of Parkinson's disease in relation to age at onset. Mov Disord 2011;26:457–463. [DOI] [PubMed] [Google Scholar]
- 17.Mehanna R, Moore S, Hou JG, Sarwar AI, Lai EC. Comparing clinical features of young onset, middle onset and late onset Parkinson's disease. Parkinsonism Relat Disord 2014;20:530–534. [DOI] [PubMed] [Google Scholar]
- 18.Szewczyk-Krolikowski K, Tomlinson P, Nithi K, et al. The influence of age and gender on motor and non-motor features of early Parkinson's disease: initial findings from the Oxford Parkinson Disease Center (OPDC) discovery cohort. Parkinsonism Relat Disord 2014;20:99–105. [DOI] [PubMed] [Google Scholar]
- 19.Parkinson Progression Marker Initiative. The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol 2011;95:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Post B, Speelman JD, de Haan RJ; CARPA-study group. Clinical heterogeneity in newly diagnosed Parkinson's disease. J Neurol 2008;255:716–722. [DOI] [PubMed] [Google Scholar]
- 21.Kang JH, Irwin DJ, Chen-Plotkin AS, et al. ; Parkinson's Progression Markers Initiative. Association of cerebrospinal fluid β-amyloid 1-42, T-tau, P-tau181, and α-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol 2013;70:1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berg D, Postuma RB, Bloem B, et al. Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson's disease. Mov Disord 2014;29:454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marras C, Lang A. Parkinson's disease subtypes: lost in translation? J Neurol Neurosurg Psychiatry 2013;844:409–415. [DOI] [PubMed] [Google Scholar]
- 24.van der Hoorn A, Burger H, Leenders KL, de Jong BM. Handedness correlates with the dominant Parkinson side: a systematic review and meta-analysis. Mov Disord 2012;27:206–210. [DOI] [PubMed] [Google Scholar]
- 25.de la Fuente-Fernández R, Kishore A, Calne DB, Ruth TJ, Stoessl AJ. Nigrostriatal dopamine system and motor lateralization. Behav Brain Res 2000;112:63–68. [DOI] [PubMed] [Google Scholar]
- 26.Vidailhet M, Bonnet AM, Marconi R, Gouider-Khouja N, Agid Y. Do parkinsonian symptoms and levodopa-induced dyskinesias start in the foot? Neurology 1994;44:1613–1616. [DOI] [PubMed] [Google Scholar]
- 27.Dickson JM, Grunewald RA. Somatic symptom progression in idiopathic Parkinson's disease. Parkinsonism Relat Disord 2004;10:487–492. [DOI] [PubMed] [Google Scholar]
- 28.Rowland T, Chan HC, Brooks DJ, Pavese N. Progression of nigrostriatal projection functional loss through the striatum in Parkinson's disease: a clinico-PET correlation. MDS 17th International Congress of Parkinson's Disease and Movement Disorders, Volume 28; June 2013. Abstract Supplement.
- 29.Yurkovich M, Avina-Zubieta JA, Thomas J, Gorenchtein M, Lacaille D. A systematic review identifies valid comorbidity indices derived from administrative health data. J Clin Epidemiol 2015;68:3–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.